
Effects of Sample Size on Plant Single-Cell RNA Profiling

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Overview
2.2. Data Integration and Sampling
2.3. Determination of Significant PC Number
2.4. Cell Clustering and Assignment of Clusters to Known Cell Types
2.5. Identifying High-Specificity Genes
2.6. Pseudotime Analysis
2.7. Statistical Analyses for Estimation of Cluster Similarity
2.7.1. Rand Index
2.7.2. Morey and Agresti’s Adjusted Rand Index (MA)
2.7.3. Hubert and Arabie’s Adjusted Rand Index (HA)
2.7.4. Fowlkes and Mallows Index (FM)
2.7.5. Jaccard Index (JI)
3. Results
3.1. Overview of 1244 Reported Single-Cell Transcriptome Studies
3.2. Effect of Cell Numbers on Significant PC Numbers
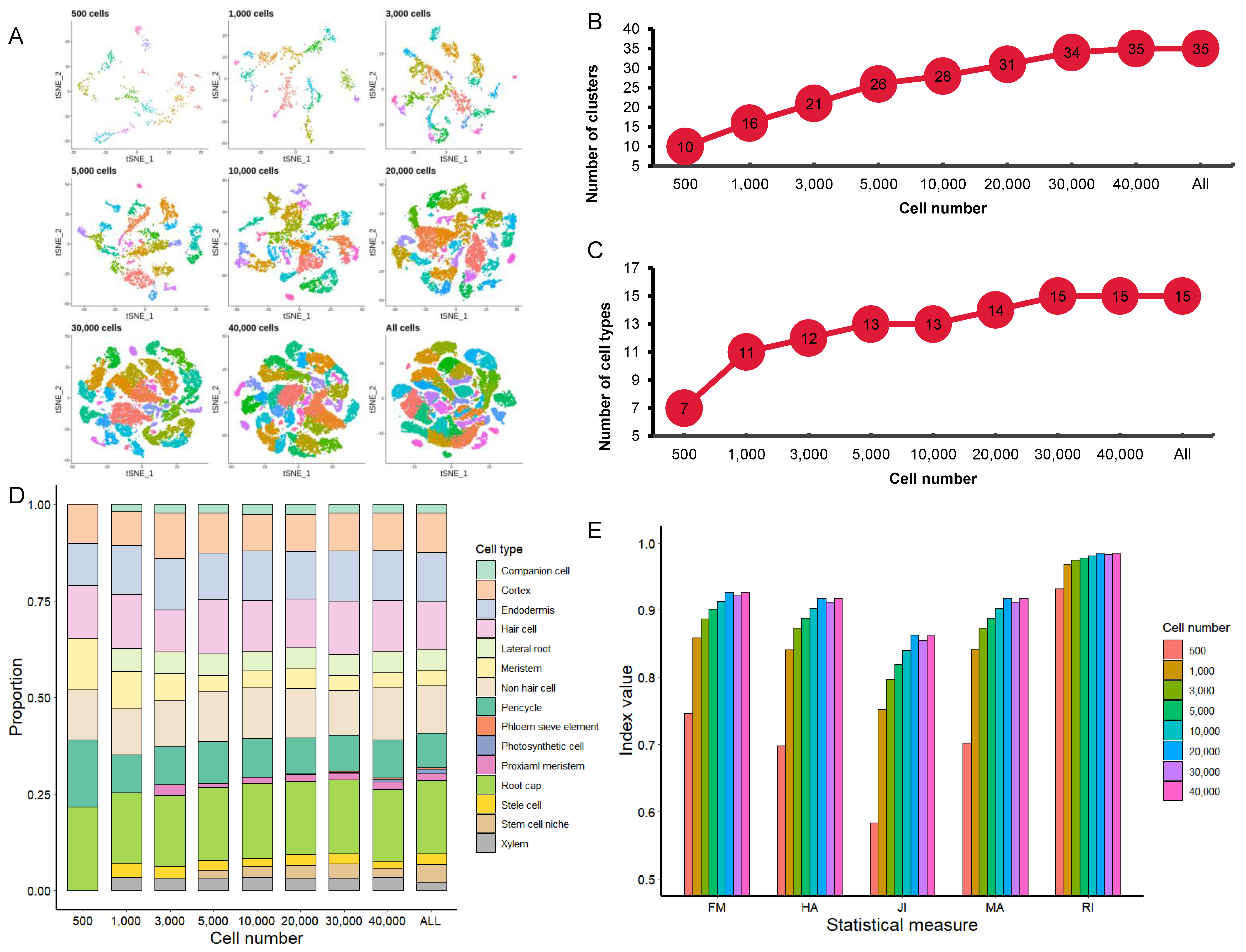
3.3. Effect of Cell Numbers on Cell Clustering
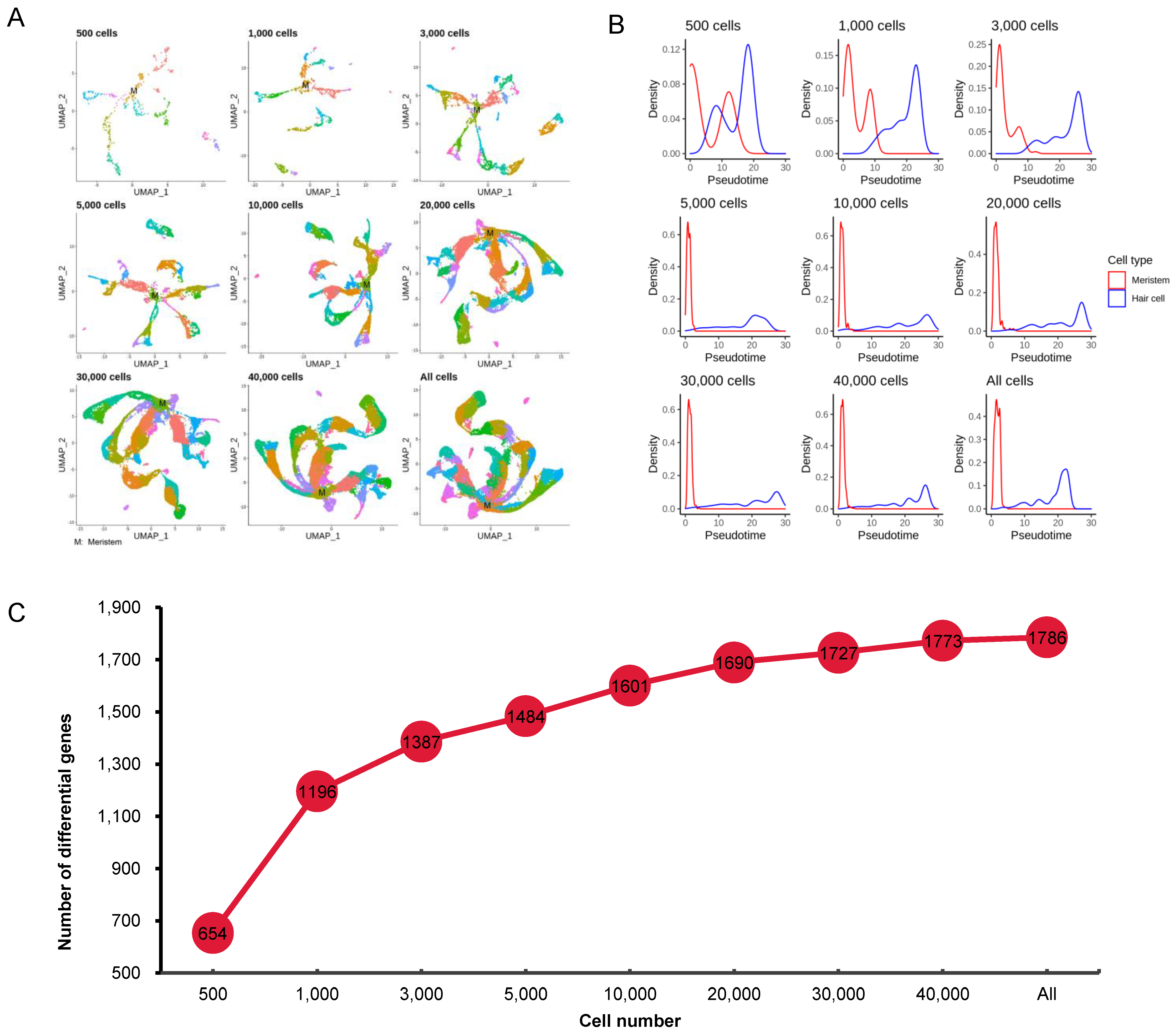
3.4. Effect of Cell Numbers on Identification of Differentially Expressed Genes
3.5. Effect of Cell Numbers on Cell Trajectory Inference
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hong, L.; Dumond, M.; Zhu, M.; Tsugawa, S.; Li, C.-B.; Boudaoud, A.; Hamant, O.; Roeder, A. Heterogeneity and robustness in plant morphogenesis: From cells to organs. Annu. Rev. Plant Biol. 2018, 69, 469–495. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, I.C.; Ponting, C.P.; Voet, T. Single-cell multiomics: Multiple measurements from single cells. Trends Genet. 2017, 33, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.-A.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 2017, 169, 750–765.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, M.L.; Davis, A.; Gao, R.; Casasent, A.; Wang, Y.; Sei, E.; Vilar, E.; Maru, D.; Kopetz, S.; Navin, N.E. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res. 2017, 27, 1287–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Li, C.; Fan, Z.; Liu, H.; Zhang, X.; Cai, Z.; Xu, L.; Luo, J.; Huang, Y.; He, L.; et al. Single-cell sequencing reveals variants in ARID1A, GPRC5A and MLL2 criving self-renewal of human bladder cancer stem cells. Eur. Urol. 2017, 71, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Casasent, A.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, N.E. Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell 2018, 172, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Touch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Islam, S.; Kjällquist, U.; Moliner, A.; Zajac, P.; Fan, J.B.; Lönnerberg, P.; Linnarsson, S. Characterization of the single-cell tran-scriptional landscape by highly multiplex RNA-seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar] [CrossRef] [Green Version]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-Cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular architecture of the mouse nervous system. Cell 2018, 174, 999–1014.e22. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Liu, H.; Xie, F.; Fischer, S.; Booeshaghi, A.S.; Adkins, R.S.; Aldridge, A.I.; Ament, S.A.; Pinto-Duarte, A.; Bartlett, A.; et al. An integrated transcriptomic and epigenomic atlas of mouse primary motor cortex cell types. BioRxiv 2020. [Google Scholar]
- Young, M.D.; Mitchell, T.J.; Braga, F.A.V.; Tran, M.G.; Stewart, B.J.; Ferdinand, J.R.; Collord, G.; Botting, R.A.; Popescu, D.M.; Loudon, K.W.; et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 2018, 361, 594–599. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.Y.; Birnbaum, K.D.; Ehrhardt, D.W. Towards building a plant cell atlas. Trends Plant Sci. 2019, 24, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efroni, I.; Birnbaum, K.D. The potential of single-cell profiling in plants. Genome Biol. 2016, 17, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efroni, I.; Ip, P.-L.; Nawy, T.; Mello, A.; Birnbaum, K.D. Quantification of cell identity from single-cell gene expression profiles. Genome Biol. 2015, 16, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efroni, I.; Mello, A.; Nawy, T.; Ip, P.-L.; Rahni, R.; DelRose, N.; Powers, A.; Satija, R.; Birnbaum, K.D. Root regeneration triggers an embryo-like sequence guided by hormonal interactions. Cell 2016, 165, 1721–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelms, B.; Walbot, V. Defining the developmental program leading to meiosis in maize. Science 2019, 364, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liang, Z.; Feng, D.; Jiang, S.; Wang, Y.; Du, Z.; Li, R.; Hu, G.; Zhang, P.; Ma, Y.; et al. Transcriptional landscape of rice roots at the single-cell resolution. Mol. Plant 2021, 14, 384–394. [Google Scholar] [CrossRef]
- Satterlee, J.W.; Strable, J.; Scanlon, M.J. Plant stem-cell organization and differentiation at single-cell resolution. Proc. Natl. Acad. Sci. USA 2020, 117, 33689–33699. [Google Scholar] [CrossRef]
- Bezrutczyk, M.; Zöllner, N.R.; Kruse, C.P.S.; Hartwig, T.; Lautwein, T.; Köhrer, K.; Frommer, W.B.; Kim, J.-Y. Evidence for phloem loading via the abaxial bundle sheath cells in maize leaves. Plant Cell 2021, 33, 531–547. [Google Scholar] [CrossRef]
- Xu, X.; Crow, M.; Rice, B.R.; Li, F.; Harris, B.; Liu, L.; Demesa-Arevalo, E.; Lu, Z.; Wang, L.; Fox, N.; et al. Single-cell RNA sequenc-ing of developing maize ears facilitates functional analysis and trait candidate gene discovery. Dev. Cell 2021, 56, 557–568. [Google Scholar] [CrossRef]
- Denyer, T.; Ma, X.; Klesen, S.; Scacchi, E.; Nieselt, K.; Timmermans, M.C. Spatiotemporal developmental trajectories in the ara-bidopsis root revealed using high-throughput single-cell RNA sequencing. Dev. Cell 2019, 48, 840–852. [Google Scholar] [CrossRef] [Green Version]
- Jean-Baptiste, K.; McFaline-Figueroa, J.L.; Alexandre, C.M.; Dorrity, M.W.; Saunders, L.; Bubb, K.L.; Trapnell, C.; Fields, S.; Queitsch, C.; Cuperus, J.T. Dynamics of gene expression in single root cells of Arabidopsis thaliana. Plant Cell 2019, 31, 993–1011. [Google Scholar] [CrossRef] [Green Version]
- Ryu, K.H.; Huang, L.; Kang, H.M.; Schiefelbein, J. Single-cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiol. 2019, 179, 1444–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulse, C.N.; Cole, B.J.; Ciobanu, D.; Lin, J.; Yoshinaga, Y.; Gouran, M.; Turco, G.M.; Zhu, Y.; O’Malley, R.C.; Brady, S.M.; et al. High-throughput single-cell transcriptome profiling of plant cell types. Cell Rep. 2019, 27, 2241–2247.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.-Q.; Xu, Z.-G.; Shang, G.; Wang, J.-W. A single-cell RNA sequencing profiles the developmental landscape of arabidopsis root. Mol. Plant 2019, 12, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Lafzi, A.; Moutinho, C.; Picelli, S.; Heyn, H. Tutorial: Guidelines for the experimental design of single-cell RNA sequencing studies. Nat. Protoc. 2018, 13, 2742–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.; Gao, R.; Navin, N.E. SCOPIT: Sample size calculations for single-cell sequencing experiments. BMC Bioinform. 2019, 20, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, V.; Beltrame, E.D.V.; Pachter, L. A curated database reveals trends in single-cell transcriptomics. Database 2020, 2020, baaa073. [Google Scholar] [CrossRef]
- Brady, S.M.; Orlando, D.A.; Lee, J.-Y.; Wang, J.Y.; Koch, J.; Dinneny, J.R.; Mace, D.; Ohler, U.; Benfey, P.N. A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 2007, 318, 801–806. [Google Scholar] [CrossRef]
- Diaz-Papkovich, A.; Anderson-Trocmé, L.; Ben-Eghan, C.; Gravel, S. UMAP reveals cryptic population structure and phenotype heterogeneity in large genomic cohorts. PLoS Genet. 2019, 15, e1008432. [Google Scholar] [CrossRef] [Green Version]
- Suner, A. Clustering methods for single-cell RNA-sequencing expression data: Performance evaluation with varying sample sizes and cell compositions. Stat. Appl. Genet. Mol. Biol. 2019, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri, A.; Nowakowski, T.J.; A Pollen, A.; Kriegstein, A.R. Identification of cell types in a mouse brain single-cell atlas using low sampling coverage. BMC Biol. 2018, 16, 113. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Yuan, G.-C. GiniClust3: A fast and memory-efficient tool for rare cell type identification. BMC Bioinform. 2020, 21, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Grün, D.; Lyubimova, A.; Kester, L.; Wiebrands, K.; Basak, O.; Sasaki, N.; Clevers, H.; van Oudenaarden, A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nat. Cell Biol. 2015, 525, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Jindal, A.; Gupta, P.; Jayadeva; Sengupta, D. Discovery of rare cells from voluminous single cell expression data. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Lv, Y.; Yin, X.; Chen, X.; Chu, Q.; Zhu, Q.-H.; Fan, L.; Guo, L. Effects of Sample Size on Plant Single-Cell RNA Profiling. Curr. Issues Mol. Biol. 2021, 43, 1685-1697. https://doi.org/10.3390/cimb43030119
Chen H, Lv Y, Yin X, Chen X, Chu Q, Zhu Q-H, Fan L, Guo L. Effects of Sample Size on Plant Single-Cell RNA Profiling. Current Issues in Molecular Biology. 2021; 43(3):1685-1697. https://doi.org/10.3390/cimb43030119
Chicago/Turabian StyleChen, Hongyu, Yang Lv, Xinxin Yin, Xi Chen, Qinjie Chu, Qian-Hao Zhu, Longjiang Fan, and Longbiao Guo. 2021. "Effects of Sample Size on Plant Single-Cell RNA Profiling" Current Issues in Molecular Biology 43, no. 3: 1685-1697. https://doi.org/10.3390/cimb43030119