
Single Circulating-Tumor-Cell-Targeted Sequencing to Identify Somatic Variants in Liquid Biopsies in Non-Small-Cell Lung Cancer Patients

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Enrollment of Subjects
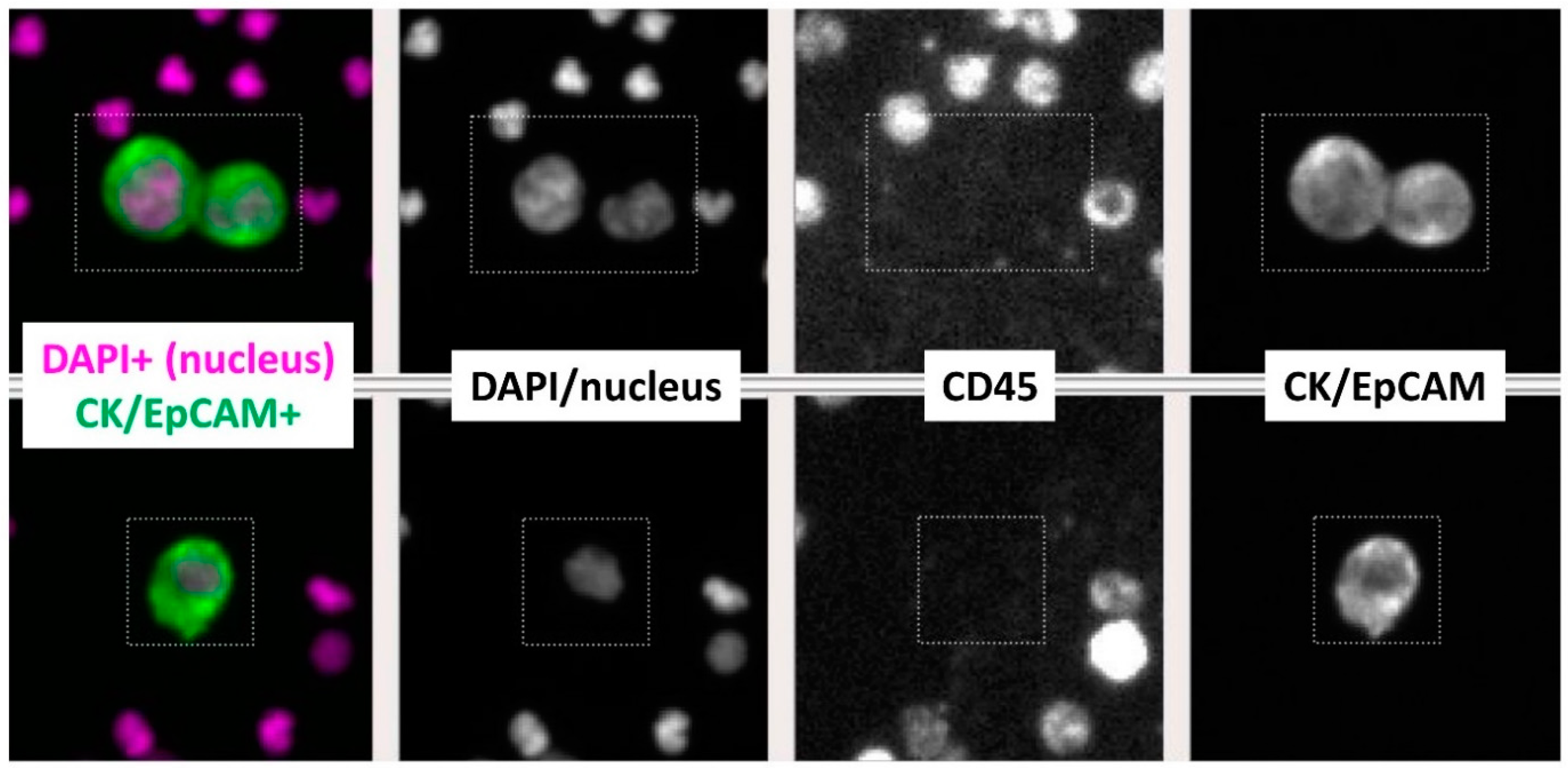
2.2. CTC Enumeration with NSCLC Cell Line Cells Spiked into Healthy Human Blood and Study Subjects’ Blood Samples
2.3. Targeted NGS of Single CTCs
2.4. Somatic Variant (SNVs and Indels) Analysis
2.5. Statistical Analysis
3. Results
3.1. Clinical Characteristics of Subjects
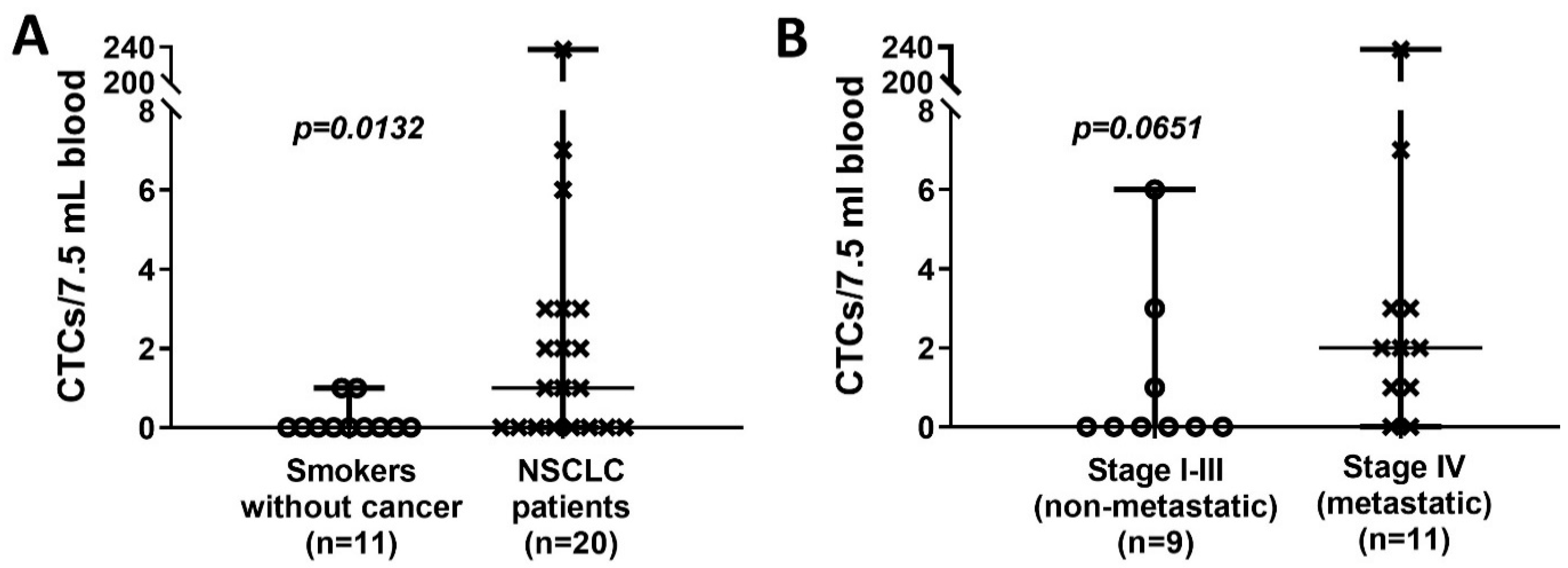
3.2. CTC Enumeration in NSCLC Patients and Chronic Smokers without Cancer
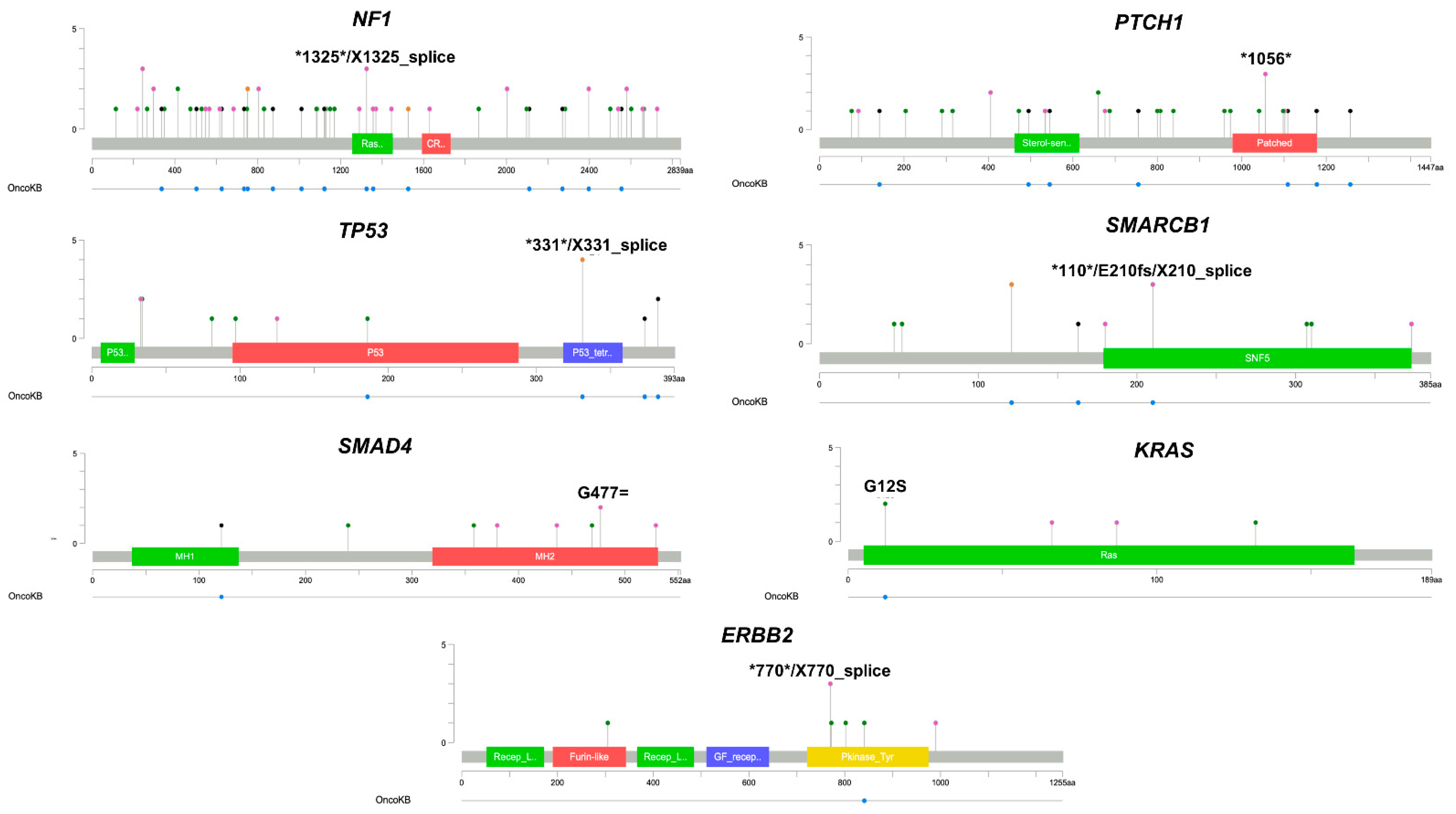
3.3. Characterization of Single CTCs Somatic Variants in NSCLC Patients
3.4. Single CTC Somatic Variants Detected in Oncogenes and Tumor-Suppressor Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Aberle, D.R.; Adams, A.M.; Berg, C.D.; Black, W.C.; Clapp, J.D.; Fagerstrom, R.M.; Gareen, I.F.; Gatsonis, C.; Marcus, P.M.; Sicks, J.D. Reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detterbeck, F.C.; Chansky, K.; Groome, P.; Bolejack, V.; Crowley, J.; Shemanski, L.; Kennedy, C.; Krasnik, M.; Peake, M.; Rami-Porta, R.; et al. The IASLC Lung Cancer Staging Project: Methodology and Validation Used in the Development of Proposals for Revision of the Stage Classification of NSCLC in the Forthcoming (Eighth) Edition of the TNM Classification of Lung Cancer. J. Thorac. Oncol. 2016, 11, 1433–1446. [Google Scholar] [CrossRef] [Green Version]
- National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology: Non-Small Cell Lung Cancer (Version 6.2020). Available online: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (accessed on 1 July 2021).
- Horn, L.; Whisenant, J.G.; Wakelee, H.; Reckamp, K.L.; Qiao, H.; Leal, T.A.; Du, L.; Hernandez, J.; Huang, V.; Blumenschein, G.R.; et al. Monitoring Therapeutic Response and Resistance: Analysis of Circulating Tumor DNA in Patients With ALK+ Lung Cancer. J. Thorac. Oncol. 2019, 14, 1901–1911. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Brannon, A.R.; Ferris, L.A.; Campbell, C.D.; Lin, J.J.; Schultz, K.R.; Ackil, J.; Stevens, S.; Dardaei, L.; Yoda, S.; et al. Tracking the Evolution of Resistance to ALK Tyrosine Kinase Inhibitors through Longitudinal Analysis of Circulating Tumor DNA. JCO Precis. Oncol. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Thompson, J.C.; Chien, A.L.; Quinn, K.J.; Hwang, W.T.; Black, T.A.; Yee, S.S.; Christensen, T.E.; LaRiviere, M.J.; Silva, B.A.; et al. Baseline Plasma Tumor Mutation Burden Predicts Response to Pembrolizumab-based Therapy in Patients with Metastatic Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2354–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alix-Panabieres, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef]
- Rolfo, C.; Mack, P.; Scagliotti, G.V.; Aggarwal, C.; Arcila, M.E.; Barlesi, F.; Bivona, T.; Diehn, M.; Dive, C.; Dziadziuszko, R.; et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement From the International Association for the Study of Lung Cancer. J. Thorac. Oncol. 2021, 16, 1647–1662. [Google Scholar] [CrossRef]
- Powles, T.; Assaf, Z.J.; Davarpanah, N.; Banchereau, R.; Szabados, B.E.; Yuen, K.C.; Grivas, P.; Hussain, M.; Oudard, S.; Gschwend, J.E.; et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature 2021, 595, 432–437. [Google Scholar] [CrossRef]
- Rostami, A.; Lambie, M.; Yu, C.W.; Stambolic, V.; Waldron, J.N.; Bratman, S.V. Senescence, Necrosis, and Apoptosis Govern Circulating Cell-free DNA Release Kinetics. Cell Rep. 2020, 31, 107830. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Revythis, A.; Shah, S.; Kutka, M.; Moschetta, M.; Ozturk, M.A.; Pappas-Gogos, G.; Ioannidou, E.; Sheriff, M.; Rassy, E.; Boussios, S. Unraveling the Wide Spectrum of Melanoma Biomarkers. Diagnostics 2021, 11, 1341. [Google Scholar] [CrossRef] [PubMed]
- Morrow, C.J.; Trapani, F.; Metcalf, R.L.; Bertolini, G.; Hodgkinson, C.L.; Khandelwal, G.; Kelly, P.; Galvin, M.; Carter, L.; Simpson, K.L.; et al. Tumourigenic non-small-cell lung cancer mesenchymal circulating tumour cells: A clinical case study. Ann. Oncol. 2016, 27, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Onidani, K.; Shoji, H.; Kakizaki, T.; Yoshimoto, S.; Okaya, S.; Miura, N.; Sekikawa, S.; Furuta, K.; Lim, C.T.; Shibahara, T.; et al. Monitoring of cancer patients via next-generation sequencing of patient-derived circulating tumor cells and tumor DNA. Cancer Sci. 2019, 110, 2590–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alix-Panabieres, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Campton, D.E.; Ramirez, A.B.; Nordberg, J.J.; Drovetto, N.; Clein, A.C.; Varshavskaya, P.; Friemel, B.H.; Quarre, S.; Breman, A.; Dorschner, M.; et al. High-recovery visual identification and single-cell retrieval of circulating tumor cells for genomic analysis using a dual-technology platform integrated with automated immunofluorescence staining. BMC Cancer 2015, 15, 360. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/proj (accessed on 15 January 2021).
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra, 1st ed.; O’Reilly Media: Sebastopol, VA, USA, 2020. [Google Scholar]
- Benjamin, D.; Sato, T.; Cibulskis, K.; Getz, G.; Stewart, C.; Lichtenstein, L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic. Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic. Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic. Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 30 January 2021).
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Hodgkinson, C.L.; Morrow, C.J.; Li, Y.; Metcalf, R.L.; Rothwell, D.G.; Trapani, F.; Polanski, R.; Burt, D.J.; Simpson, K.L.; Morris, K.; et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat. Med. 2014, 20, 897–903. [Google Scholar] [CrossRef]
- Gray, P.N.; Dunlop, C.L.; Elliott, A.M. Not All Next Generation Sequencing Diagnostics are Created Equal: Understanding the Nuances of Solid Tumor Assay Design for Somatic Mutation Detection. Cancers 2015, 7, 1313–1332. [Google Scholar] [CrossRef]
- Redig, A.J.; Capelletti, M.; Dahlberg, S.E.; Sholl, L.M.; Mach, S.; Fontes, C.; Shi, Y.; Chalasani, P.; Janne, P.A. Clinical and Molecular Characteristics of NF1-Mutant Lung Cancer. Clin. Cancer Res. 2016, 22, 3148–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Sun, Q. TP53 mutations, expression and interaction networks in human cancers. Oncotarget 2017, 8, 624–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, J.S.; Alnoor, F.; Sadiq, Q.; Solares, J.; Gradowski, J.F. SMARCA4 (BRG1) and SMARCB1 (INI1) expression in TTF-1 negative neuroendocrine carcinomas including merkel cell carcinoma. Pathol. Res. Pract. 2021, 219, 153341. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, H.; Ranaweera, R.S.; Izumchenko, E.; Makarev, E.; Zhavoronkov, A.; Fertig, E.J.; Howard, J.D.; Markovic, A.; Bedi, A.; Ravi, R.; et al. SMAD4 Loss Is Associated with Cetuximab Resistance and Induction of MAPK/JNK Activation in Head and Neck Cancer Cells. Clin. Cancer Res. 2017, 23, 5162–5175. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Li, M.; Wang, X.; Pan, Y.; Li, J. Correlation between loss of Smad4 and clinical parameters of non-small cell lung cancer: An observational cohort study. BMC Pulm. Med. 2021, 21, 111. [Google Scholar] [CrossRef]
- Haeger, S.M.; Thompson, J.J.; Kalra, S.; Cleaver, T.G.; Merrick, D.; Wang, X.J.; Malkoski, S.P. Smad4 loss promotes lung cancer formation but increases sensitivity to DNA topoisomerase inhibitors. Oncogene 2016, 35, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tan, X.; Tang, Y.; Zhang, C.; Xu, J.; Zhou, J.; Cheng, X.; Hou, N.; Liu, W.; Yang, G.; et al. Dysregulated Tgfbr2/ERK-Smad4/SOX2 Signaling Promotes Lung Squamous Cell Carcinoma Formation. Cancer Res. 2019, 79, 4466–4479. [Google Scholar] [CrossRef] [Green Version]
- Kartolo, A.; Feilotter, H.; Hopman, W.; Fung, A.S.; Robinson, A. A single institution study evaluating outcomes of PD-L1 high KRAS-mutant advanced non-small cell lung cancer (NSCLC) patients treated with first line immune checkpoint inhibitors. Cancer Treat. Res. Commun. 2021, 27, 100330. [Google Scholar] [CrossRef]
- Byeon, S.; Lee, B.; Park, W.Y.; Choi, Y.L.; Jung, H.A.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Lee, S.H. Benefit of Targeted DNA Sequencing in Advanced Non-Small-Cell Lung Cancer Patients without EGFR and ALK Alterations on Conventional Tests. Clin. Lung Cancer 2020, 21, e182–e190. [Google Scholar] [CrossRef]
- Liberelle, M.; Jonckheere, N.; Melnyk, P.; Van Seuningen, I.; Lebegue, N. EGF-Containing Membrane-Bound Mucins: A Hidden ErbB2 Targeting Pathway? J. Med. Chem. 2020, 63, 5074–5088. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, L.; Xia, L.; Zhu, X. MicroRNA-133a-3p suppresses malignant behavior of non-small cell lung cancer cells by negatively regulating ERBB2. Oncol. Lett. 2021, 21, 457. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| N | |
|---|---|
| Total number of of subjects | 31 |
| NSCLC patients | 20 (65%) |
| Median age (range) | 66 (55–75) |
Gender
| 14 (70%) 6 (30%) |
| Smoking history Never-smokers Smokers
| 3 (15%) 17 (85%) 1 (6%) 2 (12%) 14 (82%) |
Tumor stage (TNM/AJCC 8th ed.)
| 9 (45%) 11 (55%) |
Histologic subtype
| 13 (65%) 7 (35%) |
| Smokers without cancer | 11 (35%) |
| Median age (range) | 67 (52–76) |
Gender
| 7 (64%) 4 (36%) |
Smoking history
| 11 (100%) |
| N | Circulating Tumor Cells/7.5 mL Blood | p Value | |||
|---|---|---|---|---|---|
| Present (%) | Mean (±SEM) | Median (Range) | |||
| Smokers without cancer | 11 | 2 (18%) | 0.18 (±0.12) | 0 (0–1) | |
| NSCLC patients | 20 | 12 (60%) | 13.40 (± 11.78) | 1 (0–237) | 0.0132 * |
NSCLC tumor stage
| 9 11 | 3 (33%) 9 (82%) | 1.11 (±0.70) 23.45 (±21.36) | 0 (0–6) 2 (0–237) | 0.0651 † |
| Patient ID | NSCLC Stage | Number of CTCs Detected | Number of CTCs Sequenced | Number of WBCs Sequenced | Total Number of Shared Variants Detected in All Sequenced CTCs within the Same Subject | Total Number of Variants Detected in ≥1 Sequenced CTC within the Same Subject |
|---|---|---|---|---|---|---|
| RL13 | I | 6 | 4 | 2 | 1 | 125 |
| RL5 | I | 3 | 2 | 1 | 85 | 148 |
| RL14 | IV | 3 | 3 | 2 | 72 | 121 |
| RL16 | IV | 237 | 6 | 2 | 130 | 441 |
| RL17 | IV | 7 | 3 | 2 | 91 | 147 |
| RL19 | IV | 2 | 2 | 2 | 137 | 194 |
| RL20 | IV | 3 | 3 | 2 | 101 | 245 |
| Gene | Number of Variants |
|---|---|
| NF1 | 69 |
| BRCA2 | 33 |
| PTCH1 | 33 |
| NF2 | 27 |
| ATM | 25 |
| EGFR | 24 |
| ERBB3 | 24 |
| PIK3CA | 19 |
| APC | 17 |
| BRCA1 | 17 |
| RB1 | 13 |
| SMARCB1 | 13 |
| TP53 | 13 |
| CDH1 | 12 |
| CSF1R | 11 |
| NOTCH1 | 11 |
| SMO | 11 |
| TERT | 11 |
| ABL1 | 10 |
| MLH1 | 10 |
| EZH2 | 9 |
| FGFR2 | 9 |
| SMAD4 | 9 |
| DNMT3A | 8 |
| FBXW7 | 8 |
| MTOR | 8 |
| CTNNB1 | 7 |
| ERBB2 | 7 |
| HNF1A | 7 |
| KIT | 7 |
| PIK3R1 | 7 |
| PTEN | 7 |
| ALK | 6 |
| CDKN2A | 6 |
| PTPN11 | 6 |
| ERBB4 | 5 |
| JAK2 | 5 |
| JAK3 | 5 |
| RET | 5 |
| BRAF | 4 |
| KRAS | 4 |
| LOC100507346 | 4 |
| STK11 | 4 |
| VHL | 4 |
| FGFR3 | 3 |
| FLT3 | 3 |
| HRAS | 3 |
| IDH1 | 3 |
| IDH2 | 3 |
| KDR | 3 |
| MET | 3 |
| NPM1 | 3 |
| TSC1 | 3 |
| AKT1 | 2 |
| FGFR1 | 2 |
| GNAQ | 2 |
| GNAS | 2 |
| NRAS | 2 |
| PDGFRA | 2 |
| ATM; C11orf65 | 1 |
| FBXW7-AS1 | 1 |
| GNA11 | 1 |
| MAP2K1 | 1 |
| MSH6 | 1 |
| Gene | Number of Variants |
|---|---|
| TP53 | 4 |
| NF1 | 2 |
| SMARCB1 | 1 |
| SMAD4 | 1 |
| PTEN | 1 |
| PTCH1 | 1 |
| MAP2K1 | 1 |
| KRAS | 1 |
| JAK3 | 1 |
| ERBB2 | 1 |
| DNMT3A | 1 |
| CTNNB1 | 1 |
| ABL1 | 1 |
| ALK | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbirou, M.; Miller, A.; Manjunath, Y.; Ramirez, A.B.; Ericson, N.G.; Staveley-O’Carroll, K.F.; Mitchem, J.B.; Warren, W.C.; Chaudhuri, A.A.; Huang, Y.; et al. Single Circulating-Tumor-Cell-Targeted Sequencing to Identify Somatic Variants in Liquid Biopsies in Non-Small-Cell Lung Cancer Patients. Curr. Issues Mol. Biol. 2022, 44, 750-763. https://doi.org/10.3390/cimb44020052
Barbirou M, Miller A, Manjunath Y, Ramirez AB, Ericson NG, Staveley-O’Carroll KF, Mitchem JB, Warren WC, Chaudhuri AA, Huang Y, et al. Single Circulating-Tumor-Cell-Targeted Sequencing to Identify Somatic Variants in Liquid Biopsies in Non-Small-Cell Lung Cancer Patients. Current Issues in Molecular Biology. 2022; 44(2):750-763. https://doi.org/10.3390/cimb44020052
Chicago/Turabian StyleBarbirou, Mouadh, Amanda Miller, Yariswamy Manjunath, Arturo B. Ramirez, Nolan G. Ericson, Kevin F. Staveley-O’Carroll, Jonathan B. Mitchem, Wesley C. Warren, Aadel A. Chaudhuri, Yi Huang, and et al. 2022. "Single Circulating-Tumor-Cell-Targeted Sequencing to Identify Somatic Variants in Liquid Biopsies in Non-Small-Cell Lung Cancer Patients" Current Issues in Molecular Biology 44, no. 2: 750-763. https://doi.org/10.3390/cimb44020052
APA StyleBarbirou, M., Miller, A., Manjunath, Y., Ramirez, A. B., Ericson, N. G., Staveley-O’Carroll, K. F., Mitchem, J. B., Warren, W. C., Chaudhuri, A. A., Huang, Y., Li, G., Tonellato, P. J., & Kaifi, J. T. (2022). Single Circulating-Tumor-Cell-Targeted Sequencing to Identify Somatic Variants in Liquid Biopsies in Non-Small-Cell Lung Cancer Patients. Current Issues in Molecular Biology, 44(2), 750-763. https://doi.org/10.3390/cimb44020052