
Nonsense-Mediated mRNA Decay, a Finely Regulated Mechanism


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. NMD Reaction and Regulation
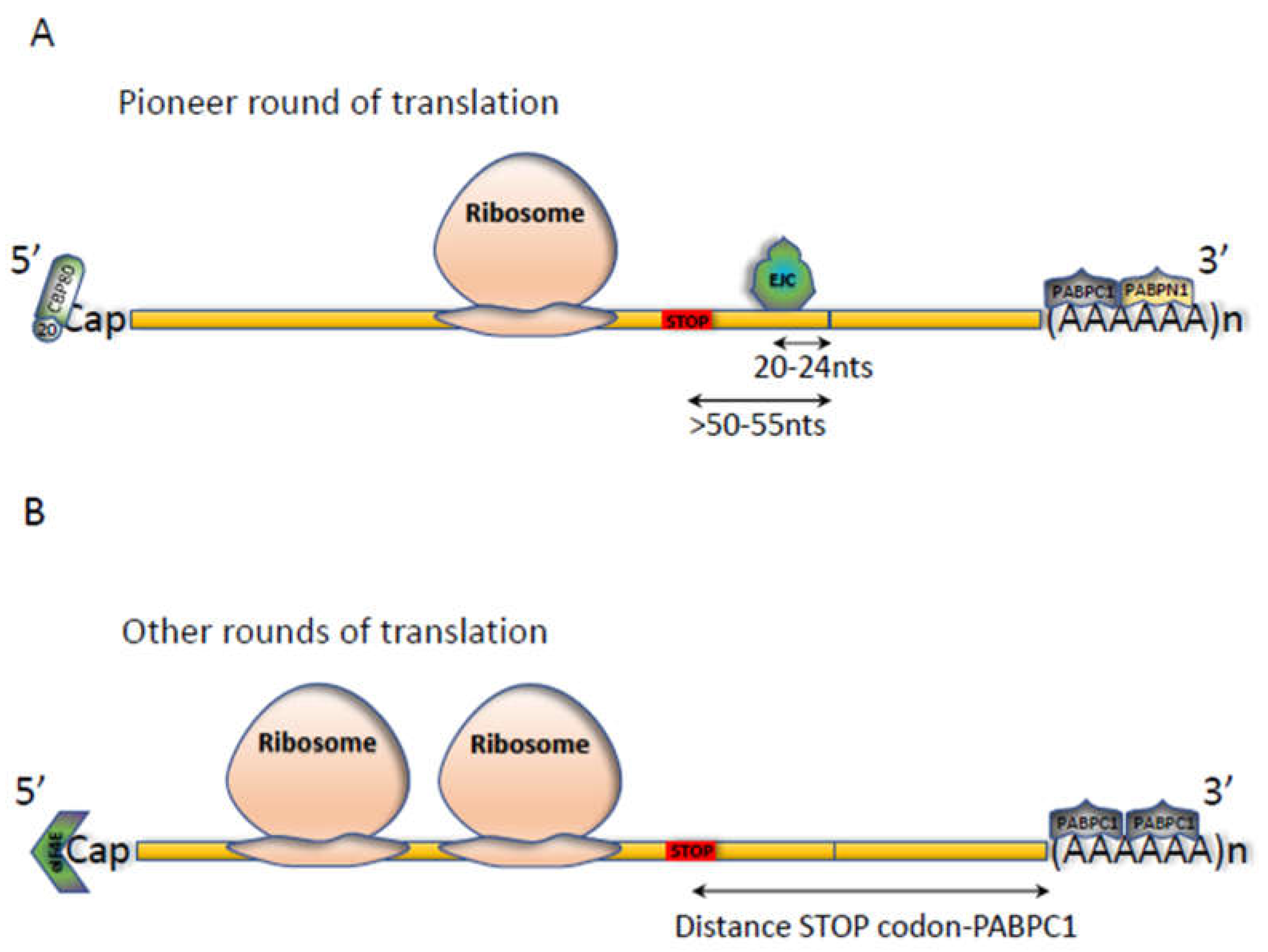
2.1. The NMD Reaction
2.2. Tissue and Cell-Type Specificity
2.3. Regulation during Differentiation
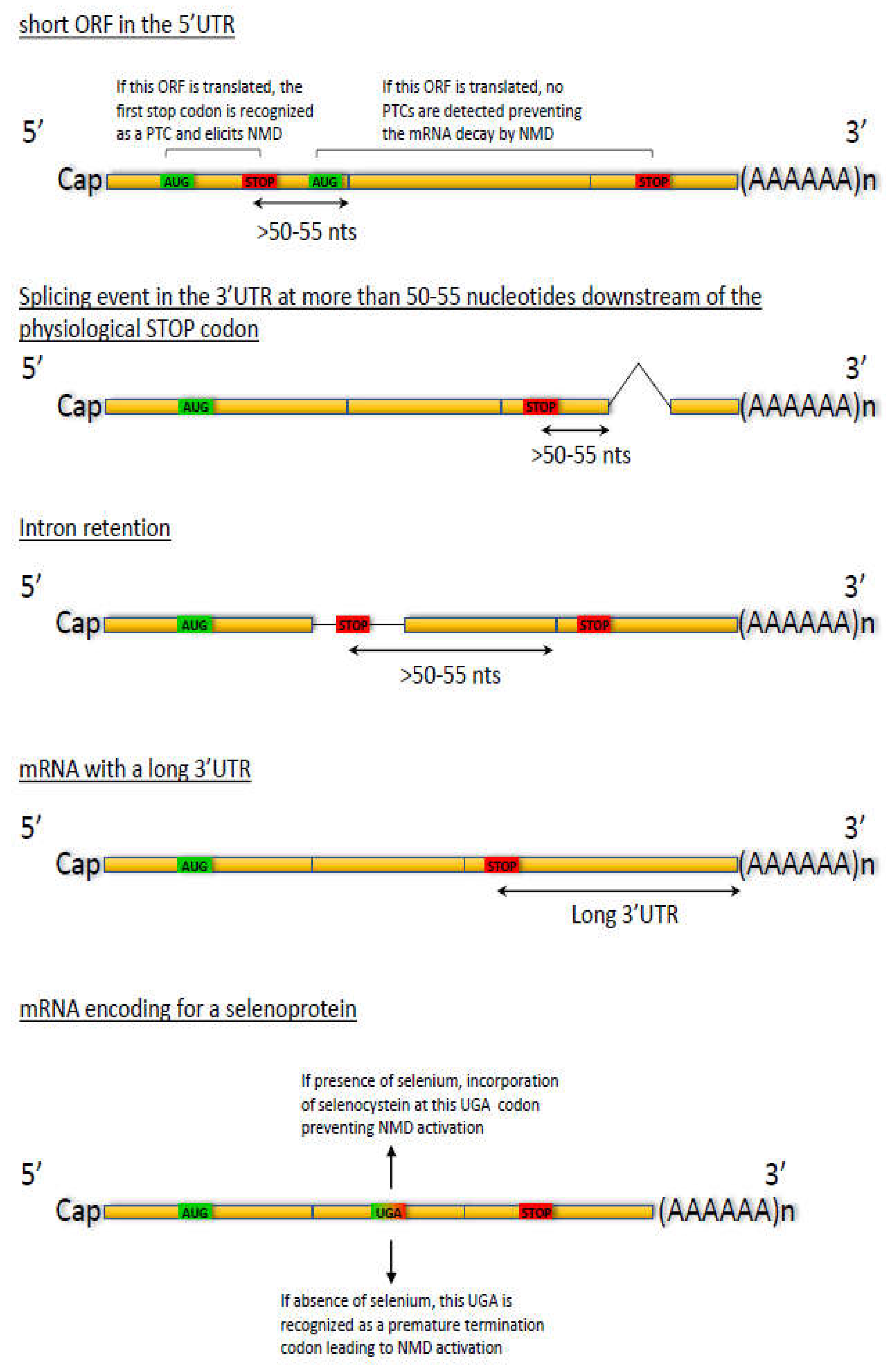
2.4. Regulation through Splicing
2.5. Regulation Via the Endoplasmic Reticulum (ER)
2.6. Regulation by Calcium
2.7. Regulation during Apoptosis
2.8. Regulation in Cancer
2.9. Autoregulation
3. Conclusions
4. Prospects for Therapy Development
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hwang, H.J.; Park, Y.; Kim, Y.K. UPF1: From mRNA Surveillance to Protein Quality Control. Biomedicines 2021, 9, 995. [Google Scholar] [CrossRef]
- Karousis, E.D.; Nasif, S.; Muhlemann, O. Nonsense-mediated mRNA decay: Novel mechanistic insights and biological impact. Wiley Interdiscip Rev. RNA 2016, 7, 661–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, G.; Fernandes, R.; Garcia-Moreno, J.F.; Romao, L. Nonsense-mediated RNA decay and its bipolar function in cancer. Mol. Cancer 2021, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Maquat, L.E. Nonsense-mediated mRNA Decay and Cancer. Curr Opin Genet. Dev. 2018, 48, 44–50. [Google Scholar] [CrossRef]
- Ishigaki, Y.; Li, X.; Serin, G.; Maquat, L.E. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell 2001, 106, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Eberle, A.B.; Lykke-Andersen, S.; Muhlemann, O.; Jensen, T.H. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat. Struct Mol. Biol 2009, 16, 49–55. [Google Scholar] [CrossRef]
- Huntzinger, E.; Kashima, I.; Fauser, M.; Sauliere, J.; Izaurralde, E. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 2008, 14, 2609–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lejeune, F.; Li, X.; Maquat, L.E. Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol. Cell 2003, 12, 675–687. [Google Scholar] [CrossRef]
- Buhler, M.; Steiner, S.; Mohn, F.; Paillusson, A.; Muhlemann, O. EJC-independent degradation of nonsense immunoglobulin-mu mRNA depends on 3’ UTR length. Nat. Struct Mol. Biol 2006, 13, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Rebbapragada, I.; Lykke-Andersen, J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol 2008, 6, e111. [Google Scholar] [CrossRef]
- Mendell, J.T.; Sharifi, N.A.; Meyers, J.L.; Martinez-Murillo, F.; Dietz, H.C. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 2004, 36, 1073–1078. [Google Scholar] [CrossRef] [Green Version]
- Lejeune, F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 2017, 50, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Sun, X.; Qian, Y.; LaDuca, J.P.; Maquat, L.E. At least one intron is required for the nonsense-mediated decay of triosephosphate isomerase mRNA: A possible link between nuclear splicing and cytoplasmic translation. Mol Cell Biol 1998, 18, 5272–5283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Hir, H.; Izaurralde, E.; Maquat, L.E.; Moore, M.J. The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. EMBO J 2000, 19, 6860–6869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoda, N.; Kim, Y.K.; Lejeune, F.; Maquat, L.E. CBP80 promotes interaction of Upf1 with Upf2 during nonsense-mediated mRNA decay in mammalian cells. Nat. Struct Mol. Biol 2005, 12, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Neu-Yilik, G.; Raimondeau, E.; Eliseev, B.; Yeramala, L.; Amthor, B.; Deniaud, A.; Huard, K.; Kerschgens, K.; Hentze, M.W.; Schaffitzel, C.; et al. Dual function of UPF3B in early and late translation termination. EMBO J. 2017, 36, 2968–2986. [Google Scholar] [CrossRef]
- Kashima, I.; Yamashita, A.; Izumi, N.; Kataoka, N.; Morishita, R.; Hoshino, S.; Ohno, M.; Dreyfuss, G.; Ohno, S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006, 20, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Franks, T.M.; Singh, G.; Lykke-Andersen, J. Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense- mediated mRNA decay. Cell 2010, 143, 938–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, B.; Jonas, S.; Izaurralde, E. The SMG5-SMG7 heterodimer directly recruits the CCR4-NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev. 2013, 27, 2125–2138. [Google Scholar] [CrossRef] [Green Version]
- Eberle, A.B.; Stalder, L.; Mathys, H.; Orozco, R.Z.; Muhlemann, O. Posttranscriptional gene regulation by spatial rearrangement of the 3’ untranslated region. PLoS Biol 2008, 6, e92. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Maquat, L.E. Rules that govern UPF1 binding to mRNA 3’ UTRs. Proc. Natl. Acad. Sci. USA 2013, 110, 3357–3362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeoka, T.; Kato, S.; Kawaichi, M.; Ishida, Y. Evidence that the Upf1-related molecular motor scans the 3’-UTR to ensure mRNA integrity. Nucleic Acids Res. 2012, 40, 6887–6897. [Google Scholar] [CrossRef] [Green Version]
- Hogg, J.R.; Goff, S.P. Upf1 senses 3’UTR length to potentiate mRNA decay. Cell 2010, 143, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Serin, G.; Gersappe, A.; Black, J.D.; Aronoff, R.; Maquat, L.E. Identification and characterization of human orthologues to Saccharomyces cerevisiae Upf2 protein and Upf3 protein (Caenorhabditis elegans SMG-4). Mol. Cell Biol 2001, 21, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Bateman, J.F.; Freddi, S.; Nattrass, G.; Savarirayan, R. Tissue-specific RNA surveillance? Nonsense-mediated mRNA decay causes collagen X haploinsufficiency in Schmid metaphyseal chondrodysplasia cartilage. Hum. Mol. Genet. 2003, 12, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Viegas, M.H.; Gehring, N.H.; Breit, S.; Hentze, M.W.; Kulozik, A.E. The abundance of RNPS1, a protein component of the exon junction complex, can determine the variability in efficiency of the Nonsense Mediated Decay pathway. Nucleic Acids Res. 2007, 35, 4542–4551. [Google Scholar] [CrossRef] [Green Version]
- Zetoune, A.B.; Fontaniere, S.; Magnin, D.; Anczukow, O.; Buisson, M.; Zhang, C.X.; Mazoyer, S. Comparison of nonsense-mediated mRNA decay efficiency in various murine tissues. BMC Genet. 2008, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Addington, A.M.; Gauthier, J.; Piton, A.; Hamdan, F.F.; Raymond, A.; Gogtay, N.; Miller, R.; Tossell, J.; Bakalar, J.; Inoff-Germain, G.; et al. A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol. Psychiatry 2011, 16, 238–239. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Raymond, F.L.; Nguyen, L.S.; Rodriguez, J.; Hackett, A.; Vandeleur, L.; Smith, R.; Shoubridge, C.; Edkins, S.; Stevens, C.; et al. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet. 2007, 39, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.A.; Nguyen, L.S.; Ng, L.Y.; Waldron, M.; McDonald, D.; Gecz, J. Broadening the phenotype associated with mutations in UPF3B: Two further cases with renal dysplasia and variable developmental delay. Eur J. Med. Genet. 2012, 55, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Laumonnier, F.; Shoubridge, C.; Antar, C.; Nguyen, L.S.; Van Esch, H.; Kleefstra, T.; Briault, S.; Fryns, J.P.; Hamel, B.; Chelly, J.; et al. Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol. Psychiatry 2010, 15, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.L.; Stoica, L.; Liu, Y.; Zhu, P.J.; Bhattacharya, A.; Buffington, S.A.; Huq, R.; Eissa, N.T.; Larsson, O.; Porse, B.T.; et al. Inhibition of Upf2-Dependent Nonsense-Mediated Decay Leads to Behavioral and Neurophysiological Abnormalities by Activating the Immune Response. Neuron 2019, 104, 665–679 e668. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Berg, J.S.; Scaglia, F.; Belmont, J.; Bacino, C.A.; Sahoo, T.; Lalani, S.R.; Graham, B.; Lee, B.; Shinawi, M.; et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008, 40, 1466–1471. [Google Scholar] [CrossRef]
- Sartor, F.; Anderson, J.; McCaig, C.; Miedzybrodzka, Z.; Muller, B. Mutation of genes controlling mRNA metabolism and protein synthesis predisposes to neurodevelopmental disorders. Biochem Soc. Trans. 2015, 43, 1259–1265. [Google Scholar] [CrossRef]
- Shaheen, R.; Anazi, S.; Ben-Omran, T.; Seidahmed, M.Z.; Caddle, L.B.; Palmer, K.; Ali, R.; Alshidi, T.; Hagos, S.; Goodwin, L.; et al. Mutations in SMG9, Encoding an Essential Component of Nonsense-Mediated Decay Machinery, Cause a Multiple Congenital Anomaly Syndrome in Humans and Mice. Am. J. Hum. Genet. 2016, 98, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.S.; Kim, H.G.; Rosenfeld, J.A.; Shen, Y.; Gusella, J.F.; Lacassie, Y.; Layman, L.C.; Shaffer, L.G.; Gecz, J. Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum. Mol. Genet. 2013, 22, 1816–1825. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.; Jones, S.H.; Lake, B.B.; Dumdie, J.N.; Shum, E.Y.; Zhang, L.; Chen, S.; Sohni, A.; Pandya, S.; Gallo, R.L.; et al. The role of the NMD factor UPF3B in olfactory sensory neurons. Elife 2020, 9. [Google Scholar] [CrossRef]
- Colak, D.; Ji, S.J.; Porse, B.T.; Jaffrey, S.R. Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell 2013, 153, 1252–1265. [Google Scholar] [CrossRef] [Green Version]
- Bruno, I.G.; Karam, R.; Huang, L.; Bhardwaj, A.; Lou, C.H.; Shum, E.Y.; Song, H.W.; Corbett, M.A.; Gifford, W.D.; Gecz, J.; et al. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol. Cell 2011, 42, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4. [Google Scholar] [CrossRef]
- Lou, C.H.; Shao, A.; Shum, E.Y.; Espinoza, J.L.; Huang, L.; Karam, R.; Wilkinson, M.F. Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep. 2014, 6, 748–764. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Jiang, B.; Jia, C.; Chai, B.; Liang, A. MicroRNA 125 represses nonsense-mediated mRNA decay by regulating SMG1 expression. Biochem Biophys Res. Commun 2013, 435, 16–20. [Google Scholar] [CrossRef]
- Gong, C.; Kim, Y.K.; Woeller, C.F.; Tang, Y.; Maquat, L.E. SMD and NMD are competitive pathways that contribute to myogenesis: Effects on PAX3 and myogenin mRNAs. Genes Dev. 2009, 23, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, C.F.; Lejeune, F.; Stevenin, J. Broad specificity of SR (serine/arginine) proteins in the regulation of alternative splicing of pre-messenger RNA. Prog Nucleic Acid Res. Mol. Biol. 2004, 78, 37–88. [Google Scholar] [CrossRef]
- Braunschweig, U.; Gueroussov, S.; Plocik, A.M.; Graveley, B.R.; Blencowe, B.J. Dynamic integration of splicing within gene regulatory pathways. Cell 2013, 152, 1252–1269. [Google Scholar] [CrossRef] [Green Version]
- Chabot, B.; Shkreta, L. Defective control of pre-messenger RNA splicing in human disease. J. Cell Biol 2016, 212, 13–27. [Google Scholar] [CrossRef]
- Ohnishi, T.; Yamashita, A.; Kashima, I.; Schell, T.; Anders, K.R.; Grimson, A.; Hachiya, T.; Hentze, M.W.; Anderson, P.; Ohno, S. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol. Cell 2003, 12, 1187–1200. [Google Scholar] [CrossRef]
- Gowravaram, M.; Bonneau, F.; Kanaan, J.; Maciej, V.D.; Fiorini, F.; Raj, S.; Croquette, V.; Le Hir, H.; Chakrabarti, S. A conserved structural element in the RNA helicase UPF1 regulates its catalytic activity in an isoform-specific manner. Nucleic Acids Res. 2018, 46, 2648–2659. [Google Scholar] [CrossRef] [Green Version]
- Padariya, M.; Fahraeus, R.; Hupp, T.; Kalathiya, U. Molecular Determinants and Specificity of mRNA with Alternatively-Spliced UPF1 Isoforms, Influenced by an Insertion in the ‘Regulatory Loop’. Int. J. Mol. Sci. 2021, 22, 12744. [Google Scholar] [CrossRef]
- Longman, D.; Jackson-Jones, K.A.; Maslon, M.M.; Murphy, L.C.; Young, R.S.; Stoddart, J.J.; Hug, N.; Taylor, M.S.; Papadopoulos, D.K.; Caceres, J.F. Identification of a localized nonsense-mediated decay pathway at the endoplasmic reticulum. Genes Dev. 2020, 34, 1075–1088. [Google Scholar] [CrossRef]
- Chiu, S.Y.; Lejeune, F.; Ranganathan, A.C.; Maquat, L.E. The pioneer translation initiation complex is functionally distinct from but structurally overlaps with the steady-state translation initiation complex. Genes Dev 2004, 18, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Nickless, A.; Jackson, E.; Marasa, J.; Nugent, P.; Mercer, R.W.; Piwnica-Worms, D.; You, Z. Intracellular calcium regulates nonsense-mediated mRNA decay. Nat Med 2014, 20, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Tantral, L.; Malathi, K.; Kohyama, S.; Silane, M.; Berenstein, A.; Jayaraman, T. Intracellular calcium release is required for caspase-3 and -9 activation. Cell Biochem Funct 2004, 22, 35–40. [Google Scholar] [CrossRef]
- Jia, J.; Furlan, A.; Gonzalez-Hilarion, S.; Leroy, C.; Gruenert, D.C.; Tulasne, D.; Lejeune, F. Caspases shutdown nonsense-mediated mRNA decay during apoptosis. Cell Death Differ 2015, 22, 1754–1763. [Google Scholar] [CrossRef] [Green Version]
- Popp, M.W.; Maquat, L.E. Attenuation of nonsense-mediated mRNA decay facilitates the response to chemotherapeutics. Nat Commun 2015, 6, 6632. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Vuong, J.K.; Zhang, M.; Stork, C.; Zheng, S. Inhibition of nonsense-mediated RNA decay by ER stress. RNA 2017, 23, 378–394. [Google Scholar] [CrossRef]
- Chang, L.; Li, C.; Guo, T.; Wang, H.; Ma, W.; Yuan, Y.; Liu, Q.; Ye, Q.; Liu, Z. The human RNA surveillance factor UPF1 regulates tumorigenesis by targeting Smad7 in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 8. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Wang, S.; Yao, Y.; Li, Y.; Zhang, H.; Han, F.; Nie, H.; Su, J.; Wang, Z.; Yue, L.; et al. Biological and clinical significance of epigenetic silencing of MARVELD1 gene in lung cancer. Sci. Rep. 2014, 4, 7545. [Google Scholar] [CrossRef] [Green Version]
- Bokhari, A.; Jonchere, V.; Lagrange, A.; Bertrand, R.; Svrcek, M.; Marisa, L.; Buhard, O.; Greene, M.; Demidova, A.; Jia, J.; et al. Targeting nonsense-mediated mRNA decay in colorectal cancers with microsatellite instability. Oncogenesis 2018, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Palma, M.; Leroy, C.; Salome-Desnoulez, S.; Werkmeister, E.; Kong, R.; Mongy, M.; Le Hir, H.; Lejeune, F. A role for AKT1 in nonsense-mediated mRNA decay. Nucleic Acids Res. 2021, 49, 11022–11037. [Google Scholar] [CrossRef]
- Huang, L.; Lou, C.H.; Chan, W.; Shum, E.Y.; Shao, A.; Stone, E.; Karam, R.; Song, H.W.; Wilkinson, M.F. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol. Cell 2011, 43, 950–961. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Sanjeev, M.; Singh, G. The Branched Nature of the Nonsense-Mediated mRNA Decay Pathway. Trends Genet. 2021, 37, 143–159. [Google Scholar] [CrossRef]
- Lejeune, F. Triple effect of nonsense-mediated mRNA decay inhibition on cancer. Single Cell Biology 2016, 5, 136. [Google Scholar] [CrossRef] [Green Version]
- Usuki, F.; Yamashita, A.; Higuchi, I.; Ohnishi, T.; Shiraishi, T.; Osame, M.; Ohno, S. Inhibition of nonsense-mediated mRNA decay rescues the phenotype in Ullrich’s disease. Ann. Neurol 2004, 55, 740–744. [Google Scholar] [CrossRef]
- Gonzalez-Hilarion, S.; Beghyn, T.; Jia, J.; Debreuck, N.; Berte, G.; Mamchaoui, K.; Mouly, V.; Gruenert, D.C.; Deprez, B.; Lejeune, F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 2012, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Low, A.; Damle, S.S.; Keenan, M.M.; Kuntz, S.; Murray, S.F.; Monia, B.P.; Guo, S. Antisense suppression of the nonsense mediated decay factor Upf3b as a potential treatment for diseases caused by nonsense mutations. Genome Biol. 2018, 19, 4. [Google Scholar] [CrossRef] [Green Version]
- Lindeboom, R.G.H.; Vermeulen, M.; Lehner, B.; Supek, F. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 2019. [Google Scholar] [CrossRef]
- Palma, M.; Lejeune, F. Deciphering the molecular mechanism of stop codon readthrough. Biol. Rev. Camb Philos Soc. 2021, 96, 310–329. [Google Scholar] [CrossRef]
- Martins-Dias, P.; Romao, L. Nonsense suppression therapies in human genetic diseases. Cell Mol. Life Sci. 2021, 78, 4677–4701. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lejeune, F. Nonsense-Mediated mRNA Decay, a Finely Regulated Mechanism. Biomedicines 2022, 10, 141. https://doi.org/10.3390/biomedicines10010141
Lejeune F. Nonsense-Mediated mRNA Decay, a Finely Regulated Mechanism. Biomedicines. 2022; 10(1):141. https://doi.org/10.3390/biomedicines10010141
Chicago/Turabian StyleLejeune, Fabrice. 2022. "Nonsense-Mediated mRNA Decay, a Finely Regulated Mechanism" Biomedicines 10, no. 1: 141. https://doi.org/10.3390/biomedicines10010141
APA StyleLejeune, F. (2022). Nonsense-Mediated mRNA Decay, a Finely Regulated Mechanism. Biomedicines, 10(1), 141. https://doi.org/10.3390/biomedicines10010141