
Renadirsen, a Novel 2?OMeRNA/ENA® Chimera Antisense Oligonucleotide, Induces Robust Exon 45 Skipping for Dystrophin In Vivo

 ,
, 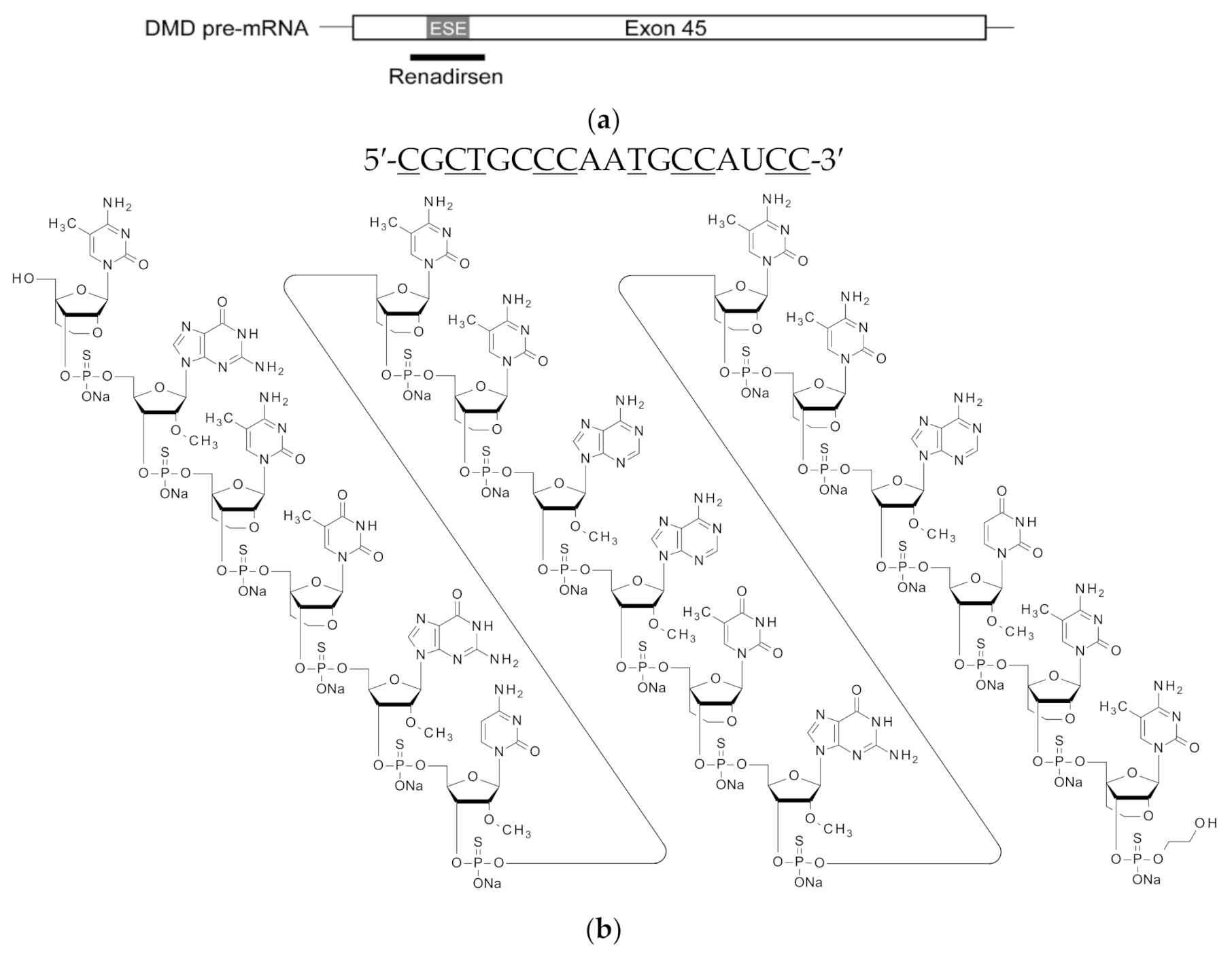{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Oligonucleotide Synthesis
2.2. Drug Administration to Mice for RNA Extraction
2.3. RNA Extraction and RT-PCR
2.4. In Vitro Stability Assay in Mouse Blood
2.5. Pharmacokinetics of Renadirsen in Mice
2.6. In Vitro Protein Binding of 14C-Labeled Renadirsen Sodium to Mouse Plasma
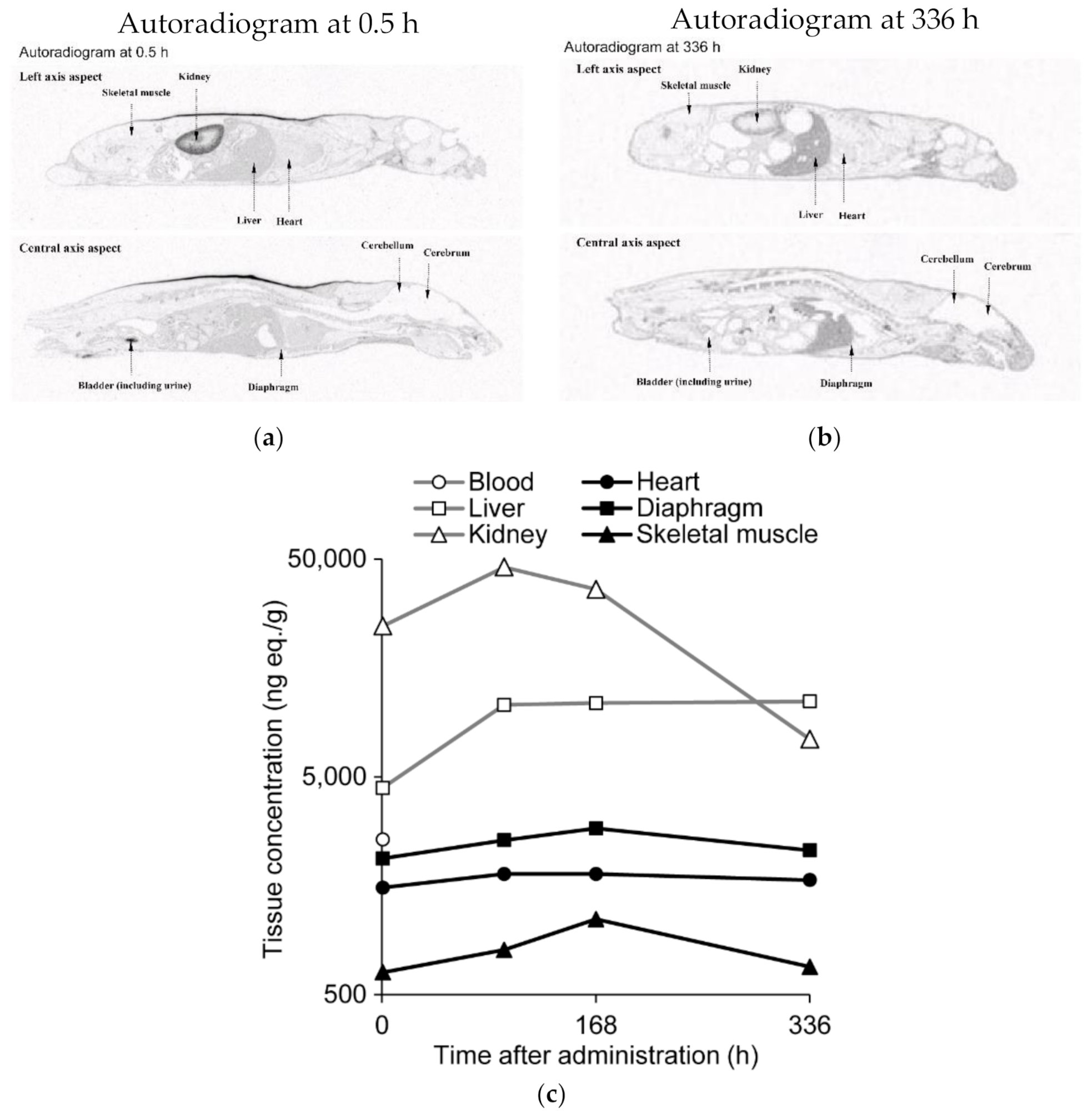
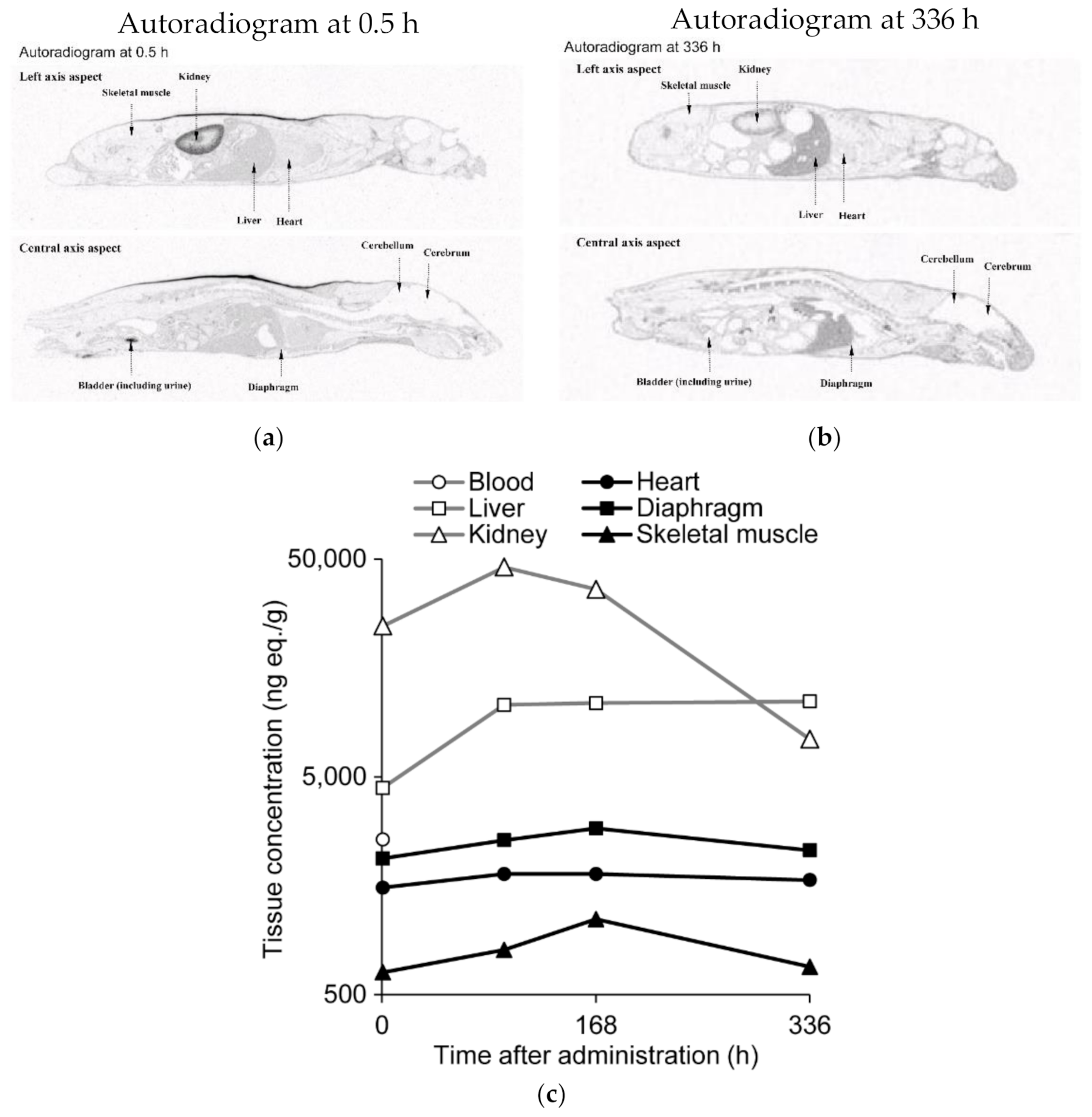
2.7. Quantitative Whole-Body Autoradiography after Administration of 14C-Labeled Renadirsen Sodium to Mice
3. Results
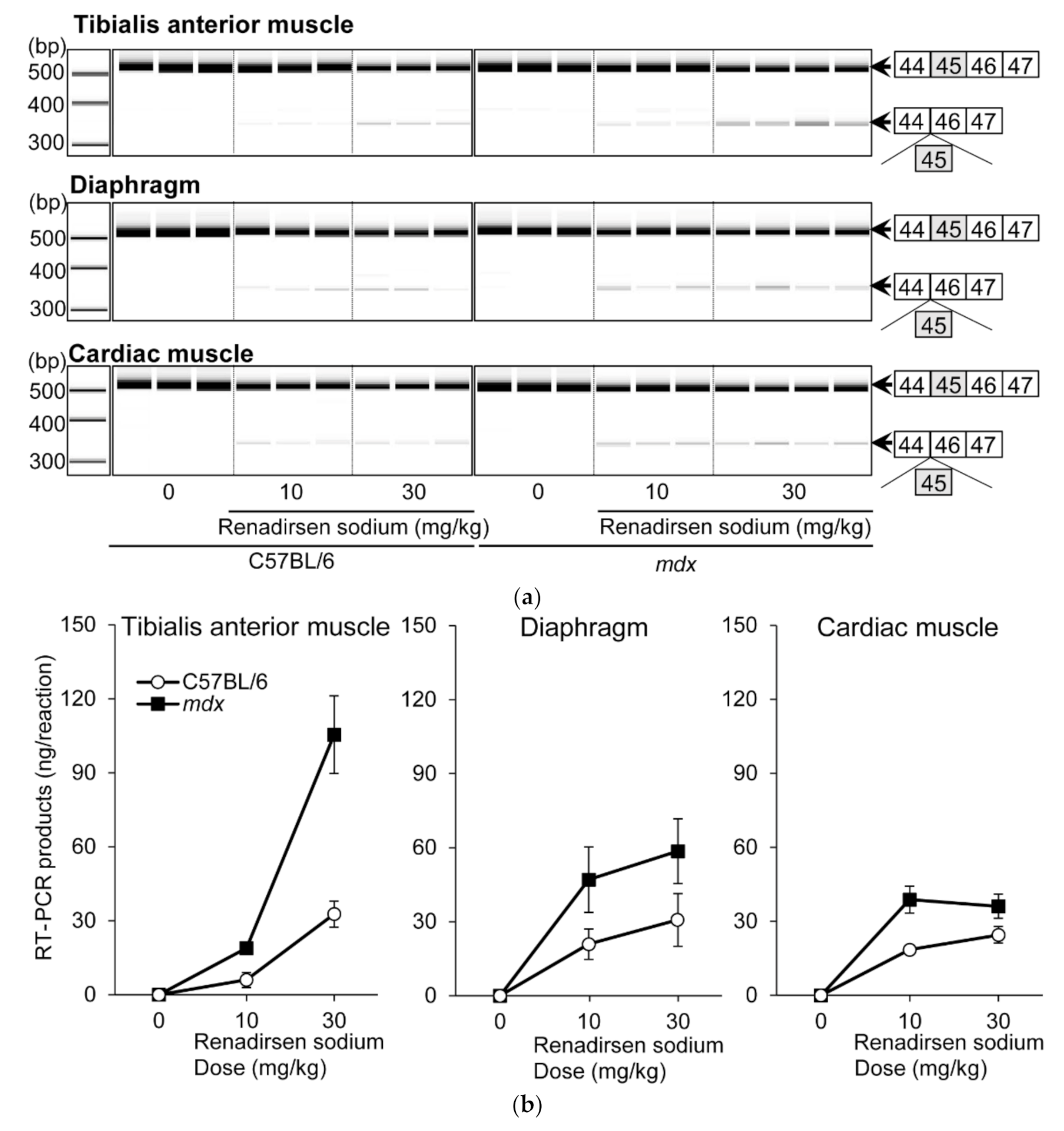
3.1. Exon Skipping in Tibialis Anterior Muscle, Diaphragm, and Cardiac Muscle in WT and Mdx Mice after Single Administration of Renadirsen Sodium
3.2. Stability and Pharmacokinetics of Renadirsen Sodium in Mice
3.3. 14C-Labeled Renadirsen Exhibited Broad Tissue Distribution and Long-Term Retention in Tissues, including the Heart, Diaphragm, and Skeletal Muscles
3.4. In Vitro Protein Binding of 14C-Labeled Renadirsen to Mouse Plasma
3.5. Renadirsen Showed Robust Exon-45-Skipping Activity in Skeletal and Cardiac Muscles Compared with 2′OMePS- and PMO-Based AO in mdx Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moat, S.J.; Bradley, D.M.; Salmon, R.; Clarke, A.; Hartley, L. Newborn bloodspot screening for Duchenne Muscular Dystrophy: 21 years experience in Wales (UK). Eur. J. Hum. Genet. 2013, 21, 1049–1053. [Google Scholar] [CrossRef] [Green Version]
- Nowak, K.J.; Davies, K.E. Duchenne muscular dystrophy and dystrophin: Pathogenesis and opportunities for treatment: Third in molecular medicine review series. EMBO Rep. 2004, 5, 872–876. [Google Scholar] [CrossRef]
- Ahn, A.H.; Kunkel, L.M. The structural and functional diversity of dystrophin. Nat. Genet. 1993, 3, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Melacini, P.; Vianello, A.; Villanova, C.; Fanin, M.; Miorin, M.; Angelini, C.; Dalla Volta, S. Cardiac and respiratory involvement in advanced stage Duchenne muscular dystrophy. Neuromuscul. Disord. 1996, 6, 367–376. [Google Scholar] [CrossRef]
- White, S.; Kalf, M.; Liu, Q.; Villerius, M.; Engelsma, D.; Kriek, M.; Vollebregt, E.; Bakker, B.; Van Ommen, G.J.B.; Breuning, M.H.; et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am. J. Hum. Genet. 2002, 71, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshima, Y.; Nishio, H.; Sakamoto, H.; Nakamura, H.; Matsuo, M. Modulation of in vitro splicing of the upstream intron by modifying an intra-exon sequence which is deleted from the dystrophin gene in dystrophin Kobe. J. Clin. Investig. 1995, 95, 515–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aartsma-Rus, A.; Janson, A.A.M.; Kaman, W.E.; Bremmer-Bout, M.; den Dunnen, J.T.; Baas, F.; van Ommen, G.J.B.; van Deutekom, J.C.T. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum. Mol. Genet. 2003, 12, 907–914. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. Antisense-mediated modulation of splicing: Therapeutic implications for Duchenne muscular dystrophy. RNA Biol. 2010, 7, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef]
- Chen, S.; Le, B.T.; Chakravarthy, M.; Kosbar, T.R.; Veedu, R.N. Systematic evaluation of 2′-Fluoro modified chimeric antisense oligonucleotide-mediated exon skipping in vitro. Sci. Rep. 2019, 9, 6078. [Google Scholar] [CrossRef] [PubMed]
- Kesselheim, A.S.; Avorn, J. Approving a problematic muscular dystrophy drug: Implications for FDA policy. JAMA—J. Am. Med. Assoc. 2016, 316, 2357–2358. [Google Scholar] [CrossRef]
- Townsend, D.W.; Yasuda, S.; Li, S.; Chamberlain, J.S.; Metzger, J.M. Emergent dilated cardiomyopathy caused by targeted repair of dystrophic skeletal muscle. Mol. Ther. 2008, 16, 832–835. [Google Scholar] [CrossRef]
- Wu, B.; Xiao, B.; Cloer, C.; Shaban, M.; Sali, A.; Lu, P.; Li, J.; Nagaraju, K.; Xiao, X.; Lu, Q.L. One-year treatment of morpholino antisense oligomer improves skeletal and cardiac muscle functions in dystrophic mdx mice. Mol. Ther. 2011, 19, 576–583. [Google Scholar] [CrossRef] [Green Version]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [CrossRef]
- Qi, L.L.; Rabinowitz, A.; Yun, C.C.; Yokota, T.; Yin, H.F.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restorers dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, M. ENA® oligonucleotides as therapeutics. Curr. Opin. Mol. Ther. 2006, 8, 144–149. [Google Scholar] [PubMed]
- Morita, K.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.; Ishikawa, T.; Imanishi, T.; Koizumi, M. 2′-O,4′-C-ethylene-bridged nucleic acids (ENA) with nuclease-resistance and high affinity for RNA. Nucleic Acids Res. Suppl. 2001, 241–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, K.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.; Ishikawa, T.; Imanishi, T.; Koizumi, M. 2′-O,4′-C-ethylene-bridged nucleic acids (ENA): Highly nuclease-resistant and thermodynamically stable oligonucleotides for antisense drug. Bioorg. Med. Chem. Lett. 2002, 12, 73–76. [Google Scholar] [CrossRef]
- Morita, K.; Takagi, M.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.; Ishikawa, T.; Imanishi, T.; Koizumi, M. Synthesis and properties of 2′-O,4′-C-ethylene-bridged nucleic acids (ENA) as effective antisense oligonucleotides. Bioorg. Med. Chem. 2003, 11, 2211–2226. [Google Scholar] [CrossRef]
- Koizumi, M. 2′-O,4′-C-Ethylene-bridged nucleic acids (ENATM) as next-generation antisense and antigene agents. Biol. Pharm. Bull. 2004, 27, 453–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.; Awano, H.; Yagi, M.; Matsumoto, M.; Watanabe, N.; Goda, R.; Koizumi, M.; Takeshima, Y.; Matsuo, M. 2′-O-methyl RNA/ethylene-bridged nucleic acid chimera antisense oligonucleotides to induce dystrophin exon 45 skipping. Genes 2017, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshima, Y.; Lee, T.; Shimomura, H.; Tanaka, Y.; Awano, H.; Nishida, A.; Ojima, I.; Minami, S.; Nakagawa, A.; Iijima, K.; et al. A new antisense oligonucleotide composed of RNA/ENA chimera (AO85) against dystrophin exon 45 significantly increased six-minute walk distance in Duchenne muscular dystrophy. In Proceedings of the 5th International Congress of Myology, Lyon, France, 14–18 March 2016. [Google Scholar]
- Ghazali Malueka, R.; Dwianingsih, E.K.; Lee, T.; Yagib, M.; Nishida, A.; Iijima, K.; Takeshima, Y.; Matsuo, M. Phosphorothioate modification of chimeric 2′-O-methyl RNA/ethylene-bridged nucleic acid oligonucleotides increases dystrophin exon 45 skipping capability and reduces cytotoxicity. Kobe J. Med. Sci. 2014, 60, 86–94. [Google Scholar] [CrossRef]
- Shoji, E.; Sakurai, H.; Nishino, T.; Nakahata, T.; Heike, T.; Awaya, T.; Fujii, N.; Manabe, Y.; Matsuo, M.; Sehara-Fujisawa, A. Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci. Rep. 2015, 5, 12831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Kaneko, M.; Obika, S.; Imanishi, T.; Kitade, Y.; Koizumi, M. Biologically stable 2-5A analogues containing 3′-O,4′-C-bridged adenosine as potent RNase L agonists. ChemMedChem 2007, 2, 1703–1707. [Google Scholar] [CrossRef]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Dirin, M.; Winkler, J. Influence of diverse chemical modifications on the ADME characteristics and toxicology of antisense oligonucleotides. Expert Opin. Biol. Ther. 2013, 13, 875–888. [Google Scholar] [CrossRef]
- Butler, M.; Stecker, K.; Bennett, C.F. Cellular distribution of phosphorothioate oligodeoxynucleotides in normal rodent tissues. Lab. Investig. 1997, 77, 379–388. [Google Scholar]
- Heemskerk, H.; De Winter, C.; Van Kuik, P.; Heuvelmans, N.; Sabatelli, P.; Rimessi, P.; Braghetta, P.; Van Ommen, G.J.B.; De Kimpe, S.; Ferlini, A.; et al. Preclinical PK and PD studies on 2′-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol. Ther. 2010, 18, 1210–1217. [Google Scholar] [CrossRef]
- Kurreck, J.; Wyszko, E.; Gillen, C.; Erdmann, V.A. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res. 2002, 30, 1911–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grünweller, A.; Wyszko, E.; Bieber, B.; Jahnel, R.; Erdmann, V.A.; Kurreck, J. Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNA. Nucleic Acids Res. 2003, 31, 3185–3193. [Google Scholar] [CrossRef] [PubMed]
- Sarepta Therapeutics, I. Drug Approval Package: Exondys 51 Injection (Eteplirsen). Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=206488 (accessed on 14 May 2018).
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amantana, A.; Iversen, P.L. Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr. Opin. Pharmacol. 2005, 5, 550–555. [Google Scholar] [CrossRef]
- Lehto, T.; Alvarez, A.C.; Gauck, S.; Gait, M.J.; Coursindel, T.; Wood, M.J.A.; Lebleu, B.; Boisguerin, P. Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells. Nucleic Acids Res. 2014, 42, 3207–3217. [Google Scholar] [CrossRef]
- González-Barriga, A.; Nillessen, B.; Kranzen, J.; Van Kessel, I.D.G.; Croes, H.J.E.; Aguilera, B.; De Visser, P.C.; Datson, N.A.; Mulders, S.A.M.; Van Deutekom, J.C.T.; et al. Intracellular distribution and nuclear activity of antisense oligonucleotides after unassisted uptake in myoblasts and differentiated myotubes in vitro. Nucleic Acid Ther. 2017, 27, 144–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Novak, J.S.; Hogarth, M.W.; Boehler, J.F.; Nearing, M.; Vila, M.C.; Heredia, R.; Fiorillo, A.A.; Zhang, A.; Hathout, Y.; Hoffman, E.P.; et al. Myoblasts and macrophages are required for therapeutic morpholino antisense oligonucleotide delivery to dystrophic muscle. Nat. Commun. 2017, 8, 941. [Google Scholar] [CrossRef]
- Cao, L.; Han, G.; Gu, B.; Yin, H.F. Wild-type mouse models to screen antisense oligonucleotides for exon-skipping efficacy in Duchenne muscular dystrophy. PLoS ONE 2014, 9, e111079. [Google Scholar] [CrossRef] [Green Version]
- Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. Biophys. Acta—Biomembr. 2010, 1798, 2296–2303. [Google Scholar] [CrossRef] [Green Version]
- Goemans, N.; Mercuri, E.; Belousova, E.; Komaki, H.; Dubrovsky, A.; McDonald, C.M.; Kraus, J.E.; Lourbakos, A.; Lin, Z.; Campion, G.; et al. A randomized placebo-controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in Duchenne muscular dystrophy. Neuromuscul. Disord. 2018, 28, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spurney, C.F.; Sali, A.; Guerron, A.D.; Iantorno, M.; Yu, Q.; Gordish-Dressman, H.; Rayavarapu, S.; Van Der Meulen, J.; Hoffman, E.P.; Nagaraju, K. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ’T Hoen, P.A.C.; De Meijer, E.J.; Boer, J.M.; Vossen, R.H.A.M.; Turk, R.; Maatman, R.G.H.J.; Davies, K.E.; Van Ommen, G.J.B.; Van Deutekom, J.C.T.; Den Dunnen, J.T. Generation and characterization of transgenic mice with the full-length human DMD gene. J. Biol. Chem. 2008, 283, 5899–5907. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, K.; Takakusa, H.; Kakuta, M.; Kanda, A.; Takagi, N.; Nagase, H.; Watanabe, N.; Asano, D.; Goda, R.; Masuda, T.; et al. Renadirsen, a Novel 2?OMeRNA/ENA® Chimera Antisense Oligonucleotide, Induces Robust Exon 45 Skipping for Dystrophin In Vivo. Curr. Issues Mol. Biol. 2021, 43, 1267-1281. https://doi.org/10.3390/cimb43030090
Ito K, Takakusa H, Kakuta M, Kanda A, Takagi N, Nagase H, Watanabe N, Asano D, Goda R, Masuda T, et al. Renadirsen, a Novel 2?OMeRNA/ENA® Chimera Antisense Oligonucleotide, Induces Robust Exon 45 Skipping for Dystrophin In Vivo. Current Issues in Molecular Biology. 2021; 43(3):1267-1281. https://doi.org/10.3390/cimb43030090
Chicago/Turabian StyleIto, Kentaro, Hideo Takakusa, Masayo Kakuta, Akira Kanda, Nana Takagi, Hiroyuki Nagase, Nobuaki Watanabe, Daigo Asano, Ryoya Goda, Takeshi Masuda, and et al. 2021. "Renadirsen, a Novel 2?OMeRNA/ENA® Chimera Antisense Oligonucleotide, Induces Robust Exon 45 Skipping for Dystrophin In Vivo" Current Issues in Molecular Biology 43, no. 3: 1267-1281. https://doi.org/10.3390/cimb43030090
APA StyleIto, K., Takakusa, H., Kakuta, M., Kanda, A., Takagi, N., Nagase, H., Watanabe, N., Asano, D., Goda, R., Masuda, T., Nakamura, A., Onishi, Y., Onoda, T., Koizumi, M., Takeshima, Y., Matsuo, M., & Takaishi, K. (2021). Renadirsen, a Novel 2?OMeRNA/ENA® Chimera Antisense Oligonucleotide, Induces Robust Exon 45 Skipping for Dystrophin In Vivo. Current Issues in Molecular Biology, 43(3), 1267-1281. https://doi.org/10.3390/cimb43030090