
Targeting Mitochondrial ROS Production to Reverse the Epithelial-Mesenchymal Transition in Breast Cancer Cells

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Lines, Drug Treatment and Hypoxia Induction
2.3. Scratch Wound Healing Assay
2.4. Evaluation of ROS Levels
2.5. Western Blot Analysis
2.6. Construction of Lentiviral Vectors
2.7. Generation of Lentiviral Particles and Target Cell Infection
2.8. Evaluation of Cell Viability
2.9. Statistical Analysis
3. Results
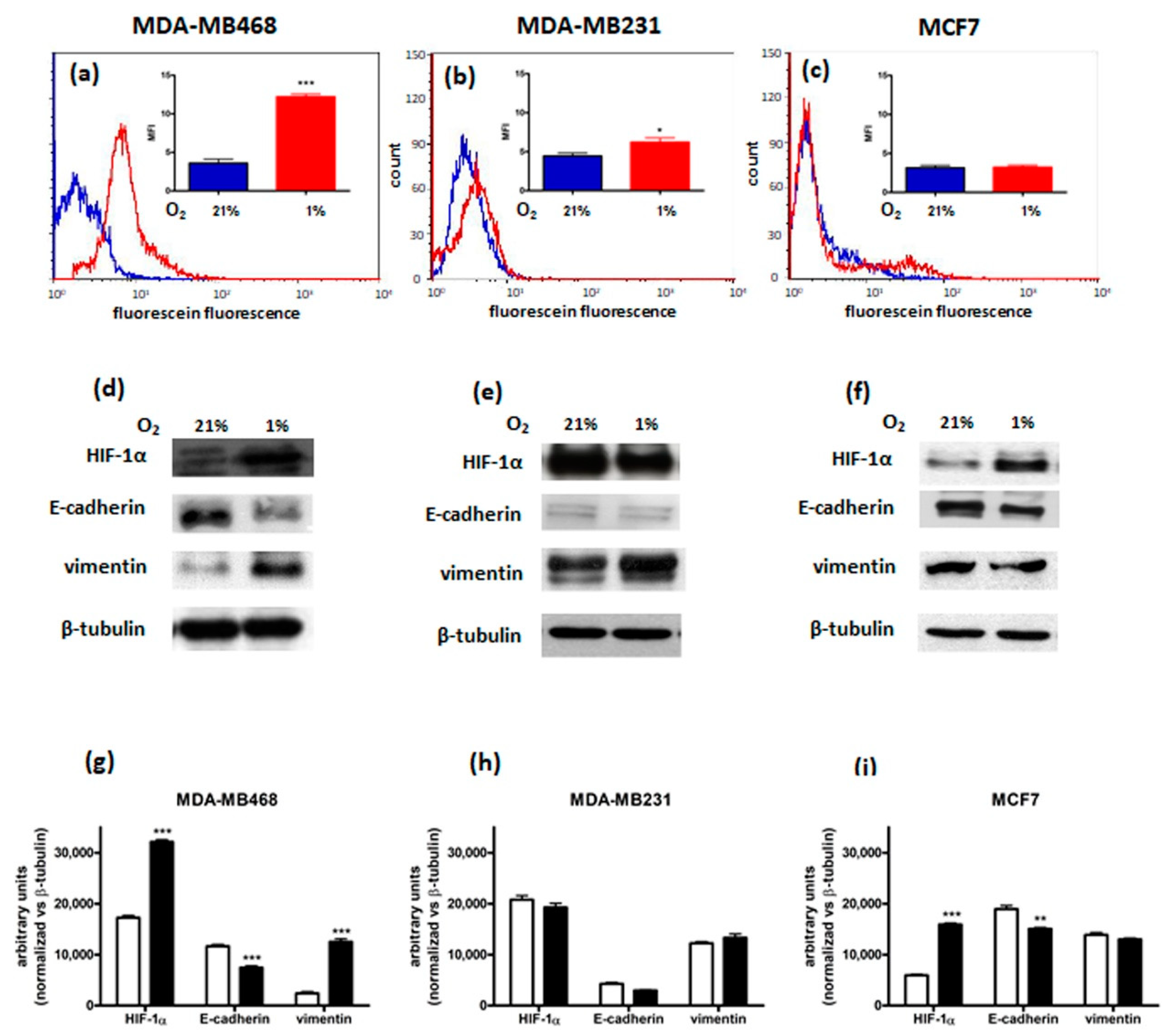
3.1. Characterization of Breast Cancer Cell Lines
3.2. Effects of the Inhibition of ROS Production on Hypoxia-Induced Responses
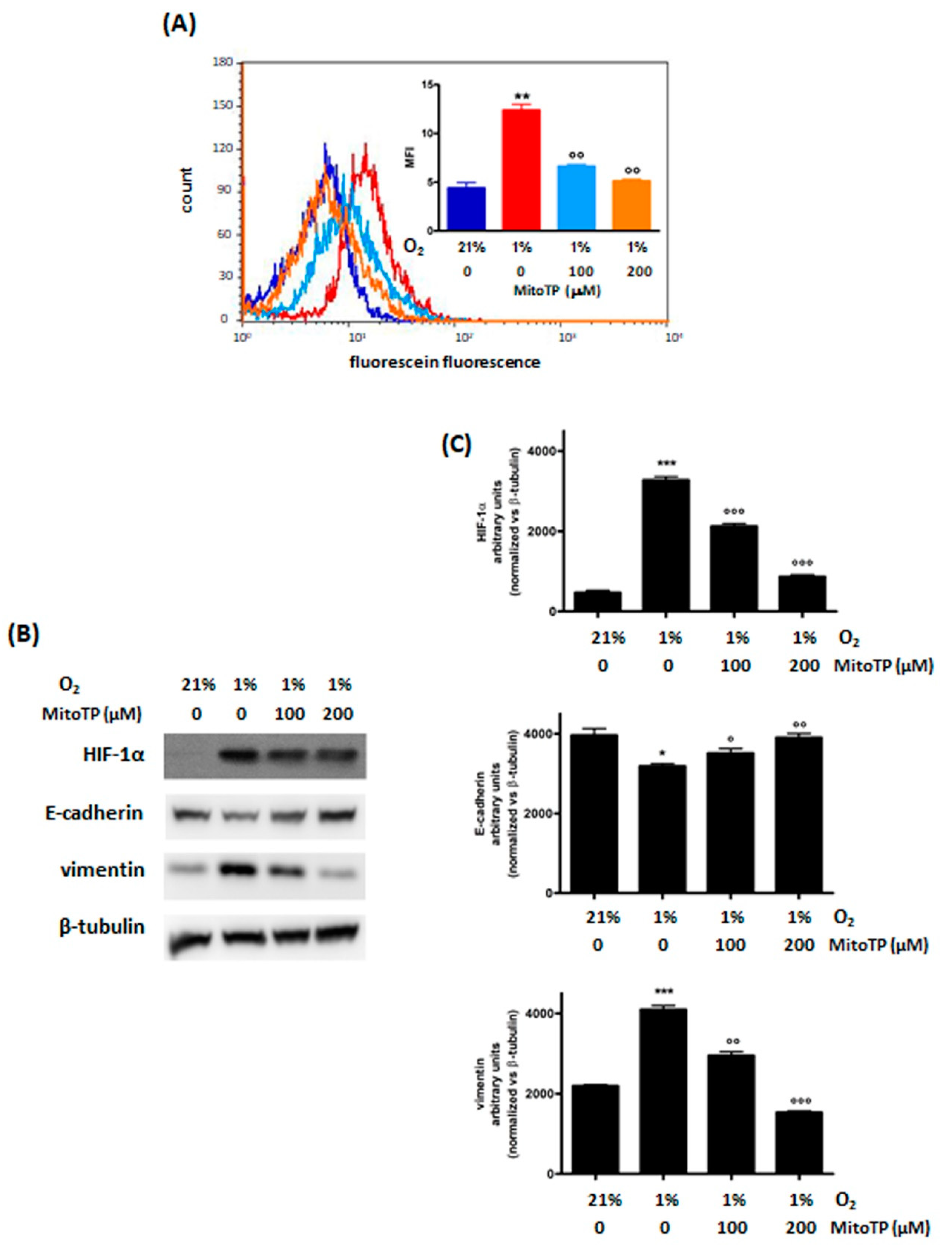
3.2.1. Decrease of ROS Levels Using MitoTP
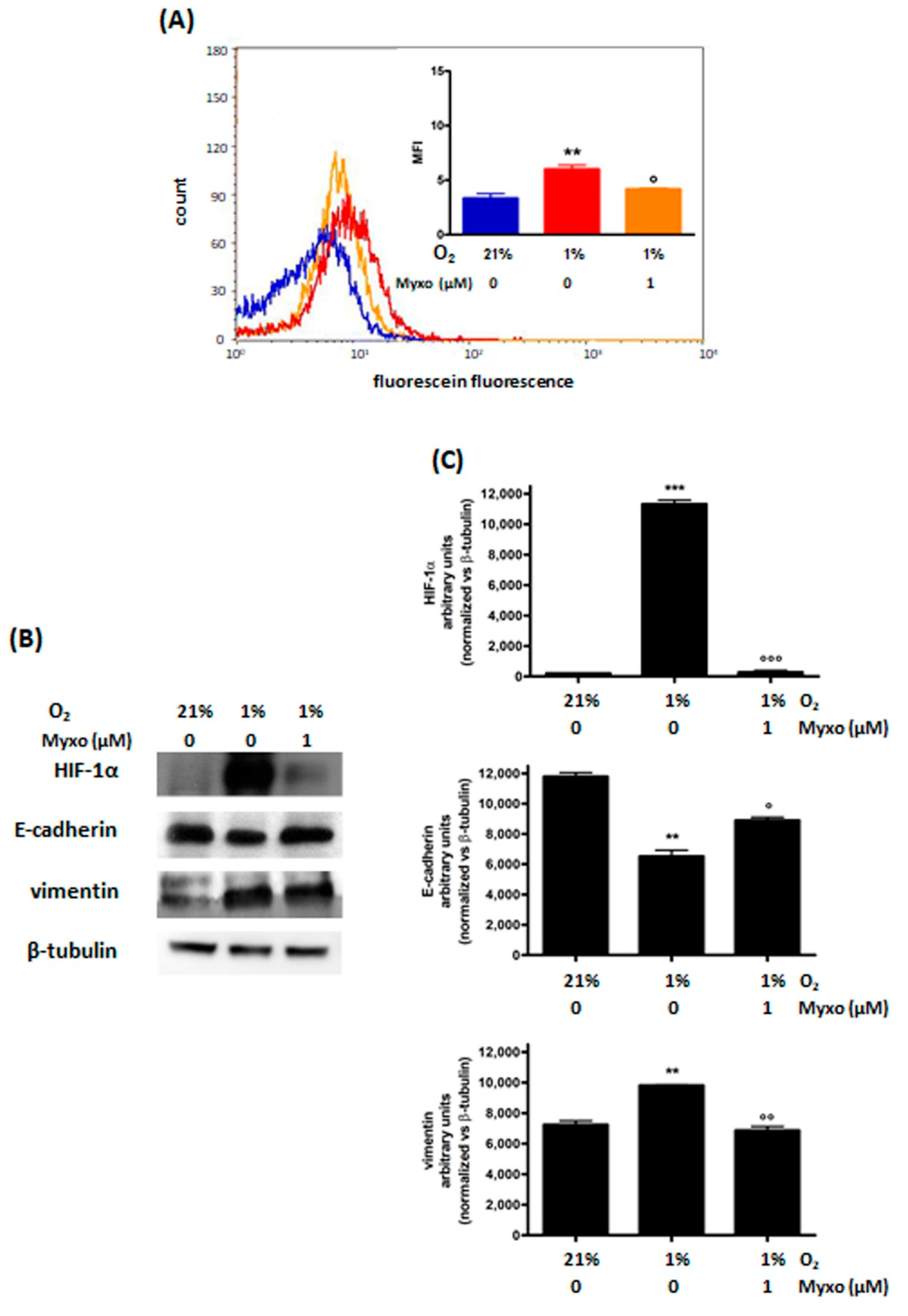
3.2.2. Decrease of ROS Levels Using Inhibitors of Mitochondrial Complex III
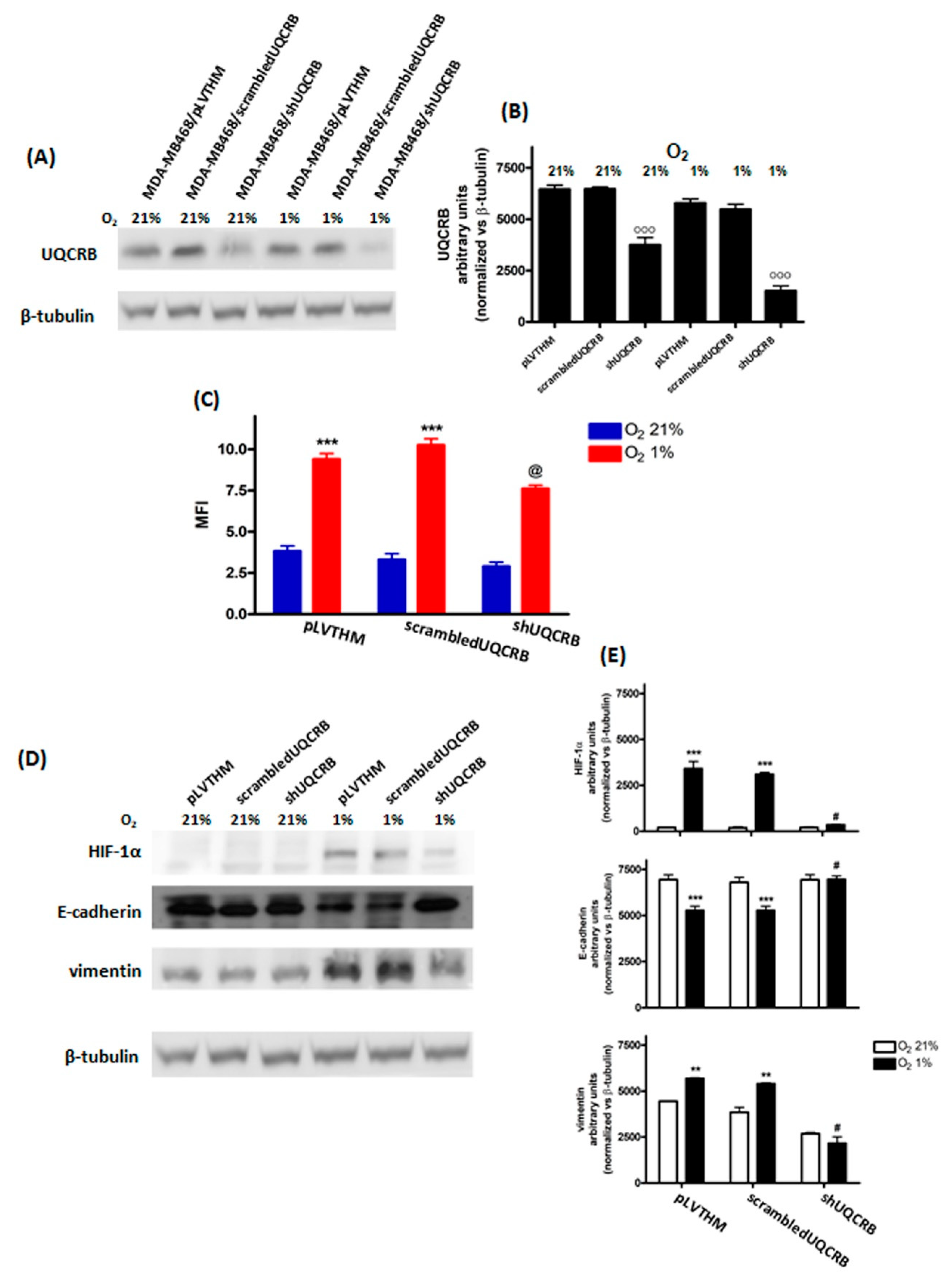
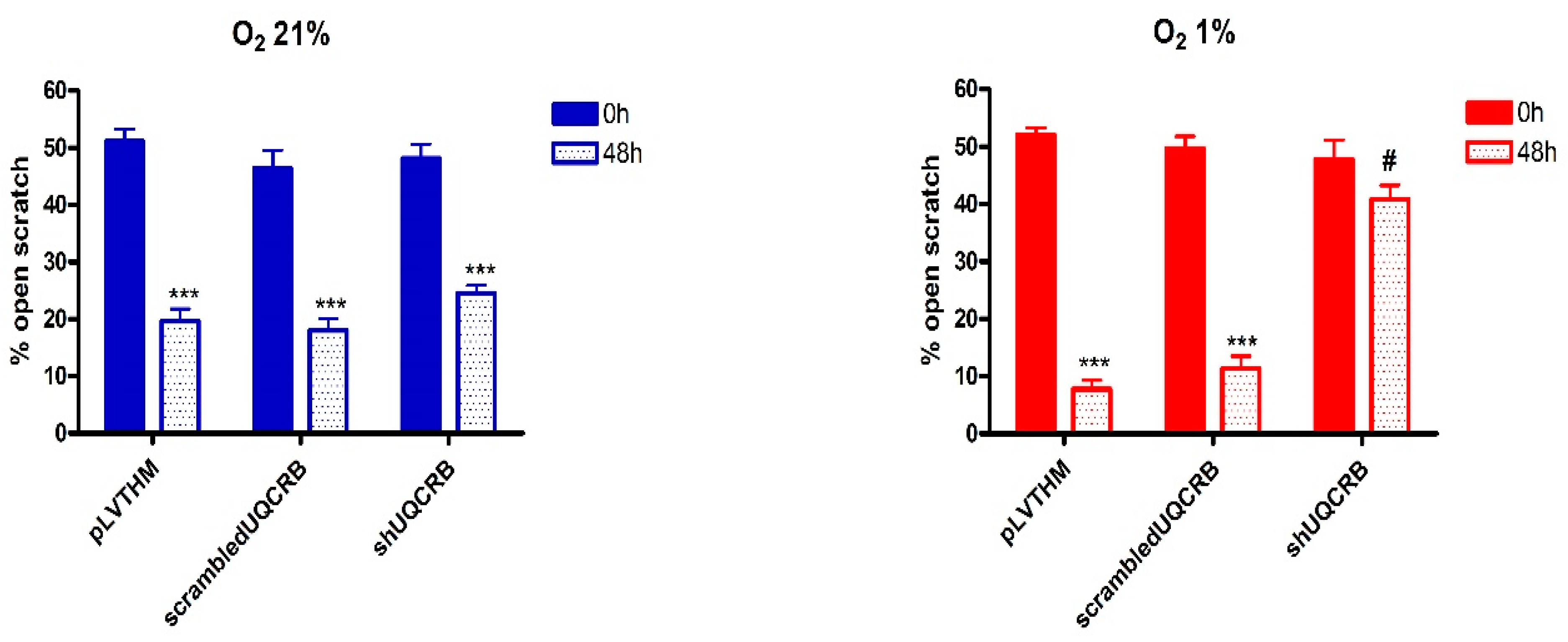
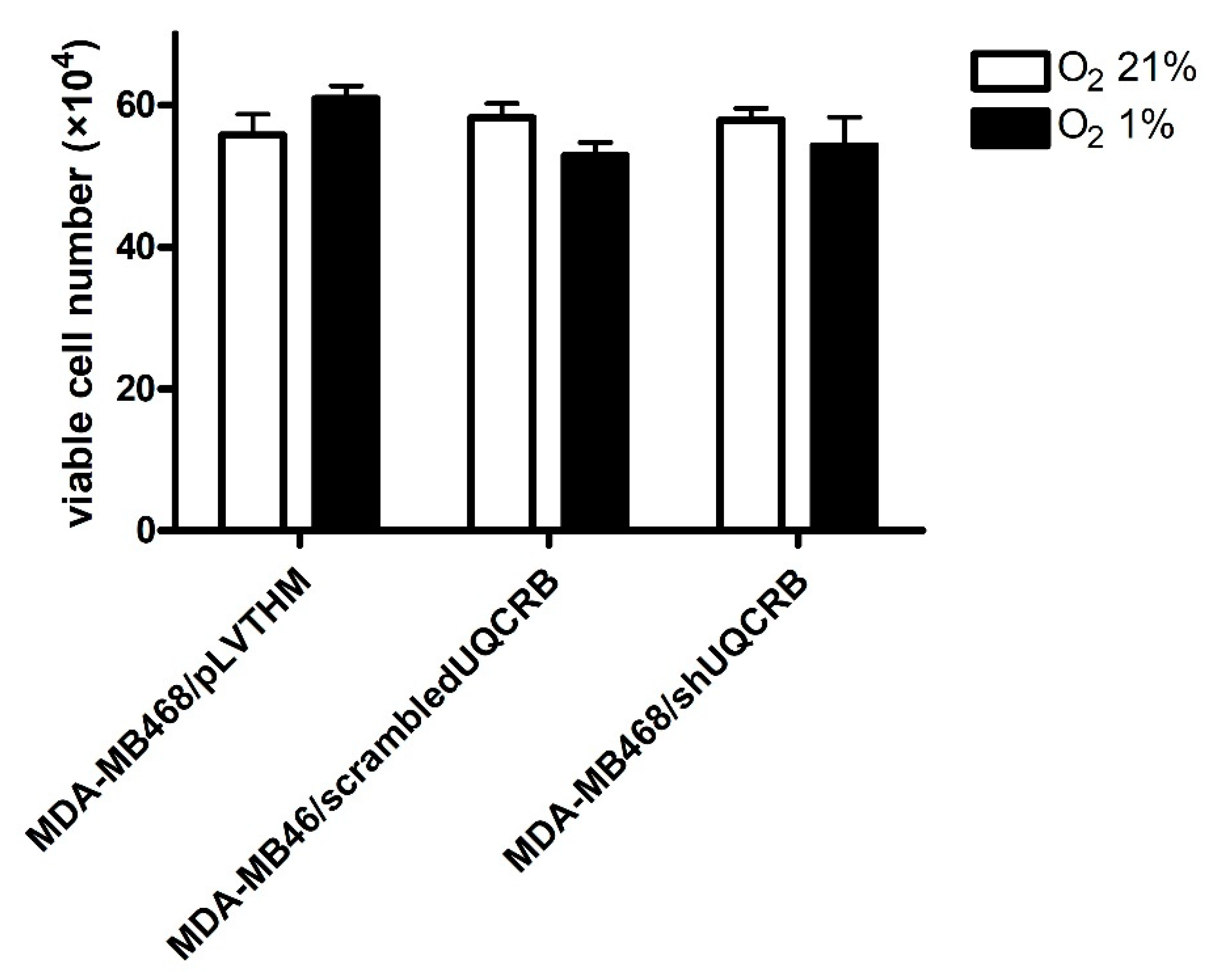
3.3. Effects of the UQCRB Silencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoni, E.; Parri, M.; Chiarugi, P. EMT and oxidative stress: A bidirectional interplay affecting tumor malignancy. Antioxid. Redox Signal 2012, 16, 1248–1263. [Google Scholar] [CrossRef]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell. 2019, 49, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1, upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salnikov, A.V.; Liu, L.; Platen, M.; Gladkich, J.; Salnikova, O.; Ryschich, E.; Mattern, J.; Moldenhauer, G.; Werner, J.; Schemmer, P.; et al. Hypoxia Induces EMT in Low and Highly Aggressive Pancreatic Tumor Cells but Only Cells with Cancer Stem Cell Characteristics Acquire Pronounced Migratory Potential. PLoS ONE 2012, 7, e46391. [Google Scholar]
- Liu, Z.-J.; Semenza, G.L.; Zhang, H.-F. Hypoxia-inducible factor 1 and breast cancer metastasis. J. Zhejiang Univ. Sci. B 2015, 16, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol. Med. 2012, 18, 534–543. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.L.; Chandel, N.S. Mitochondrial oxygen sensing: Regulation of hypoxia-inducible factor by mitochondrial generated reactive oxygen species. Essays Biochem. 2007, 43, 17–27. [Google Scholar] [PubMed]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial Complex III stabilize hypoxia-inducible factor-1α during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S. Mitochondrial complex III: An essential component of universal oxygen sensing machinery? Respir. Physiol. Neurobiol. 2010, 174, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Cooley, J.W. Protein conformational changes involved in the cytochrome bc1 complex catalytic cycle. Biochim. Biophys. Acta-Bioenerg. 2013, 1827, 1340–1345. [Google Scholar] [CrossRef] [Green Version]
- Cramer, W.A.; Hasan, S.S.; Yamashita, E. The Q cycle of cytochrome bc complexes: A structure perspective. Biochim. Biophys. Acta-Bioenerg. 2011, 1807, 788–802. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Gerencser, A.A.; Treberg, J.R.; Brand, M.D. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 2011, 286, 31361–31372. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.J.; Shim, J.S.; Lee, J.; Song, Y.M.; Park, K.C.; Choi, S.H.; Kim, N.D.; Yoon, J.H.; Mungai, P.T.; Schumacker, P.T.; et al. Terpestacin inhibits tumor angiogenesis by targeting UQCRB of mitochondrial complex III and suppressing hypoxia-induced reactive oxygen species production and cellular oxygen sensing. J. Biol. Chem. 2010, 285, 11584–11595. [Google Scholar]
- Jung, H.J.; Kim, Y.; Chang, J.; Kang, S.W.; Kim, J.H.; Kwon, H.J. Mitochondrial UQCRB regulates VEGFR2 signaling in endothelial cells. J. Mol. Med. 2013, 91, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.; Kwon, H.J.; Jung, H.J. Downregulation of mitochondrial UQCRB inhibits cancer stem cell-like properties in glioblastoma. Int. J. Oncol. 2018, 52, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast cancer cell line classification and Its relevance with breast tumor subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, E.; Marras, E.; Prini, P.; Gariboldi, M.B. Luteolin impairs hypoxia adaptation and progression in human breast and colon cancer cells. Eur. J. Pharmacol. 2020, 881, 173210. [Google Scholar] [CrossRef]
- Ye, J.; Jiang, Z.; Chen, X.; Liu, M.; Li, J.; Liu, N. Electron transport chain inhibitors induce microglia activation through enhancing mitochondrial reactive oxygen species production. Exp. Cell Res. 2016, 340, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Young, T.A.; Cunningham, C.C.; Bailey, S.M. Reactive oxygen species production by the mitochondrial respiratory chain in isolated rat hepatocytes and liver mitochondria: Studies using myxothiazol. Arch. Biochem. Biophys. 2002, 405, 65–72. [Google Scholar] [CrossRef]
- Luo, J.; Li, N.; Robinson, J.P.; Shi, R. The increase of reactive oxygen species and their inhibition in an isolated guinea pig spinal cord compression model. Spinal Cord. 2002, 40, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Zielonka, J.; Vasquez-Vivar, J.; Kalyanaraman, B. Detection of 2-hydroxyethidium in cellular systems: A unique marker product of superoxide and hydroethidine. Nat. Protoc. 2008, 3, 8–21. [Google Scholar] [CrossRef]
- Osti, D.; Marras, E.; Ceriani, I.; Grassini, G.; Rubino, T.; Viganò, D.; Parolaro, D.; Perletti, G. Comparative analysis of molecular strategies attenuating positional effects in lentiviral vectors carrying multiple genes. J. Virol. Methods 2006, 136, 93–101. [Google Scholar] [CrossRef]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.-P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, M.Y.; Spivak-Kroizman, T.R.; Powis, G. HIF-1 regulation: Not so easy come, easy go. Trends Biochem. Sci. 2008, 33, 526–534. [Google Scholar] [CrossRef]
- Naranjo-Suarez, S.; Carlson, B.A.; Tsuji, P.A.; Yoo, M.-H.; Gladyshev, V.N.; Hatfield, D.L. HIF-independent regulation of thioredoxin reductase 1 contributes to the high levels of reactive oxygen species induced by hypoxia. PLoS ONE 2012, 7, e30470. [Google Scholar] [CrossRef] [Green Version]
- Kallergi, G.; Papadaki, M.A.; Politaki, E.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res. 2011, 13, R59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medici, D.; Hay, E.D.; Olsen, B.R. Snail and slug promote epithelial-mesenchymal transition through β-catenin-T-cell factor-4-dependent expression of transforming growth factor-β3. Mol. Biol. Cell. 2008, 19, 4875–4887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; David, C.A.; Donnelly, J.B.; Michaelides, M.; Chandel, N.S.; Huang, X.; Warrior, U.; Weinberg, F.; Tormos, K.V.; Fesik, S.W.; et al. A chemical genomics screen highlights the essential role of mitochondria in HIF-1 regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Le Gal, K.; Wiel, C.; Ibrahim, M.X.; Henricsson, M.; Sayin, V.I.; Bergo, M.O. Mitochondria-targeted antioxidants MitoQ and mitoTEMPO do not influence BRAF-driven malignant melanoma and KRAS-driven lung cancer progression in mice. Antioxidants 2021, 10, 163. [Google Scholar] [CrossRef]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Dada, L.A.; Wu, M.; Kelly, A.; Trejo, H.; Zhou, Q.; Varga, J.; Sznajder, J.I. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am. J. Physiol.-Lung Cell Mol. Physiol. 2009, 297, L1120–L1130. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial–mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 17036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate N-cadherin and SERPINE1 expression, EGFR signalling and motility in MDA-MB-468 breast cancer cells. Sci. Rep. 2017, 7, 15140. [Google Scholar] [CrossRef]
- Rakowski, M.; Porębski, S.; Grzelak, A. Silver nanoparticles modulate the epithelial-to-mesenchymal transition in estrogen-dependent breast cancer cells in vitro. Int. J. Mol. Sci. 2021, 22, 9203. [Google Scholar] [CrossRef] [PubMed]
- Shagieva, G.; Domnina, L.; Makarevich, O.; Chernyak, B.; Skulachev, V.; Dugina, V. Depletion of mitochondrial reactive oxygen species downregulates epithelial-to-mesenchymal transition in cervical cancer cells. Oncotarget 2017, 8, 4901–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.-K.; Suh, Y.; Yoo, K.-C.; Cui, Y.-H.; Kim, H.; Kim, M.-J.; Kim, I.G.; Lee, S.-J. Activation of KRAS promotes the mesenchymal features of Basal-type breast cancer. Exp. Mol. Med. 2015, 47, e137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, M.; Lester, R.D.; Montel, V.; Eastman, B.; Takimoto, S.; Gonias, S.L. Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. J. Biol. Chem. 2009, 284, 22825–22833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta-Bioenerg. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Patten, D.A.; Lafleur, V.N.; Robitaille, G.A.; Chan, D.A.; Giaccia, A.J.; Richard, D.E. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: The essential role of mitochondrial-derived reactive oxygen species. Mol. Biol. Cell. 2010, 21, 3247–3257. [Google Scholar] [CrossRef] [Green Version]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.S.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell. Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Theys, J.; Jutten, B.; Habets, R.; Paesmans, K.; Groot, A.J.; Lambin, P.; Wouters, B.G.; Lammering, G.; Vooijs, M. E-Cadherin loss associated with EMT promotes radioresistance in human tumor cells. Radiother. Oncol. 2011, 99, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Zhao, Q. Hypoxia-inducible factor 1α participates in hypoxia-induced epithelial-mesenchymal transition via response gene to complement 32. Exp. Ther. Med. 2017, 14, 1825–1831. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Tianjie, L.I.U.; Wang, K.; Wang, X.; Shan, X.U.; Dalin, H.E.; Zeng, J. Transcriptional regulation of FoxM1 by HIF-1α mediates hypoxia-induced EMT in prostate cancer. Oncol. Rep. 2019, 42, 1307–1318. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Djamgoz, M.B.A. Triple negative breast cancer: Emerging therapeutic modalities and novel combination therapies. Cancer Treat. Rev. 2018, 62, 110–122. [Google Scholar] [CrossRef]
- Yu, K.-D.; Zhu, R.; Zhan, M.; Rodriguez, A.A.; Yang, W.; Wong, S.; Makris, A.; Lehmann, B.D.; Chen, X.; Mayer, I.; et al. Identification of prognosis-relevant subgroups in patients with chemoresistant triple-negative breast cancer. Clin. Cancer Res. 2013, 19, 2723–2733. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MDA-MB468 | MDA-MB231 | MCF7 | ||||
|---|---|---|---|---|---|---|
| Time (h) | O2 21% | O2 1% | O2 21% | O2 1% | O2 21% | O2 1% |
| 0 | 80.0 ± 2.0 | 75.0 ± 1.0 | 78.0 ± 2.0 | 78.0 ± 3.0 | 76.5 ± 1.5 | 78.0 ± 3.0 |
| 48 | 54.5 ± 0.5 * | 36.5 ± 4.0 *** | 59.0 ± 1.5 * | 46.5 ± 1.5 *** | 74.0 ± 1.0 | 74.5 ± 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monti, E.; Mancini, A.; Marras, E.; Gariboldi, M.B. Targeting Mitochondrial ROS Production to Reverse the Epithelial-Mesenchymal Transition in Breast Cancer Cells. Curr. Issues Mol. Biol. 2022, 44, 5277-5293. https://doi.org/10.3390/cimb44110359
Monti E, Mancini A, Marras E, Gariboldi MB. Targeting Mitochondrial ROS Production to Reverse the Epithelial-Mesenchymal Transition in Breast Cancer Cells. Current Issues in Molecular Biology. 2022; 44(11):5277-5293. https://doi.org/10.3390/cimb44110359
Chicago/Turabian StyleMonti, Elena, Alessandro Mancini, Emanuela Marras, and Marzia Bruna Gariboldi. 2022. "Targeting Mitochondrial ROS Production to Reverse the Epithelial-Mesenchymal Transition in Breast Cancer Cells" Current Issues in Molecular Biology 44, no. 11: 5277-5293. https://doi.org/10.3390/cimb44110359