
A Comparison of Doxorubicin-Resistant Colon Cancer LoVo and Leukemia HL60 Cells: Common Features, Different Underlying Mechanisms

 , , , ,
, , , ,  , and
, and 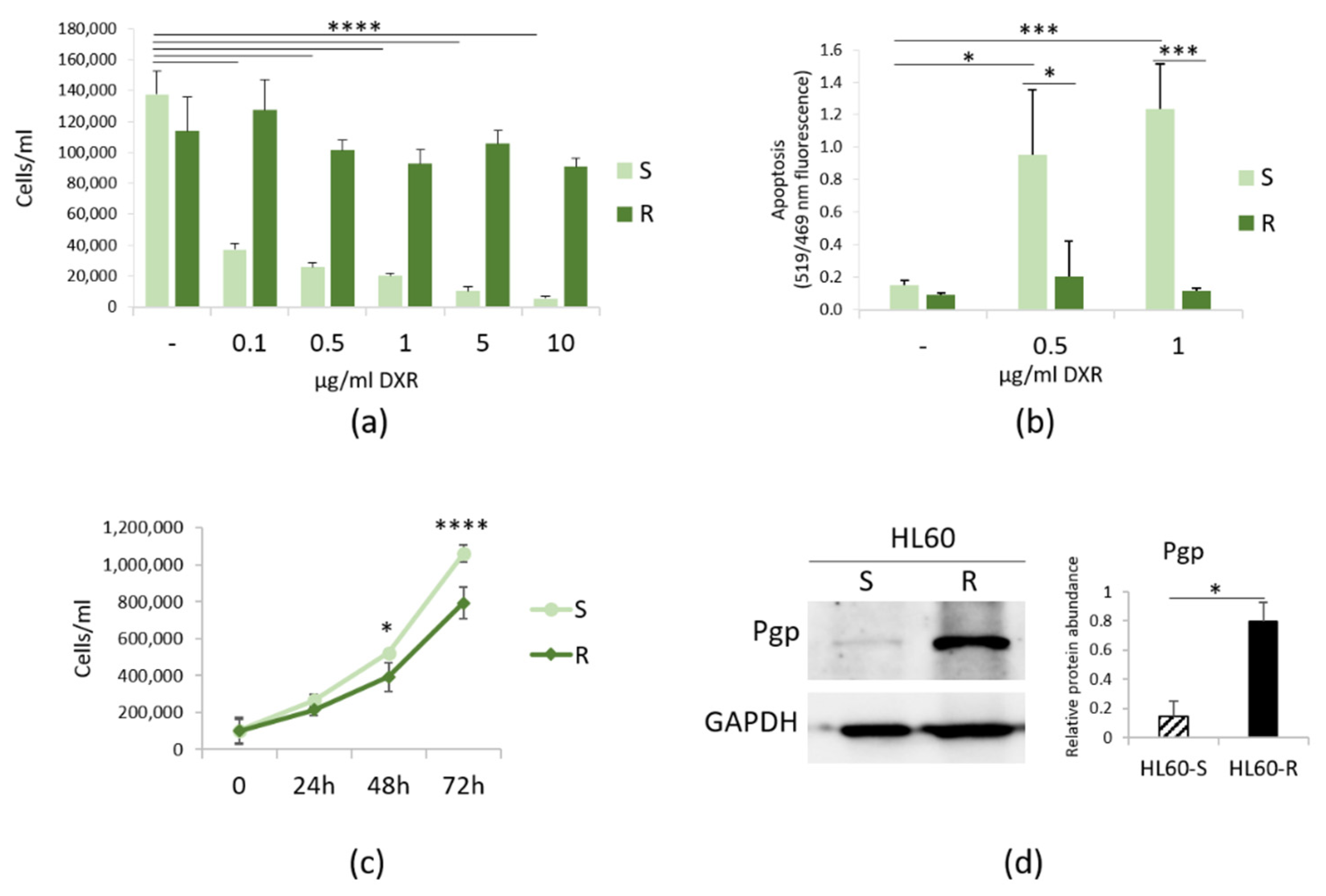{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. TUNEL Assay
2.3. Western Blot Analysis
2.4. Immunofluorescence and Confocal Imaging
2.5. Quantification of Total Intracellular Mg
2.6. Reactive Oxygen Species Production
2.7. Statistical Analysis
3. Results
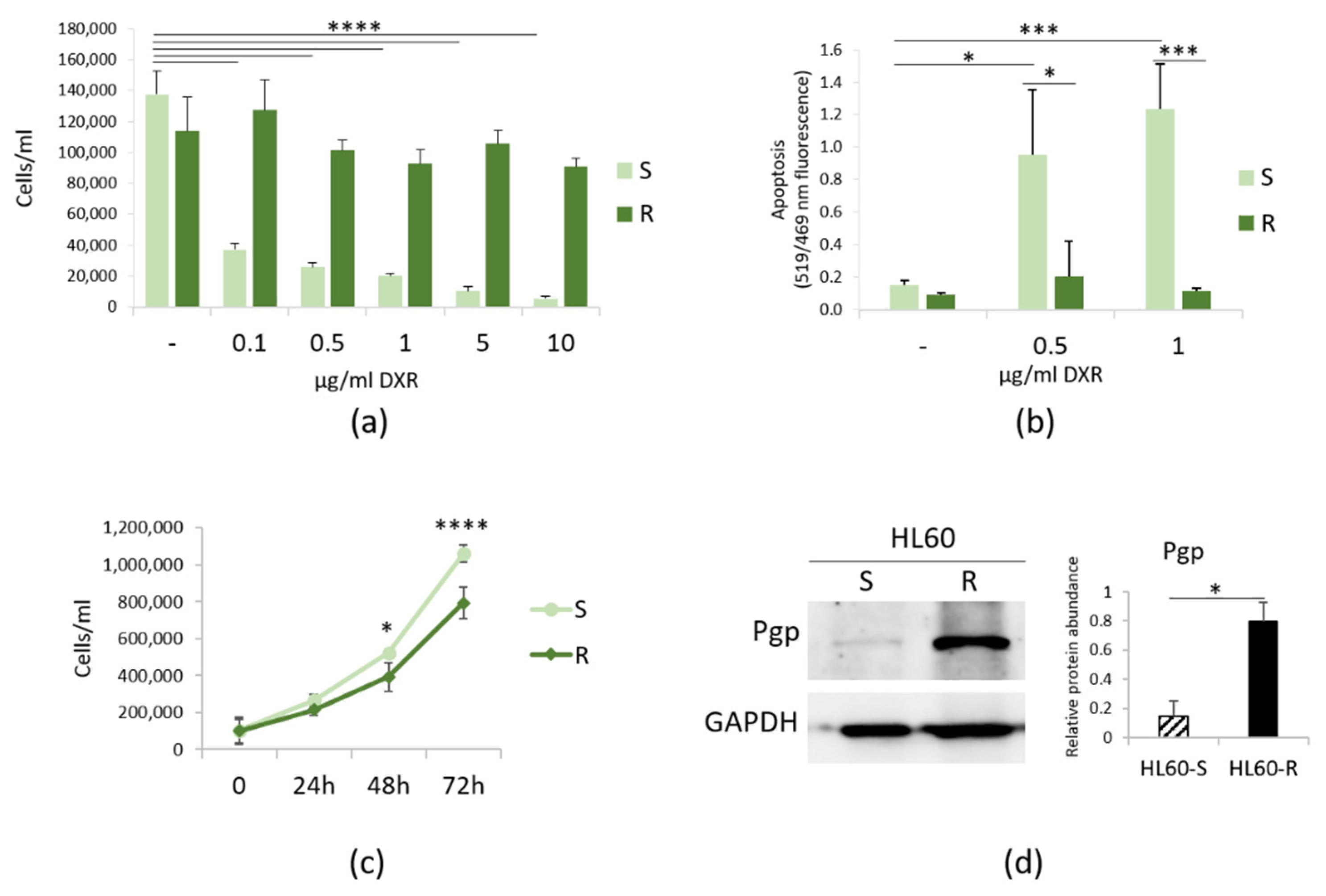
3.1. Characterization of HL60-R
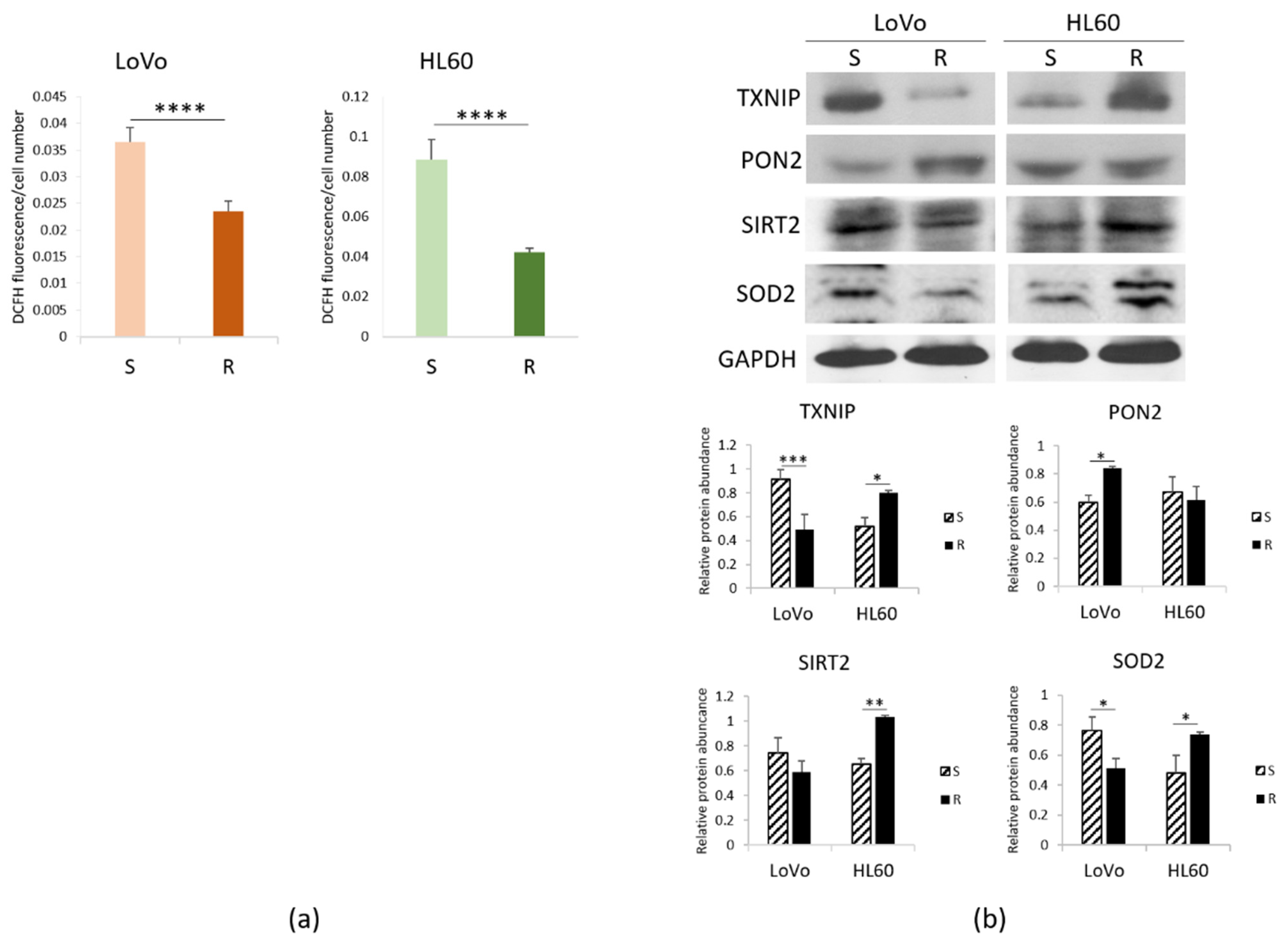
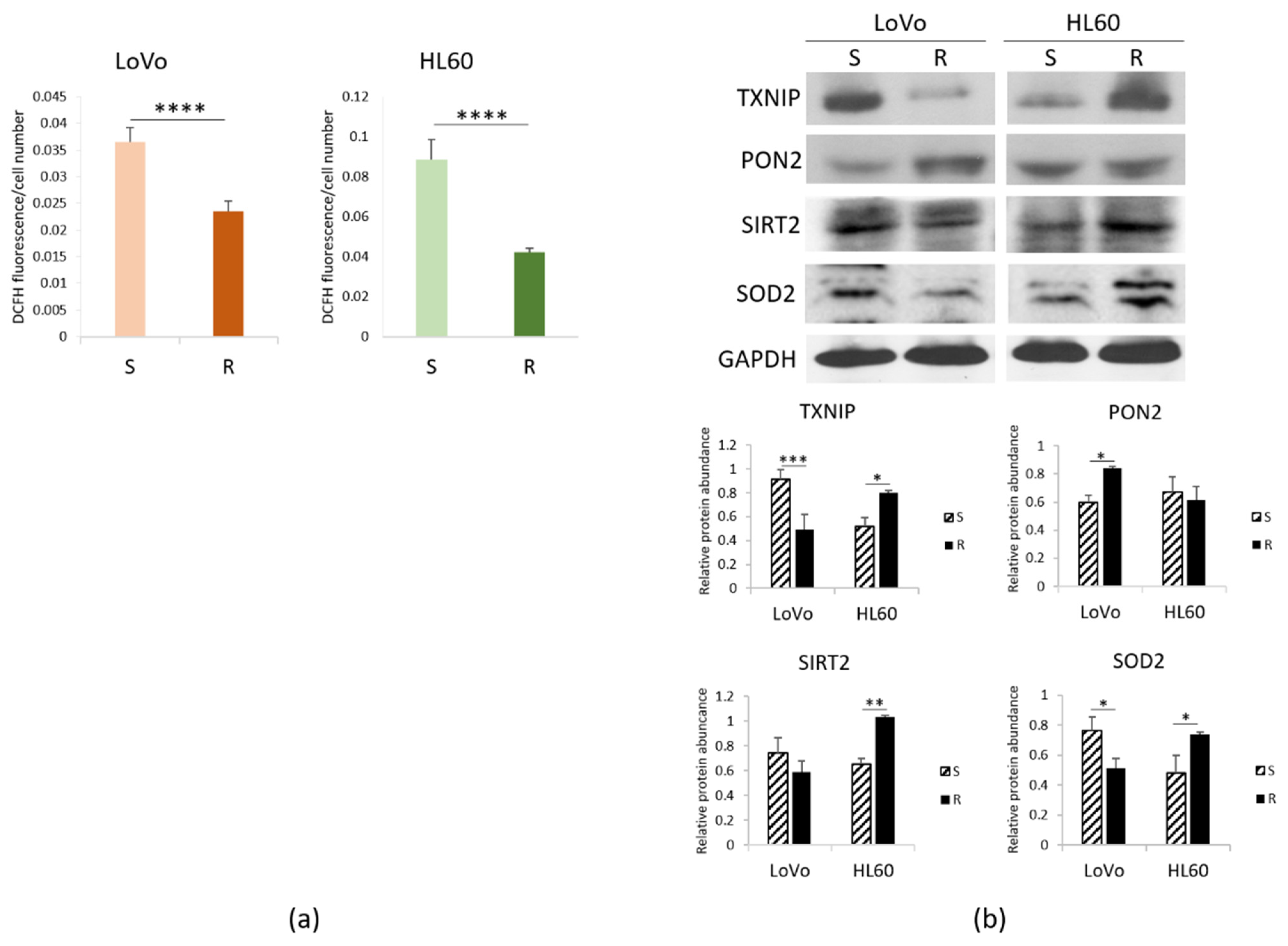
3.2. ROS in Sensitive and Resistant LoVo and HL60 Cells
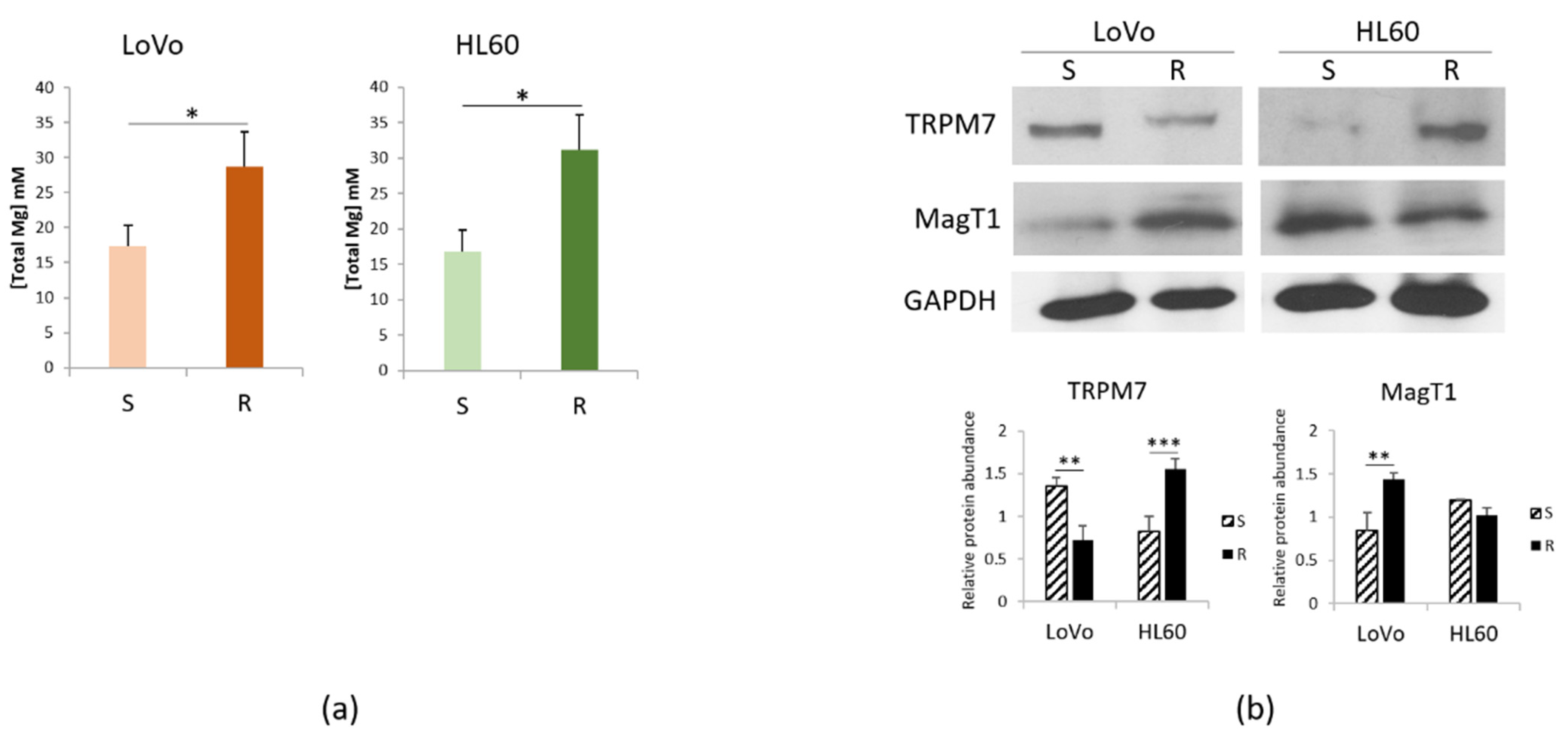
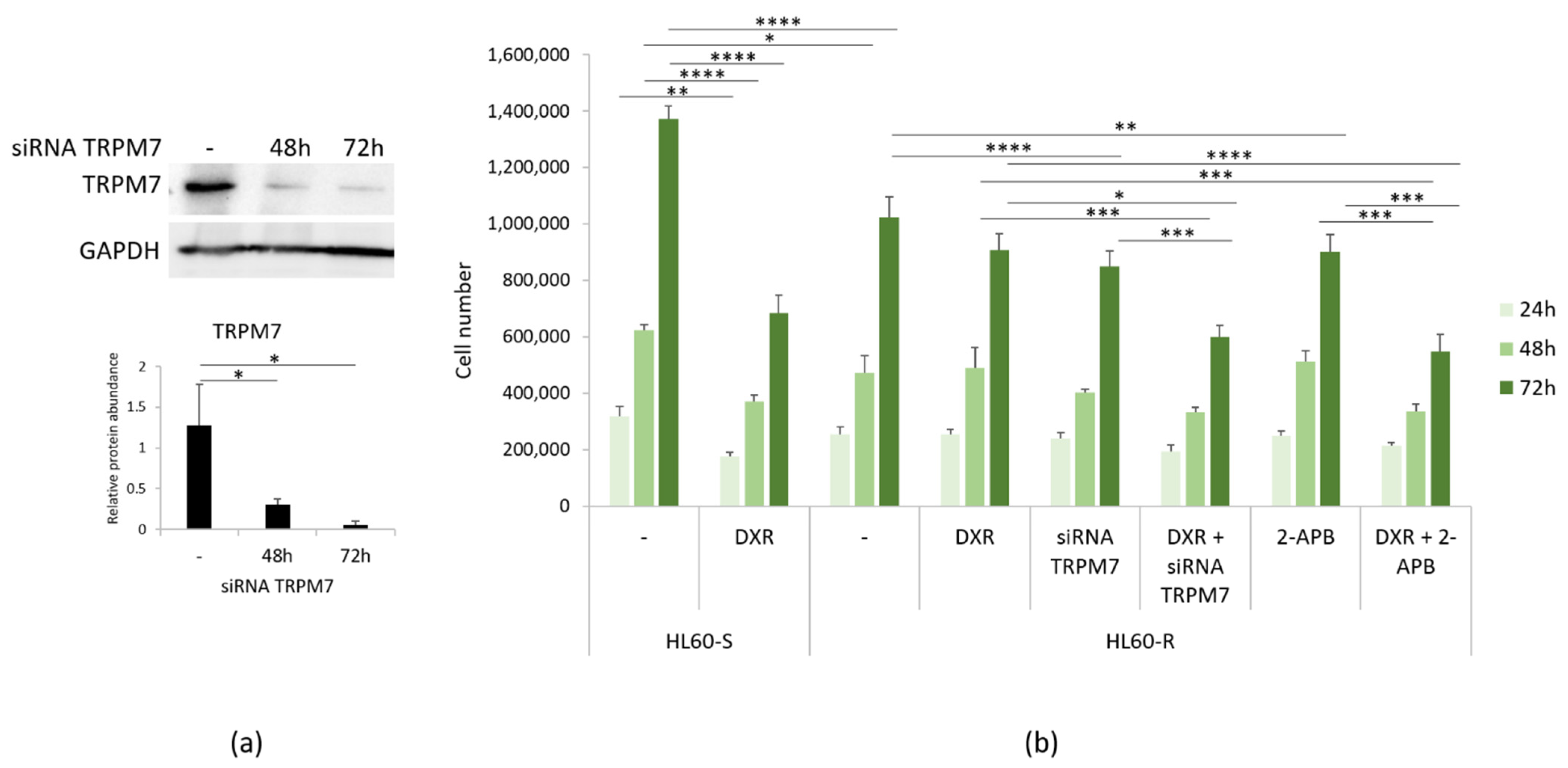
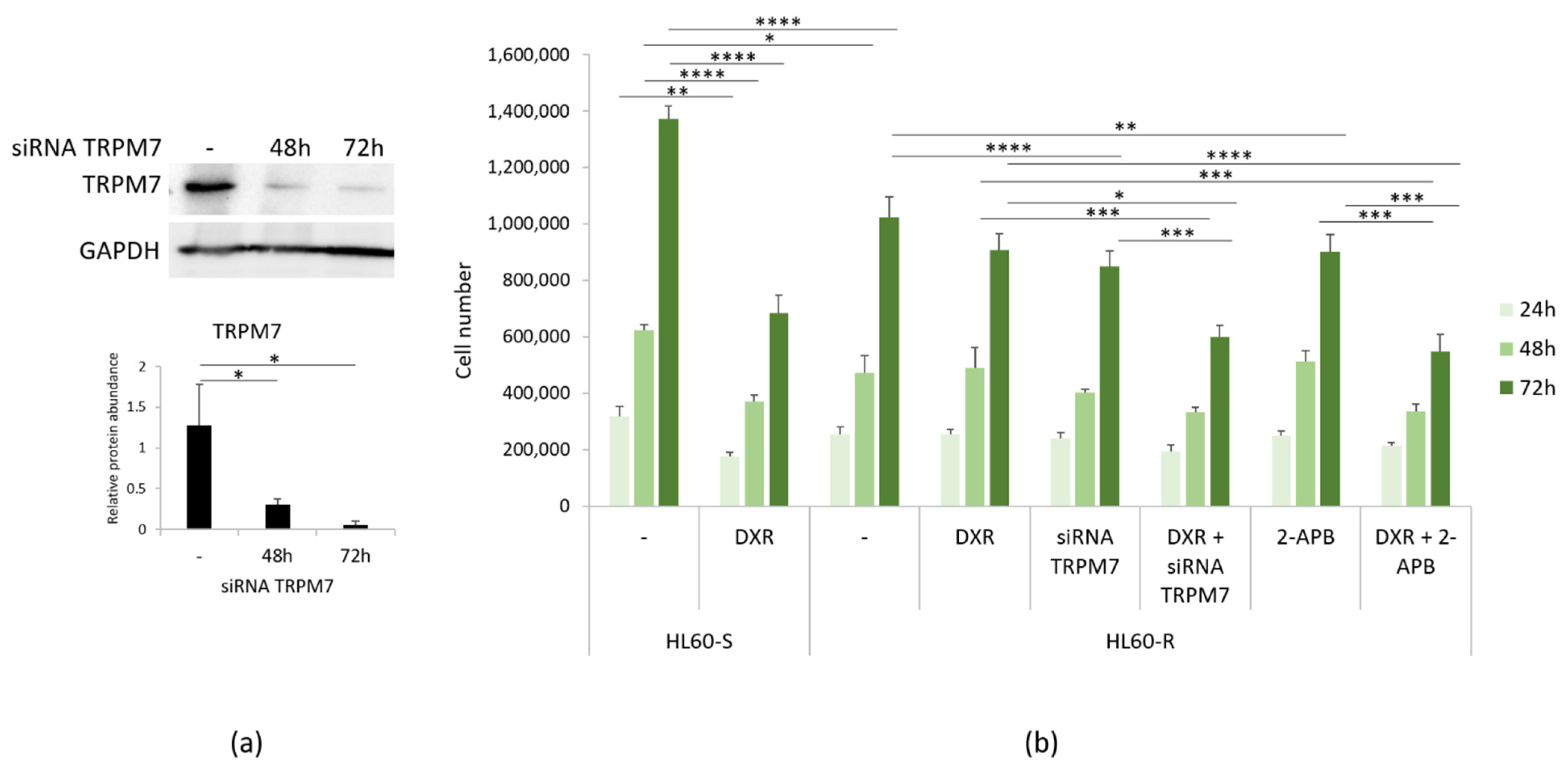
3.3. Mg Homeostasis in HL60-S and -R
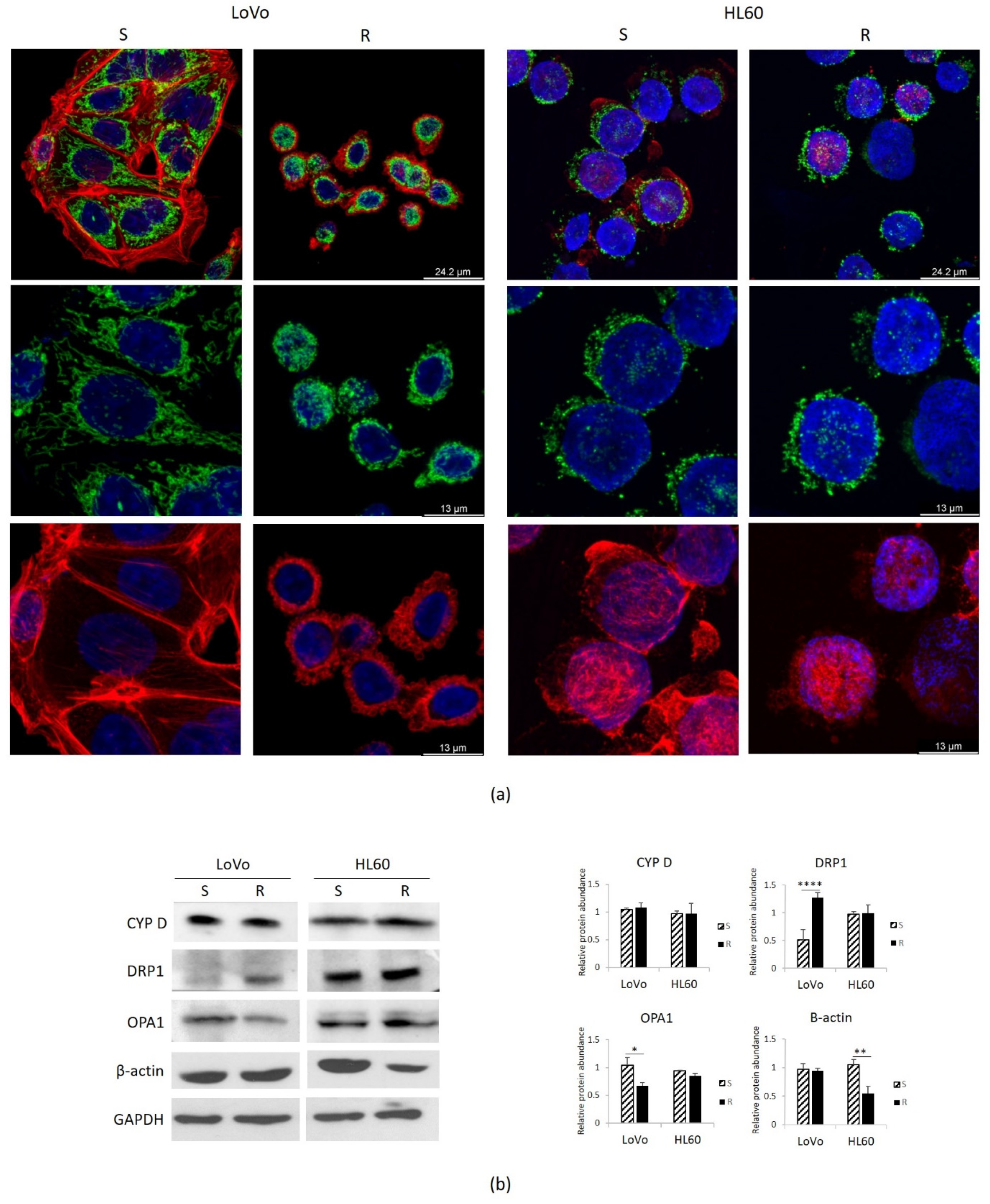
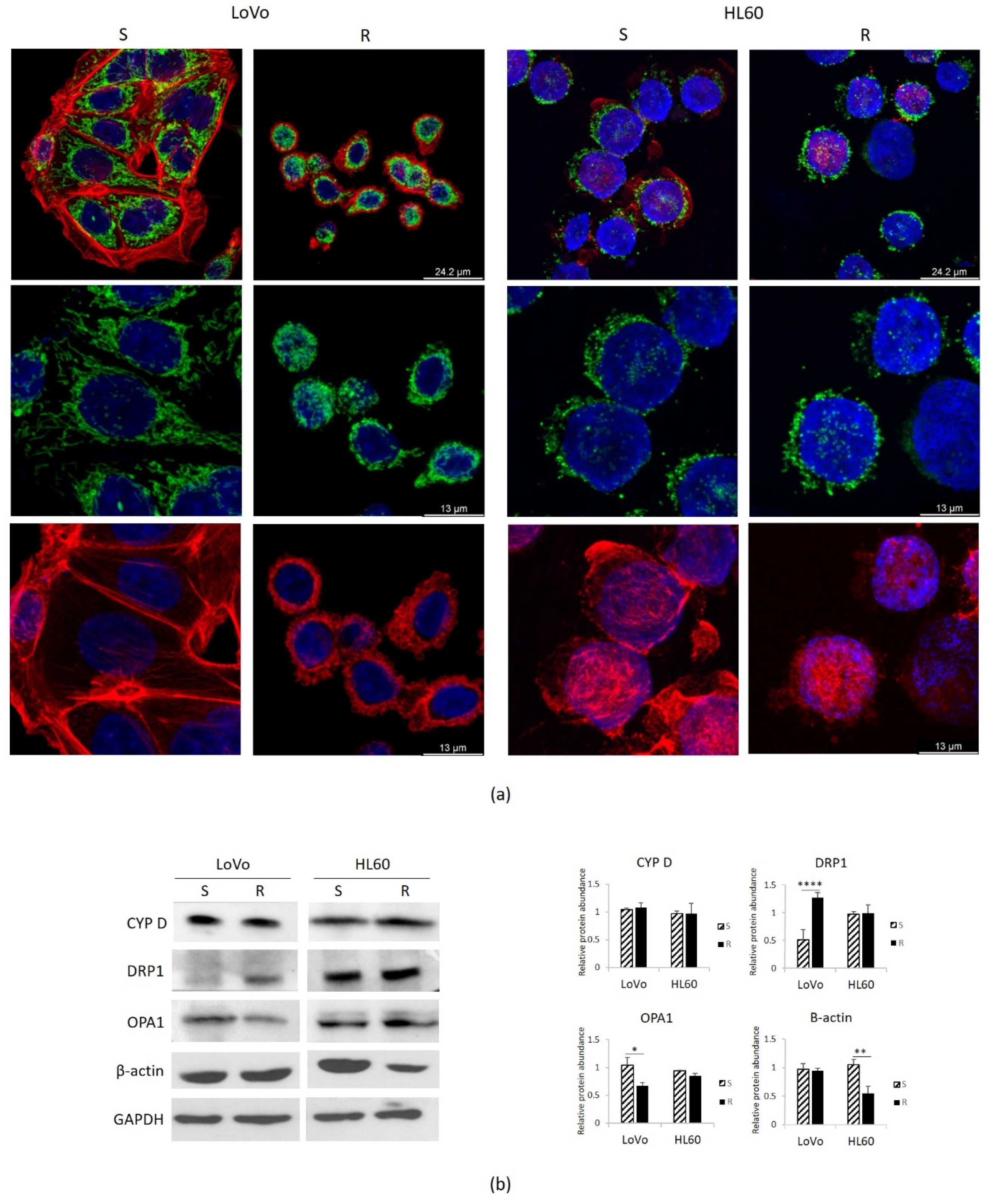
3.4. Mitochondria in DXR-Sensitive or Resistant HL60 and LoVo
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higgins, C.F. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007, 446, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Wijdeven, R.H.; Pang, B.; Assaraf, Y.G.; Neefjes, J. Old drugs, novel ways out: Drug resistance toward cytotoxic chemotherapeutics. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2016, 28, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, H.; Ashby, C.R.J.; Assaraf, Y.G.; Chen, Z.-S.; Liu, H.-M. Chemical molecular-based approach to overcome multidrug resistance in cancer by targeting P-glycoprotein (P-gp). Med. Res. Rev. 2021, 41, 525–555. [Google Scholar] [CrossRef] [PubMed]
- Cascorbi, I. P-glycoprotein: Tissue distribution, substrates, and functional consequences of genetic variations. Handb. Exp. Pharmacol. 2011, 261–283. [Google Scholar] [CrossRef]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef]
- Chen Jianli Reactive Oxygen Species and Drug Resistance in Cancer Chemotherapy. Austin J. Clin. Pathol. 2014, 1, 1017.
- Moscheni, C.; Malucelli, E.; Castiglioni, S.; Procopio, A.; De Palma, C.; Sorrentino, A.; Sartori, P.; Locatelli, L.; Pereiro, E.; Maier, J.A.; et al. 3D quantitative and ultrastructural analysis of mitochondria in a model of doxorubicin sensitive and resistant human colon carcinoma cells. Cancers 2019, 11, 1254. [Google Scholar] [CrossRef] [Green Version]
- Castiglioni, S.; Cazzaniga, A.; Trapani, V.; Cappadone, C.; Farruggia, G.; Merolle, L.; Wolf, F.I.; Iotti, S.; Maier, J.A.M. Magnesium homeostasis in colon carcinoma LoVo cells sensitive or resistant to doxorubicin. Sci. Rep. 2015, 5, 16538. [Google Scholar] [CrossRef]
- de Baaij, J.H.F.; Hoenderop, J.G.J.; Bindels, R.J.M. Magnesium in man: Implications for health and disease. Physiol. Rev. 2015, 95, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Cazzaniga, A.; Moscheni, C.; Trapani, V.; Wolf, F.I.; Farruggia, G.; Sargenti, A.; Iotti, S.; Maier, J.A.M.; Castiglioni, S. The different expression of TRPM7 and MagT1 impacts on the proliferation of colon carcinoma cells sensitive or resistant to doxorubicin. Sci. Rep. 2017, 7, 40538. [Google Scholar] [CrossRef] [Green Version]
- Auwercx, J.; Rybarczyk, P.; Kischel, P.; Dhennin-Duthille, I.; Chatelain, D.; Sevestre, H.; Van Seuningen, I.; Ouadid-Ahidouch, H.; Jonckheere, N.; Gautier, M. Mg(2+) Transporters in Digestive Cancers. Nutrients 2021, 13, 210. [Google Scholar] [CrossRef]
- Kubota, T.; Shindo, Y.; Tokuno, K.; Komatsu, H.; Ogawa, H.; Kudo, S.; Kitamura, Y.; Suzuki, K.; Oka, K. Mitochondria are intracellular magnesium stores: Investigation by simultaneous fluorescent imagings in PC12 cells. Biochim. Biophys. Acta 2005, 1744, 19–28. [Google Scholar] [CrossRef] [Green Version]
- D’Onofrio, N.; Cacciola, N.A.; Martino, E.; Borrelli, F.; Fiorino, F.; Lombardi, A.; Neglia, G.; Balestrieri, M.L.; Campanile, G. ROS-Mediated Apoptotic Cell Death of Human Colon Cancer LoVo Cells by Milk δ-Valerobetaine. Sci. Rep. 2020, 10, 8978. [Google Scholar] [CrossRef]
- Collins, S.J.; Gallo, R.C.; Gallagher, R.E. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature 1977, 270, 347–349. [Google Scholar] [CrossRef]
- Birnie, G.D. The HL60 cell line: A model system for studying human myeloid cell differentiation. Br. J. Cancer. Suppl. 1988, 9, 41–45. [Google Scholar] [PubMed]
- Wolf, D.; Rotter, V. Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc. Natl. Acad. Sci. USA 1985, 82, 790–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Wei, H.; Cheng, J.; Xie, B.; Wang, B.; Yi, J.; Tian, B.; Liu, Z.; Wang, F.; Zhang, Z. Characteristics of doxorubicin-selected multidrug-resistant human leukemia HL-60 cells with tolerance to arsenic trioxide and contribution of leukemia stem cells. Oncol. Lett. 2018, 15, 1255–1262. [Google Scholar] [CrossRef]
- Sargenti, A.; Castiglioni, S.; Olivi, E.; Bianchi, F.; Cazzaniga, A.; Farruggia, G.; Cappadone, C.; Merolle, L.; Malucelli, E.; Ventura, C.; et al. Magnesium Deprivation Potentiates Human Mesenchymal Stem Cell Transcriptional Remodeling. Int. J. Mol. Sci. 2018, 19, 1410. [Google Scholar] [CrossRef] [Green Version]
- Vezmar, M.; Georges, E. Reversal of MRP-mediated doxorubicin resistance with quinoline-based drugs. Biochem. Pharmacol. 2000, 59, 1245–1252. [Google Scholar] [CrossRef]
- Shen, F.; Chu, S.; Bence, A.K.; Bailey, B.; Xue, X.; Erickson, P.A.; Montrose, M.H.; Beck, W.T.; Erickson, L.C. Quantitation of doxorubicin uptake, efflux, and modulation of multidrug resistance (MDR) in MDR human cancer cells. J. Pharmacol. Exp. Ther. 2008, 324, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Caja, S.; Enríquez, J.A. Mitochondria in endothelial cells: Sensors and integrators of environmental cues. Redox Biol. 2017, 12, 821–827. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Robinson, K.; Tiriveedhi, V. Perplexing Role of P-Glycoprotein in Tumor Microenvironment. Front. Oncol. 2020, 10, 265. [Google Scholar] [CrossRef]
- Weinländer, G.; Kornek, G.; Raderer, M.; Hejna, M.; Tetzner, C.; Scheithauer, W. Treatment of advanced colorectal cancer with doxorubicin combined with two potential multidrug-resistance-reversing agents: High-dose oral tamoxifen and dexverapamil. J. Cancer Res. Clin. Oncol. 1997, 123, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Vong, L.B.; Nagasaki, Y. Combination Treatment of Murine Colon Cancer with Doxorubicin and Redox Nanoparticles. Mol. Pharm. 2016, 13, 449–455. [Google Scholar] [CrossRef]
- Sonowal, H.; Pal, P.B.; Wen, J.-J.; Awasthi, S.; Ramana, K.V.; Srivastava, S.K. Aldose reductase inhibitor increases doxorubicin-sensitivity of colon cancer cells and decreases cardiotoxicity. Sci. Rep. 2017, 7, 3182. [Google Scholar] [CrossRef] [PubMed]
- Al-Malky, H.S.; Al Harthi, S.E.; Osman, A.-M.M. Major obstacles to doxorubicin therapy: Cardiotoxicity and drug resistance. J. Oncol. Pharm. Pract. Off. Publ. Int. Soc. Oncol. Pharm. Pract. 2020, 26, 434–444. [Google Scholar] [CrossRef]
- Amawi, H.; Sim, H.-M.; Tiwari, A.K.; Ambudkar, S.V.; Shukla, S. ABC Transporter-Mediated Multidrug-Resistant Cancer. Adv. Exp. Med. Biol. 2019, 1141, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Okon, I.S.; Zou, M.-H. Mitochondrial ROS and cancer drug resistance: Implications for therapy. Pharmacol. Res. 2015, 100, 170–174. [Google Scholar] [CrossRef] [Green Version]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Doxorubicin resistance in breast cancer cells is mediated by extracellular matrix proteins. BMC Cancer 2018, 18, 41. [Google Scholar] [CrossRef] [Green Version]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Marinello, P.C.; Panis, C.; Silva, T.N.X.; Binato, R.; Abdelhay, E.; Rodrigues, J.A.; Mencalha, A.L.; Lopes, N.M.D.; Luiz, R.C.; Cecchini, R.; et al. Metformin prevention of doxorubicin resistance in MCF-7 and MDA-MB-231 involves oxidative stress generation and modulation of cell adaptation genes. Sci. Rep. 2019, 9, 5864. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, Q.; Chng, W.-J. TXNIP (VDUP-1, TBP-2): A major redox regulator commonly suppressed in cancer by epigenetic mechanisms. Int. J. Biochem. Cell Biol. 2011, 43, 1668–1673. [Google Scholar] [CrossRef]
- Chen, Y.; Ning, J.; Cao, W.; Wang, S.; Du, T.; Jiang, J.; Feng, X.; Zhang, B. Research Progress of TXNIP as a Tumor Suppressor Gene Participating in the Metabolic Reprogramming and Oxidative Stress of Cancer Cells in Various Cancers. Front. Oncol. 2020, 10, 568574. [Google Scholar] [CrossRef] [PubMed]
- Candas, D.; Li, J.J. MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxid. Redox Signal. 2014, 20, 1599–1617. [Google Scholar] [CrossRef] [Green Version]
- Altenhöfer, S.; Witte, I.; Teiber, J.F.; Wilgenbus, P.; Pautz, A.; Li, H.; Daiber, A.; Witan, H.; Clement, A.M.; Förstermann, U.; et al. One enzyme, two functions: PON2 prevents mitochondrial superoxide formation and apoptosis independent from its lactonase activity. J. Biol. Chem. 2010, 285, 24398–24403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.S.; DeLuca, H.F. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim. Biophys. Acta 1994, 1219, 26–32. [Google Scholar] [CrossRef]
- Noura, M.; Matsuo, H.; Koyama, A.; Adachi, S.; Masutani, H. TXNIP induces growth arrest and enhances ABT263-induced apoptosis in mixed-lineage leukemia-rearranged acute myeloid leukemia cells. FEBS Open Bio 2020, 10, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, Y.; Chen, L.; Wang, C.; Wang, Q.; Zhang, H.; Lin, Y.; Li, Q.; Pang, T. SIRT2 mediates multidrug resistance in acute myelogenous leukemia cells via ERK1/2 signaling pathway. Int. J. Oncol. 2016, 48, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Aimjongjun, S.; Mahmud, Z.; Jiramongkol, Y.; Alasiri, G.; Yao, S.; Yagüe, E.; Janvilisri, T.; Lam, E.W.-F. Lapatinib sensitivity in nasopharyngeal carcinoma is modulated by SIRT2-mediated FOXO3 deacetylation. BMC Cancer 2019, 19, 1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castiglioni, S.; Maier, J.A.M. Magnesium and cancer: A dangerous liason. Magnes. Res. 2011, 24, S92–S100. [Google Scholar] [CrossRef]
- Zheng, K.; Yang, Q.; Xie, L.; Qiu, Z.; Huang, Y.; Lin, Y.; Tu, L.; Cui, C. Overexpression of MAGT1 is associated with aggressiveness and poor prognosis of colorectal cancer. Oncol. Lett. 2019, 18, 3857–3862. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Sun, L.; Zheng, H.; Yang, D.; Zhang, J.; Zhang, Q. The association between single-nucleotide polymorphisms of TRPM7 gene and breast cancer in Han Population of Northeast China. Med. Oncol. 2014, 31, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Q.; Shrubsole, M.J.; Ness, R.M.; Schlundt, D.; Cai, Q.; Smalley, W.E.; Li, M.; Shyr, Y.; Zheng, W. The relation of magnesium and calcium intakes and a genetic polymorphism in the magnesium transporter to colorectal neoplasia risk. Am. J. Clin. Nutr. 2007, 86, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Trapani, V.; Wolf, F.I. Dysregulation of Mg(2+) homeostasis contributes to acquisition of cancer hallmarks. Cell Calcium 2019, 83, 102078. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, L.; Jia, R. The role of mitochondrial dynamics in human cancers. Am. J. Cancer Res. 2020, 10, 1278–1293. [Google Scholar]
- Wang, Y.; Liu, H.-H.; Cao, Y.-T.; Zhang, L.-L.; Huang, F.; Yi, C. The Role of Mitochondrial Dynamics and Mitophagy in Carcinogenesis, Metastasis and Therapy. Front. Cell Dev. Biol. 2020, 8, 413. [Google Scholar] [CrossRef]
- Yang, Z.; Feng, Z.; Gu, J.; Li, X.; Dong, Q.; Liu, K.; Li, Y.; OuYang, L. microRNA-488 inhibits chemoresistance of ovarian cancer cells by targeting Six1 and mitochondrial function. Oncotarget 2017, 8, 80981–80993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppins, S.; Edlich, F.; Cleland, M.M.; Banerjee, S.; McCaffery, J.M.; Youle, R.J.; Nunnari, J. The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol. Cell 2011, 41, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Tian, C.; Puszyk, W.M.; Ogunwobi, O.O.; Cao, M.; Wang, T.; Cabrera, R.; Nelson, D.R.; Liu, C. OPA1 downregulation is involved in sorafenib-induced apoptosis in hepatocellular carcinoma. Lab. Investig. 2013, 93, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Chiang, S.-F.; Chen, W.T.-L.; Ke, T.-W.; Chen, T.-W.; You, Y.-S.; Lin, C.-Y.; Chao, K.S.C.; Huang, C.-Y. HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Dis. 2018, 9, 1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Shi, Y. Mitochondria as a target in cancer treatment. MedComm 2020, 1, 129–139. [Google Scholar] [CrossRef]
- Han, X.-J.; Shi, S.-L.; Wei, Y.-F.; Jiang, L.-P.; Guo, M.-Y.; Wu, H.-L.; Wan, Y.-Y. Involvement of mitochondrial dynamics in the antineoplastic activity of cisplatin in murine leukemia L1210 cells. Oncol. Rep. 2017, 38, 985–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Locatelli, L.; Cazzaniga, A.; Fedele, G.; Zocchi, M.; Scrimieri, R.; Moscheni, C.; Castiglioni, S.; Maier, J.A. A Comparison of Doxorubicin-Resistant Colon Cancer LoVo and Leukemia HL60 Cells: Common Features, Different Underlying Mechanisms. Curr. Issues Mol. Biol. 2021, 43, 163-175. https://doi.org/10.3390/cimb43010014
Locatelli L, Cazzaniga A, Fedele G, Zocchi M, Scrimieri R, Moscheni C, Castiglioni S, Maier JA. A Comparison of Doxorubicin-Resistant Colon Cancer LoVo and Leukemia HL60 Cells: Common Features, Different Underlying Mechanisms. Current Issues in Molecular Biology. 2021; 43(1):163-175. https://doi.org/10.3390/cimb43010014
Chicago/Turabian StyleLocatelli, Laura, Alessandra Cazzaniga, Giorgia Fedele, Monica Zocchi, Roberta Scrimieri, Claudia Moscheni, Sara Castiglioni, and Jeanette A. Maier. 2021. "A Comparison of Doxorubicin-Resistant Colon Cancer LoVo and Leukemia HL60 Cells: Common Features, Different Underlying Mechanisms" Current Issues in Molecular Biology 43, no. 1: 163-175. https://doi.org/10.3390/cimb43010014
APA StyleLocatelli, L., Cazzaniga, A., Fedele, G., Zocchi, M., Scrimieri, R., Moscheni, C., Castiglioni, S., & Maier, J. A. (2021). A Comparison of Doxorubicin-Resistant Colon Cancer LoVo and Leukemia HL60 Cells: Common Features, Different Underlying Mechanisms. Current Issues in Molecular Biology, 43(1), 163-175. https://doi.org/10.3390/cimb43010014