
Genome-Wide Survey Reveals the Microsatellite Characteristics and Phylogenetic Relationships of Harpadon nehereus

Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collecting
2.2. DNA Extraction, Library Construction and Illumina Sequencing
2.3. Sequence Quality Control, Assembly and K-mer Analysis
2.4. Microsatellite Identification and Phylogenetic Analysis
3. Results
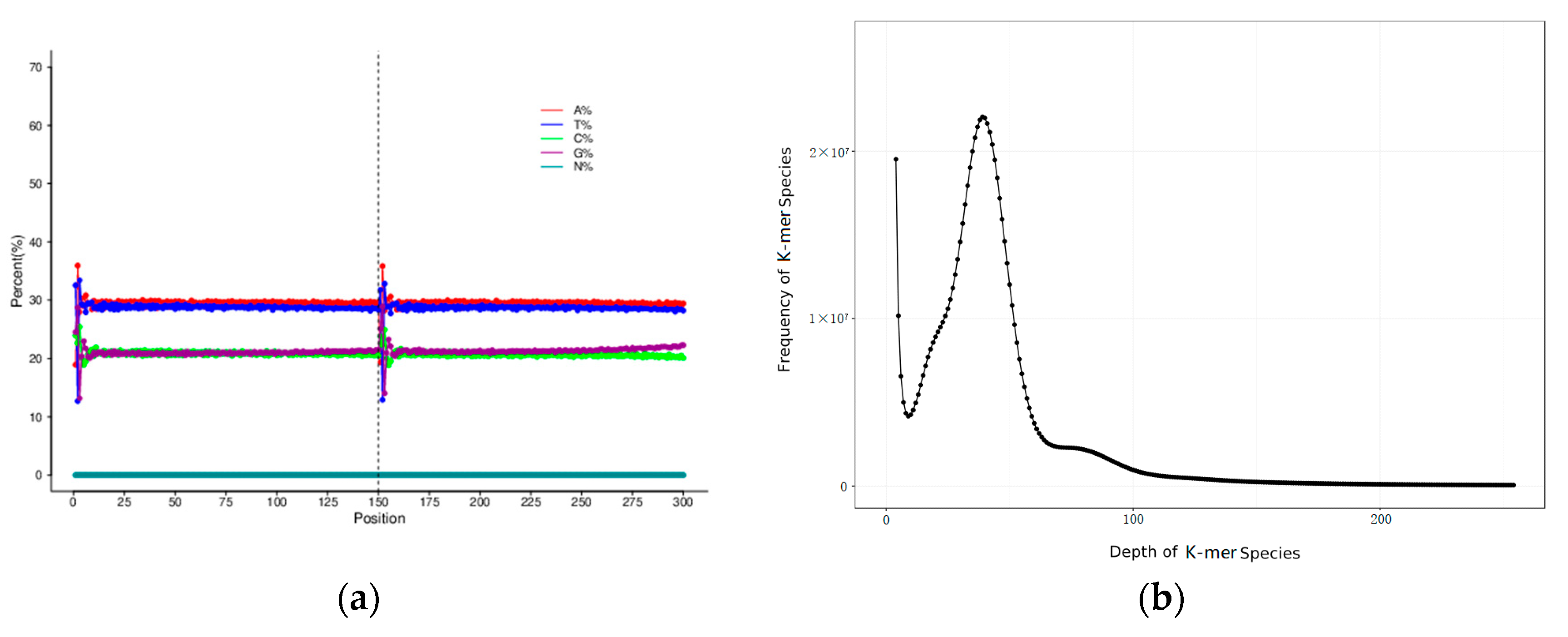
3.1. Sequencing Data Statistics and Sequence Quality Evaluation
3.2. Genome Assembly, Heterozygosity and Repeat Prediction
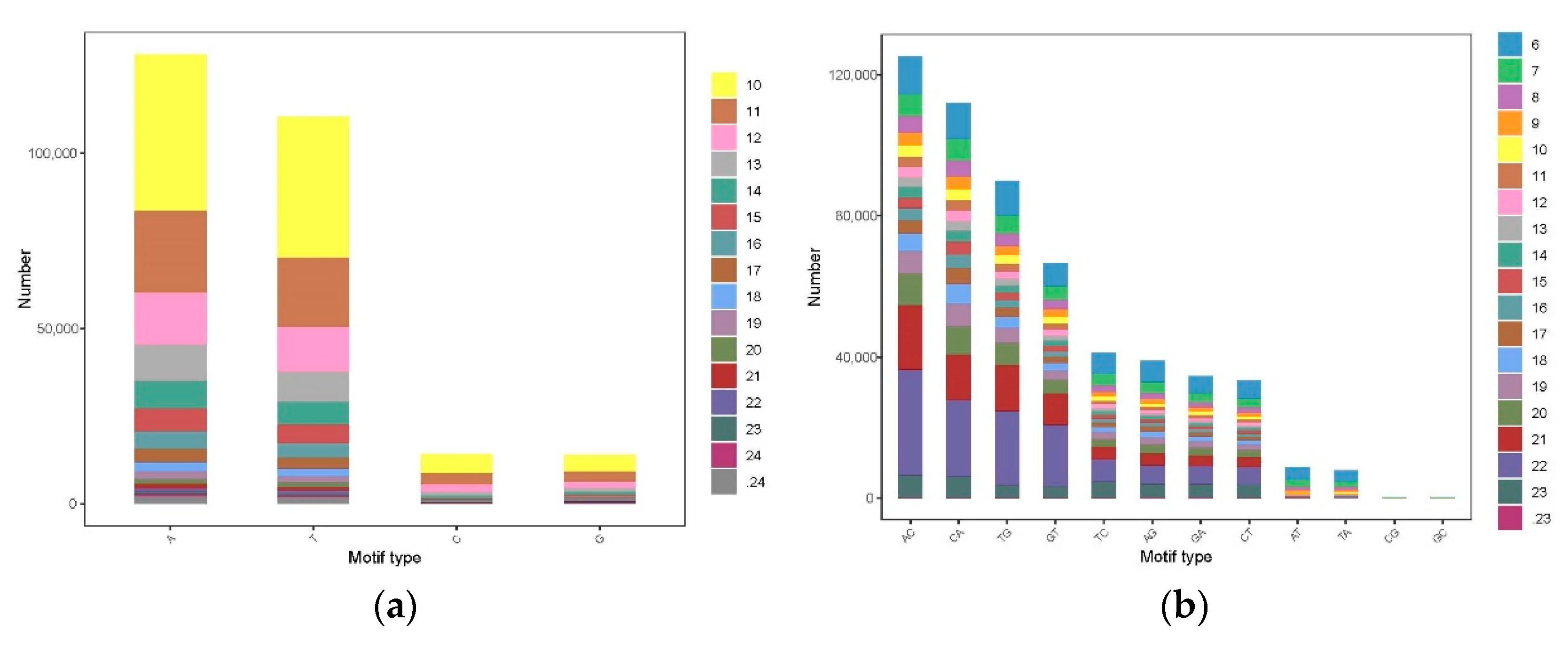
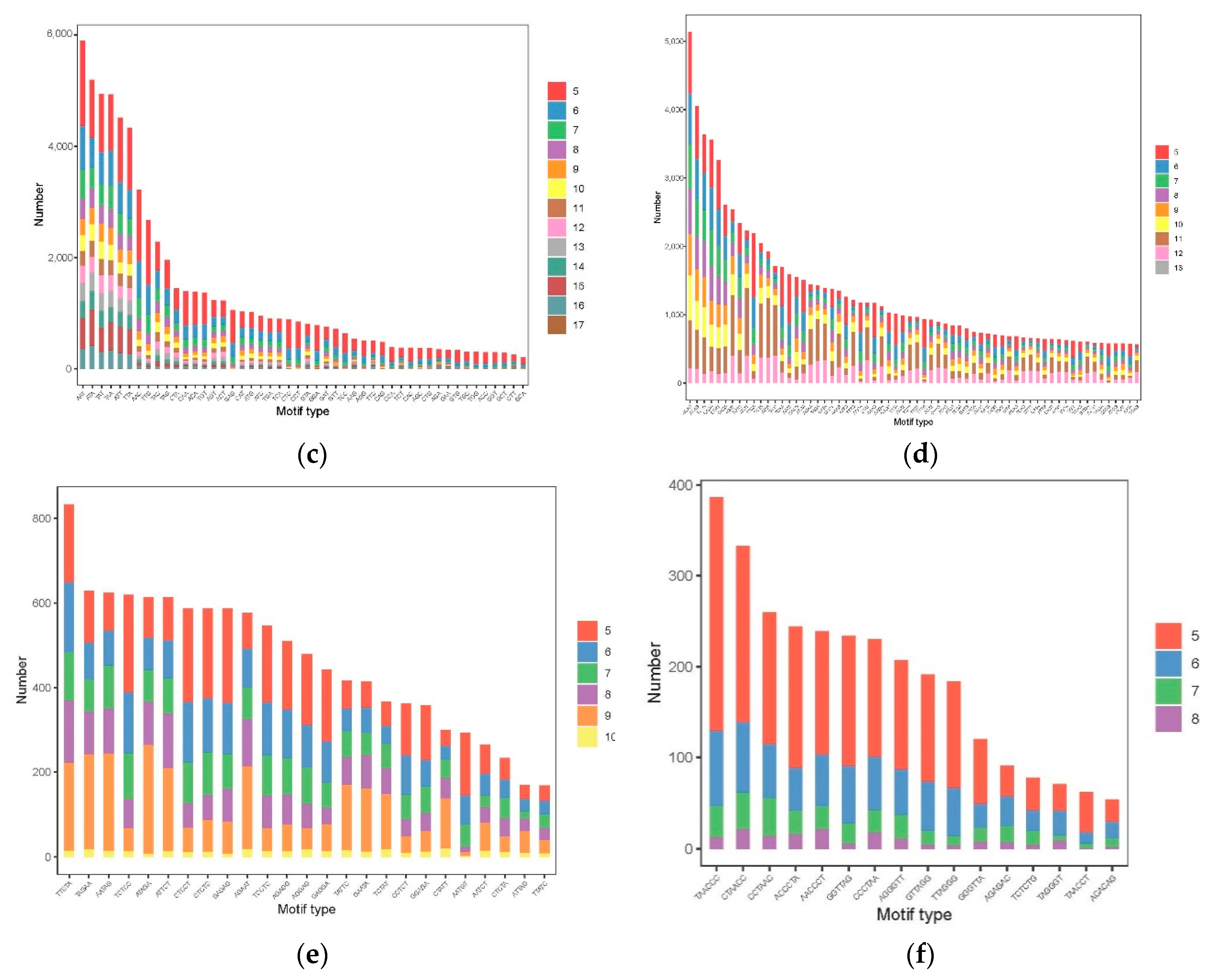
3.3. The Distribution and Characteristics of SSR Loci
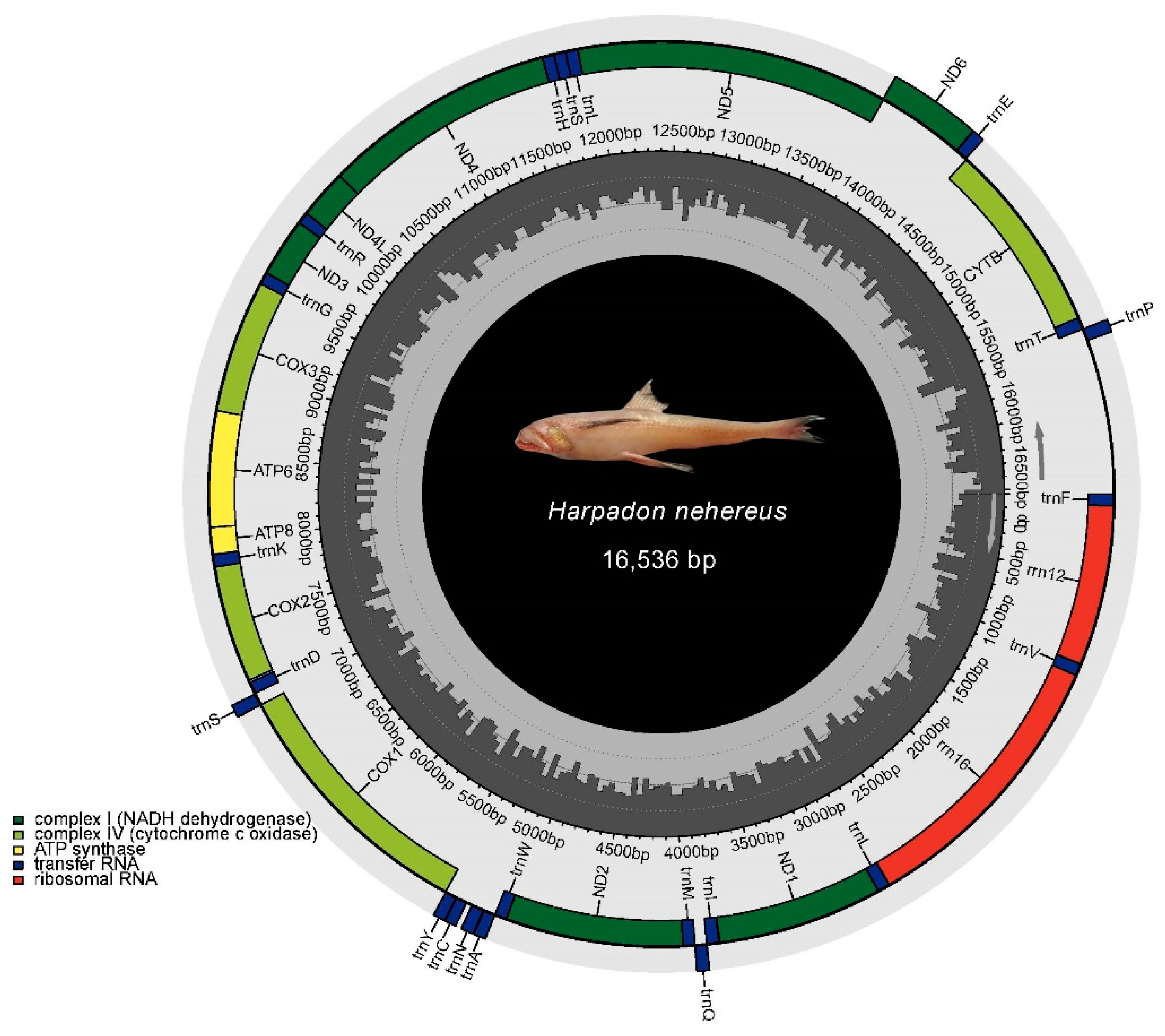
3.4. Mitochondrial DNA Assembly and Phylogenetic Relationships of H. nehereus
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nelson, J.S.; Grande, T.C.; Wilson, M.V.H. Fishes of the World, 5th ed.; John & Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Chakraborty, P.; Sahoo, S.; Bhattacharyya, D.K.; Ghosh, M. Marine lizardfish (Harpadon nehereus) meal concentrate in preparation of ready-to-eat protein and calcium rich extruded snacks. J. Food Sci. Technol. 2020, 57, 338–349. [Google Scholar] [CrossRef]
- Luo, H.Z. Study of Main Biology Character and Analysis of Resource Status on the Harpodon nehereus. Master’s Thesis, Zhejiang Ocean University, Zhoushan, China, 2012. (In Chinese). [Google Scholar]
- Kalhoro, M.A.; Liu, Q.; Memon, K.H.; Chang, M.S.; Jatt, A.N. Estimation of maximum sustainable yield of Bombay Duck, Harpodon nehereus fishery in Pakistan using the CEDA and ASPIC packages. Pakistan J. Zool. 2013, 45, 1757–1764. [Google Scholar]
- Jaiswar, A.K.; Chakraborty, S.K. A review on fishery, biology and stock parameters of Bombay duck Harpodon nehereus (Hamilton, 1822) occurring in India. J. Indian Fish. Assoc. 2016, 43, 67–96. [Google Scholar]
- Sarker, M.; Humayun, M.; Rahman, M.A.; Uddin, M.S. Population dynamics of Bombay duck Harpodon nehereus (Hamilton, 1822) of the Bay of Bengal along Bangladesh coast. Bangl. J. Zool. 2018, 45, 101–110. [Google Scholar] [CrossRef][Green Version]
- Chen, L.; Shui, B.N.; Dong, W.X. Growth characteristics and resources sustainable utilization of Harpodon nehereus. Technol. Manag. 2012, 3, 68–70. (In Chinese) [Google Scholar]
- Sudheesan, D.; Roshith, C.M.; Manna, R.K.; Das, S.K.; Koushlesh, S.K.; Chanu, T.N.; Bhakta, D. Implication towards growth overfishing of Harpadon nehereus in bag net fishing. In Proceedings of the 29th All India Congress of Zoology, ICAR-CIFRI, Kolkata, India, 9–11 June 2017. [Google Scholar]
- Utter, F.M. Biochemical genetics and fishery management: An historical perspective. J. Fish Biol. 2010, 39, 1–20. [Google Scholar] [CrossRef]
- Kurian, A.; Kurup, K.N. Stock assessment of Bombay duck Harpodon nehereus (Ham.) off Maharashtra coast. Indian J. Fish. 1992, 39, 243–248. [Google Scholar]
- Rupsankar, C. Improvement of cooking quality and gel formation capacity of Bombay duck (Harpodon nehereus) fish meat. J. Food Sci. Technol. 2010, 47, 534–540. [Google Scholar] [CrossRef]
- Firdaus, M.; Lelono, T.D.; Saleh, R.; Bintoro, G.; Salim, G. The expression of the body shape in fish species Harpadon nehereus (Hamilton, 1822) in the waters of Juata Laut, Tarakan city, North Kalimantan. AACL Bioflux 2018, 11, 613–624. [Google Scholar]
- Taqwa, A.; Burhanuddin, A.I.; Niartiningsih, A.; Nessa, M.N. Nomei fish (Harpadon nehereus, Ham. 1822) reproduction biology in Tarakan waters. In IOP Conference Series: Earth and Environmental Science; IOP Publishing: Bristol, UK, 2020; Volume 473, pp. 1–6. [Google Scholar]
- Xu, T.J.; Sun, D.Q.; Li, H.Y.; Wang, R.X. Development and characterization of microsatellite markers for the lizardfish known as the Bombay duck, Harpadon nehereus (Synodontidae). Genet. Mol. Res. 2011, 10, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.H.; Li, H.Y.; Qin, Y.; Wang, R.X. Genetic diversity and population structure in Harpadon nehereus based on sequence-related amplified polymorphism markers. Genet. Mol. Res. 2014, 13, 5974–5981. [Google Scholar] [CrossRef]
- Guo, Y.J.; Yang, T.Y.; Meng, W.; Hang, Z.Q.; Gao, T.X. The genetic structure of the Bombay duck (Harpadon nehereus) based on mitochondrial Cyt b gene. Acta Hydrobiol. Sin. 2019, 43, 945–952. (In Chinese) [Google Scholar]
- Saha, S.; Ferdous, Z.; Jahan, H.; Khandaker, A.M.; Shahjahan, M.R.; Begum, R.A. Polymorphic loci analysis of 16S ribosomal RNA gene of economically important marine lizardfish Bombay duck (Harpadon nehereus). Bangl. J. Zool. 2019, 47, 49–57. [Google Scholar] [CrossRef]
- Zhang, H.; Xian, W.W. The complete mitochondrial genome of the larval Bombay duck Harpodon nehereus (Aulopiformes, Synodontidae) from Yangtze estuary and the phylogenetic relationship of Synodontidae species. Mitochondrial DNA B 2018, 3, 657–658. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.Y.; Jiang, Y.L.; Guo, Y.J.; Zhu, L.Q.; Lin, Y.J.; Zheng, Y.J.; Gao, T.X. Molecular phylogeny of Harpadon nehereus and its close relatives based on mitochondrial Cyt b gene. Trans. Oceanol. Limn. 2020, 6, 77–85. (In Chinese) [Google Scholar]
- Zhang, H.; Audira, G.; Li, Y.; Xian, W.W.; Varikkodan, M.M.; Hsiao, C.D. Comparative study the expression of calcium cycling genes in Bombay duck (Harpadon nehereus) and beltfish (Trichiurus lepturus) with different swimming activities. Genom. Data 2017, 12, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Chen, Y.Y.; Rong, L.I.; Xiao, H.; Chen, S.Y. Research advances in whole-genome sequencing of representative fish species. J. Biol. 2017, 34, 73–77. (In Chinese) [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1982. [Google Scholar]
- Chen, Y.X.; Chen, Y.S.; Shi, C.M.; Huang, Z.B.; Zhang, Y.; Li, S.K.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Oxford Open 2018, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.B.; Liu, B.H.; Xie, Y.L.; Li, Z.Y.; Huang, W.H.; Yuan, J.Y.; He, G.Z.; Chen, Y.X.; Pan, Q.; Liu, Y.J.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Li, R.Q.; Zhu, H.M.; Ruan, J.; Qian, W.B.; Fang, X.D.; Shi, Z.B.; Li, Y.R.; Li, S.T.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef]
- Kajitani, R.; Toshimoto, K.; Noguchi, H.; Toyoda, A.; Ogura, Y.; Okuno, M.; Yabana, M.; Harada, M.; Nagayasu, E.; Nagayasu, H.; et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014, 24, 1384–1395. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Miya, M.; Kawaguchi, A.; Nishida, M. Mitogenomic exploration of higher teleostean phylogenies: A case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol. Biol. Evol. 2001, 18, 1993–2009. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.L.; Liu, S.; Gao, P.; Luan, F.S. High-throughput sequencing technology and its application. J. Northeast Agric. Univ. (Engl. Ed.) 2014, 21, 84–96. [Google Scholar]
- Reuter, J.; Spacek, D.V.; Snyder, M. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S.; Chapman, J.; Stupka, E.; Putnam, N.; Chia, J.M.; Dehal, P.; Christoffels, A.; Rash, S.; Hoon, A.; Smit, A.; et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 2002, 297, 1301–1310. [Google Scholar] [CrossRef]
- Kasahara, M.; Naruse, K.; Sasaki, S.; Nakatani, Y.; Qu, W.; Ahsan, B.; Yamada, T.; Nagayasu, Y.; Doi, K.; Kasai, Y.; et al. The medaka draft genome and insights into vertebrate genome evolution. Nature 2007, 44, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef]
- Liu, Y.J.; Xu, P.; Xu, J.M.; Huang, Y.; Liu, Y.X.; Fang, H.; Hu, Y.C.; You, X.X.; Bian, C.; Sun, M.; et al. China is initiating the aquatic 10-100-1000 genomics program. Sci. China Life Sci. 2017, 60, 329–332. [Google Scholar] [CrossRef]
- Fan, G.Y.; Song, Y.; Yang, D.L.; Huang, X.Y.; Zhang, S.Y.; Zhang, M.Q.; Yang, X.W.; Chang, Y.; Zhang, H.; Li, Y.X.; et al. Initial data release and announcement of the 10,000 Fish Genomes Project (Fish10K). GigaScience 2020, 9, 1–8. [Google Scholar] [CrossRef]
- Wardle, F.; Mueller, F. Fish genomics: Casting the net wide. Brief. Funct. Genomics 2014, 13, 79–81. [Google Scholar] [CrossRef][Green Version]
- Chen, S.L.; Xu, W.T.; Liu, Y. Fish genomic research: Decade review and prospect. J. Fish. China 2019, 43, 1–14. (In Chinese) [Google Scholar]
- Dolezel, J.; Bartos, J.; Voglmayr, H.; Greilhuber, J. Nuclean DNA content and genome size of trout and human. Cytom. Part A 2003, 51, 127–128. [Google Scholar]
- Wang, K.; Wang, J.; Zhu, C.L.; Yang, L.D.; Ren, Y.D.; Ruan, J.; Fan, G.Y.; Hu, J.; Xu, W.J.; Bi, X.P.; et al. African lungfish genome sheds light on the vertebrate water-to-land transition. Cell 2021, 184, 1362–1376. [Google Scholar] [CrossRef]
- Wu, C.W.; Zhang, D.; Kan, M.Y.; Lv, Z.M.; Zhu, A.Y.; Su, Y.Q.; Zhou, D.Z.; Zhang, J.S.; Zhang, Z.; Xu, M.Y.; et al. The draft genome of the large yellow croaker reveals well-developed innate immunity. Nat. Commun. 2014, 5, 5227. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.W.; Bao, B.L.; Xie, Z.Y.; Chen, X.Y.; Li, B.; Jia, X.D.; Yao, Q.L.; Ortí, G.; Li, W.H.; Li, X.H.; et al. The genome and transcriptome of Japanese flounder provide insights into flatfish asymmetry. Nat. Genet. 2017, 49, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.W.; Li, C.; Wang, N.; Qin, Y.T.; Xu, W.T.; Liu, Q.; Zhou, Q.; Zhao, Y.; Li, X.H.; Liu, S.S.; et al. Chromosome-level genome assembly of the spotted sea bass, Lateolabrax maculatus. GigaScience 2018, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Li, F.H.; Ge, J.L.; Wang, P.F.; Chang, Q.; Zhang, S.N.; Li, J.; Liu, C.L.; Liu, K.; Liu, X.T.; et al. Chromosome-level genome assembly of the greenfin horse-faced filefish (Thamnaconus septentrionalis) using Oxford Nanopore PromethION sequencing and Hi-C technology. Mol. Ecol. Resour. 2020, 20, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Aird, D.; Ross, M.G.; Chen, W.S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Q.; Jiang, D.N.; Li, M.; Mustapha, U.F.; Tian, C.X.; Chen, H.P.; Huang, Y.; Deng, S.P.; Wu, T.L.; Zhu, C.H.; et al. Genome survey of male and female spotted scat (Scatophagus argus). Animals 2019, 9, 1117. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Y.; Song, N.; Xiao, S.J.; Gao, T.X. Whole genome survey analysis and microsatellite motif identification of Sebastiscus marmoratus. Biosci. Rep. 2020, 40, 1–7. [Google Scholar] [CrossRef]
- Chen, B.J.; Sun, Z.C.; Lou, F.R.; Gao, T.X.; Song, N. Genomic characteristics and profile of microsatellite primers for Acanthogobius ommaturus by genome survey sequencing. Biosci. Rep. 2020, 40, 1–8. [Google Scholar] [CrossRef]
- Li, Z.Y.; Tian, C.X.; Huang, Y.; Lin, X.H.; Wang, Y.R.; Jiang, D.N.; Zhu, C.H.; Chen, H.P.; Li, G.L. A first insight into a draft genome of silver sillago (Sillago sihama) via genome survey sequencing. Animals 2019, 9, 756. [Google Scholar] [CrossRef] [PubMed]
- Miya, M.; Takeshima, H.; Endo, H.; Ishiguro, N.B.; Inoue, J.G.; Mukai, T.; Satoh, T.P.; Yamaguchi, M.; Kawaguchi, A.; Mabuchi, K.; et al. Major patterns of higher teleostean phylogenies: A new perspective based on 100 complete mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2003, 26, 121–138. [Google Scholar] [CrossRef]
- Rosen, D.E. Interrelationships of higher euteleostean fishes. Zool. J. Linn. Soc. 1973, 53, 397–513. [Google Scholar]
- Nelson, J.S. Fishes of the World, 2nd ed.; John & Wiley & Sons: Hoboken, NJ, USA, 1984. [Google Scholar]
- Stiassny, M.L.J. Basal ctenosquamate relationships and the interrelationships of the Myctophiform (Scopelomorph) fishes. In Interrelationships of Fishes; Stiassny, M.L.J., Parenti, L.R., Johnson, G.D., Eds.; Academic Press: Cambridge, UK, 1996; pp. 405–426. [Google Scholar]
- Poulsen, J.Y.; Byrkjedal, I.; Willassen, E.; Rees, D.; Takeshima, H.; Satoh, T.P.; Shinohara, G.; Nishida, M.; Miya, M. Mitogenomic sequences and evidence from unique gene rearrangements corroborate evolutionary relationships of Myctophiformes (Neoteleostei). BMC Evol. Biol. 2013, 13, 1–21. [Google Scholar] [CrossRef]
- Denton, J.S.S. Seven-locus molecular phylogeny of Myctophiformes (Teleostei; Scopelomorpha) highlights the utility of the order for studies of deep-sea evolution. Mol. Phylogenet. Evol. 2014, 76, 270–292. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.P.; Davis, M.P. The evolution of specialized dentition in the deep-sea lanternfishes (Myctophiformes). J. Morphol. 2020, 281, 536–555. [Google Scholar] [CrossRef] [PubMed]
- Nugroho, E.D.; Nawir, D.; Amin, M.; Lestari, U. DNA barcoding of nomei fish (Synodontidae: Harpadon sp.) in Tarakan Island, Indonesia. AACL Bioflux 2017, 10, 1466–1474. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Length (bp) | Total Number | Total Number (≥2 kb) | Max Length (bp) | N50 (bp) | N90 (bp) | |
|---|---|---|---|---|---|---|---|
| H. nehereus | contigs | 1,352,668,147 | 3,766,078 | 73,281 | 30,284 | 596 | 138 |
| scaffolds | 1,363,443,545 | 2,539,084 | 133,089 | 92,118 | 1568 | 162 |
| SSR Type | Number | Percent (%) | Sequence Number | Total SSR Length (bp) |
|---|---|---|---|---|
| p1 | 266,761 | 25.96 | 208,219 | 3,369,363 |
| p2 | 558,543 | 54.35 | 400,985 | 17,745,246 |
| p3 | 66,270 | 6.45 | 62,046 | 1,638,864 |
| p4 | 105,426 | 10.26 | 97,016 | 3,527,488 |
| p5 | 25,182 | 2.45 | 23,958 | 883,000 |
| p6 | 5469 | 0.53 | 5374 | 191,406 |
| Total | 1,027,651 | 100 | 797,598 | 27,355,367 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.; Huang, X.; Ning, Z.; Gao, T. Genome-Wide Survey Reveals the Microsatellite Characteristics and Phylogenetic Relationships of Harpadon nehereus. Curr. Issues Mol. Biol. 2021, 43, 1282-1292. https://doi.org/10.3390/cimb43030091
Yang T, Huang X, Ning Z, Gao T. Genome-Wide Survey Reveals the Microsatellite Characteristics and Phylogenetic Relationships of Harpadon nehereus. Current Issues in Molecular Biology. 2021; 43(3):1282-1292. https://doi.org/10.3390/cimb43030091
Chicago/Turabian StyleYang, Tianyan, Xinxin Huang, Zijun Ning, and Tianxiang Gao. 2021. "Genome-Wide Survey Reveals the Microsatellite Characteristics and Phylogenetic Relationships of Harpadon nehereus" Current Issues in Molecular Biology 43, no. 3: 1282-1292. https://doi.org/10.3390/cimb43030091
APA StyleYang, T., Huang, X., Ning, Z., & Gao, T. (2021). Genome-Wide Survey Reveals the Microsatellite Characteristics and Phylogenetic Relationships of Harpadon nehereus. Current Issues in Molecular Biology, 43(3), 1282-1292. https://doi.org/10.3390/cimb43030091