Isoform-Specific Effects of Apolipoprotein E on Markers of Inflammation and Toxicity in Brain Glia and Neuronal Cells In Vitro


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Other Reagents
2.2. Cell Culture and Treatments
2.3. Cell Viability and Toxicity
2.4. Western Blot
2.5. Statistical Analysis
3. Results
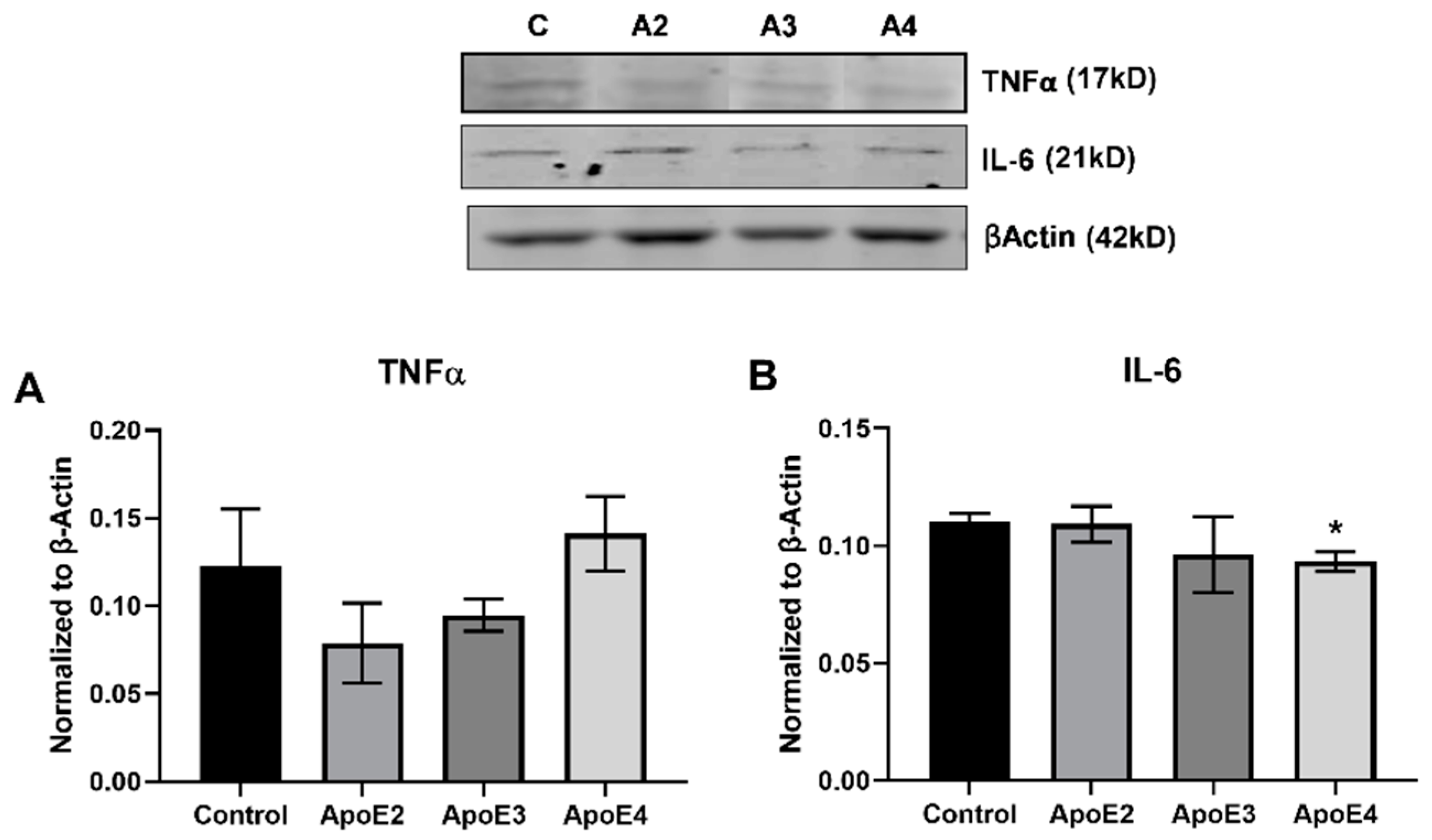
3.1. ApoE4 Increases Inflammatory Cytokine Expression in Human Astrocytes
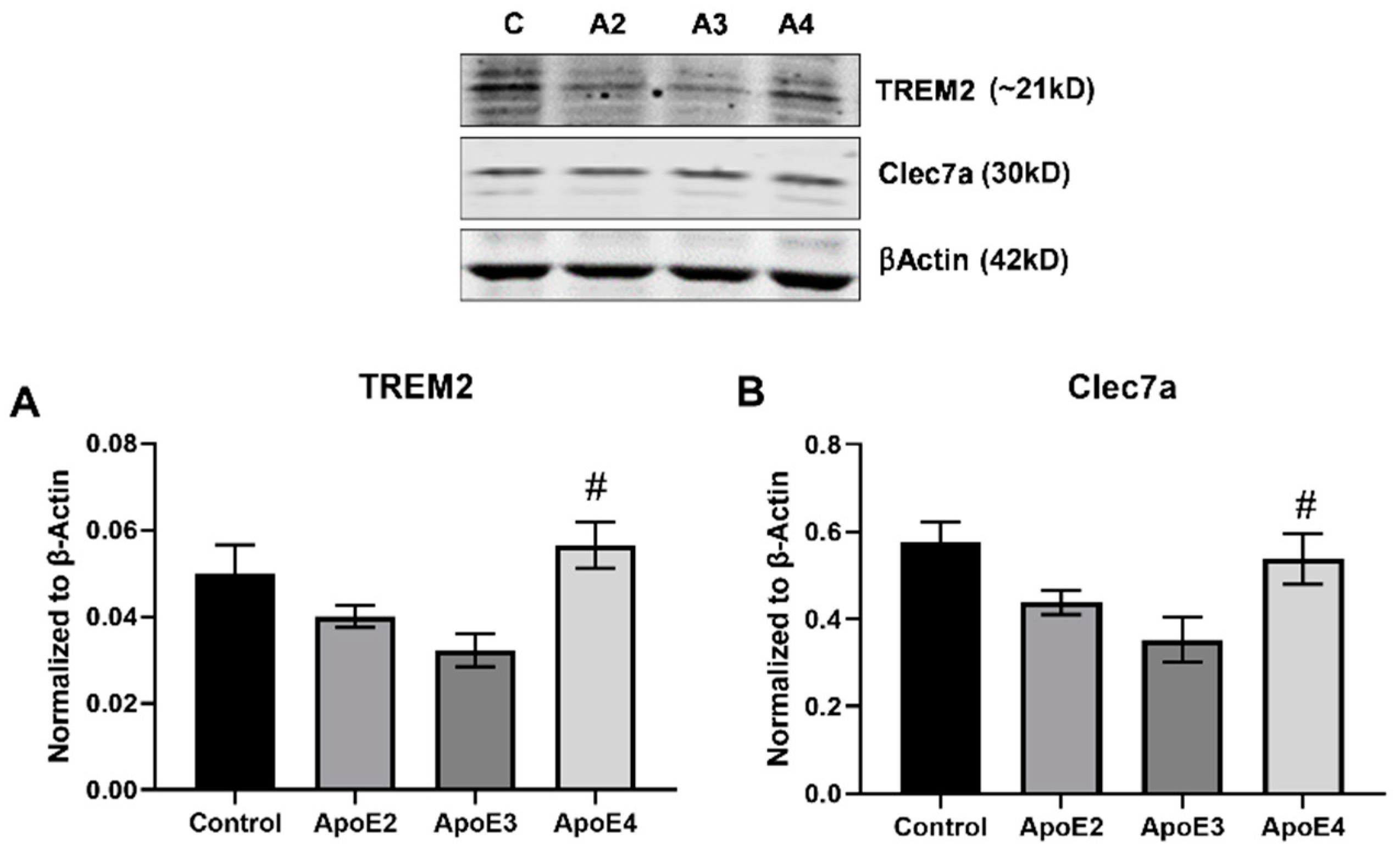
3.2. ApoE4 Enhances the Inflammatory Activation of Microglia
3.3. ApoE2 Promotes Neuron Survival While ApoE4 Reduces Neuronal Viability
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Liao, F.; Yoon, H.; Kim, J. Apolipoprotein E metabolism and functions in brain and its role in Alzheimer’s disease. Curr. Opin. Lipidol. 2016, 28, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Dorey, E.; Chang, N.; Liu, Q.Y.; Yang, Z.; Zhang, W. Apolipoprotein E, amyloid-beta, and neuroinflammation in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Gearing, M.; Lah, J.J.; Levey, A.I.; Wingo, T.S.; Seyfried, N.T. Effects of APOE Genotype on Brain Proteomic Network and Cell Type Changes in Alzheimer’s Disease. Front. Mol. Neurosci. 2018, 11, 454. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.M.; Wright, E.; Colton, C.A.; Sullivan, P.M.; Laskowitz, D.T.; Vitek, M.P. Apolipoprotein E isoform mediated regulation of nitric oxide release 1,2 1Guest Editors: Mark, A. Smith and George Perry 2This article is part of a series of reviews on “Causes and Consequences of Oxidative Stress in Alzheimer’s Disease.” The full list of papers may be found on the homepage of the journal. Free Radic. Biol. Med. 2002, 32, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A.; Brown, C.; Cook, D.; Needham, L.K.; Xu, Q.; Czapiga, M.; Saunders, A.M.; Schmechel, D.E.; Rasheed, K.; Vitek, M.P. APOE and the regulation of microglial nitric oxide production: A link between genetic risk and oxidative stress. Neurobiol. Aging 2002, 23, 777–785. [Google Scholar] [CrossRef]
- Keene, C.D.; Cudaback, E.; Li, X.; Montine, K.S.; Montine, T.J. Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Curr. Opin. Neurobiol. 2011, 21, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Vitek, M.P.; Brown, C.M.; Colton, C.A. APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 2009, 30, 1350–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konttinen, H.; Silva, M.E.C.C.D.; Ohtonen, S.; Wojciechowski, S.; Shakirzyanova, A.; Caligola, S.; Giugno, R.; Ishchenko, Y.; Hernández, D.; Fazaludeen, M.F.; et al. PSEN1ΔE9, APPswe, and APOE4 Confer Disparate Phenotypes in Human iPSC-Derived Microglia. Stem Cell Rep. 2019, 13, 669–683. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-W.A.; Zhou, B.; Nabet, A.M.; Wernig, M.; Südhof, T.C. Differential Signaling Mediated by ApoE2, ApoE3, and ApoE4 in Human Neurons Parallels Alzheimer’s Disease Risk. J. Neurosci. 2019, 39, 7408–7427. [Google Scholar] [CrossRef] [Green Version]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Tesseur, I.; Kekonius, L.; Wyss-Coray, T.; Fish, J.D.; Masliah, E.; Hopkins, P.C.; Scearce-Levie, K.; et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10966–10971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.-S.; Verghese, P.B.; Chakraborty, C.; Joung, J.; Hyman, B.T.; Ulrich, J.D.; Holtzman, D.M.; Barres, B.A. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 10186–10191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C.-M. Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer’s Disease (AD): A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE isoforms differentially regulates cleavage and secretion of BDNF. Mol. Brain 2017, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and A Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J. Neurosci. 2015, 35, 7538–7551. [Google Scholar] [CrossRef]
- Sen, A.; Alkon, D.L.; Nelson, T.J. Apolipoprotein E3 (ApoE3) but Not ApoE4 Protects against Synaptic Loss through Increased Expression of Protein Kinase Cϵ. J. Biol. Chem. 2012, 287, 15947–15958. [Google Scholar] [CrossRef] [Green Version]
- Rao, H.V.; Bihaqi, S.W.; Iannucci, J.; Sen, A.; Grammas, P. Thrombin Signaling Contributes to High Glucose-Induced Injury of Human Brain Microvascular Endothelial Cells. J. Alzheimer’s Dis. 2021, 79, 211–224. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, L.-M.; Wu, J. Cross-Talk between Apolipoprotein E and Cytokines. Mediat. Inflamm. 2011, 2011, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rebeck, G.W. The role of APOE on lipid homeostasis and inflammation in normal brains. J. Lipid Res. 2017, 58, 1493–1499. [Google Scholar] [CrossRef] [Green Version]
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer’s Disease. Mol. Cells 2019, 42, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Sidoryk-Wegrzynowicz, M.; Wegrzynowicz, M.; Lee, E.; Bowman, A.B.; Aschner, M. Role of Astrocytes in Brain Function and Disease. Toxicol. Pathol. 2010, 39, 115–123. [Google Scholar] [CrossRef]
- Lau, L.T.; Yu, A.C.-H. Astrocytes Produce and Release Interleukin-1, Interleukin-6, Tumor Necrosis Factor Alpha and Interferon-Gamma Following Traumatic and Metabolic Injury. J. Neurotrauma 2001, 18, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.A.; Alleva, L.M.; Vissel, B. The roles of TNF in brain dysfunction and disease. Pharmacol. Ther. 2010, 128, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta—A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, H.-L.; Yang, C.-M. Role of Redox Signaling in Neuroinflammation and Neurodegenerative Diseases. BioMed Res. Int. 2013, 2013, 1–18. [Google Scholar] [CrossRef] [PubMed]
- West, P.K.; Viengkhou, B.; Campbell, I.L.; Hofer, M.J. Microglia responses to interleukin-6 and type I interferons in neuroinflammatory disease. Glia 2019, 67, 1821–1841. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S. Molecular and Cellular Basis of Neurodegeneration in Alzheimer’s Disease. Mol. Cells 2017, 40, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, G.; Beiser, A.S.; Choi, S.H.; Preis, S.R.; Chen, T.C.; Vorgas, D.; Au, R.; Pikula, A.; Wolf, P.A.; DeStefano, A.L.; et al. Serum Brain-Derived Neurotrophic Factor and the Risk for Dementia. JAMA Neurol. 2014, 71, 55–61. [Google Scholar] [CrossRef]
- Chen, S.-D.; Wu, C.-L.; Hwang, W.-C.; Yang, D.-I. More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression of Autophagy. Int. J. Mol. Sci. 2017, 18, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, R.O.; Perestrelo, T.; Almeida, R.D. PROneurotrophins and CONSequences. Mol. Neurobiol. 2018, 55, 2934–2951. [Google Scholar] [CrossRef] [PubMed]
- Guttikonda, S.R.; Sikkema, L.; Tchieu, J.; Saurat, N.; Walsh, R.M.; Harschnitz, O.; Ciceri, G.; Sneeboer, M.; Mazutis, L.; Setty, M.; et al. Fully defined human pluripotent stem cell-derived microglia and tri-culture system model C3 production in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Jiang, H.; Serrano, J.R.; Gonzales, E.R.; Wang, C.; Gratuze, M.; Hoyle, R.; Bien-Ly, N.; Silverman, A.P.; Sullivan, P.M.; et al. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci. Transl. Med. 2021, 13, eabd7522. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Group | ApoE2 | ApoE3 | ApoE4 |
|---|---|---|---|
| Cellular Viability (MTS) | +14.9 ± 0.663 ** | +9.7 ± 2.780 | −9.7 ± 4.202 ^^^,### |
| Cellular Toxicity (LDH) | +2.6 ± 3.046 | +2.7 ± 4.156 | +14 ± 4.164 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iannucci, J.; Sen, A.; Grammas, P. Isoform-Specific Effects of Apolipoprotein E on Markers of Inflammation and Toxicity in Brain Glia and Neuronal Cells In Vitro. Curr. Issues Mol. Biol. 2021, 43, 215-225. https://doi.org/10.3390/cimb43010018
Iannucci J, Sen A, Grammas P. Isoform-Specific Effects of Apolipoprotein E on Markers of Inflammation and Toxicity in Brain Glia and Neuronal Cells In Vitro. Current Issues in Molecular Biology. 2021; 43(1):215-225. https://doi.org/10.3390/cimb43010018
Chicago/Turabian StyleIannucci, Jaclyn, Abhik Sen, and Paula Grammas. 2021. "Isoform-Specific Effects of Apolipoprotein E on Markers of Inflammation and Toxicity in Brain Glia and Neuronal Cells In Vitro" Current Issues in Molecular Biology 43, no. 1: 215-225. https://doi.org/10.3390/cimb43010018