
Use of Next-Generation Sequencing for Identifying Mitochondrial Disorders
 , , , , , ,
, , , , , , 
Abstract
:1. Introduction to Mitochondria and Mitochondrial Disorders
2. Clinical Features and Burden of Mitochondrial Disorders
3. Traditional Diagnostic Approach for Mitochondrial Disorders
4. Limitations Associated with Traditional Diagnostic Methods
5. Role of Sequencing in Mitochondrial Disorder Detection
6. Sanger Sequencing
7. Significance of NGS for the Detection of Mitochondrial Disorders
8. Whole-Genome Sequencing
9. Whole-Exome Sequencing
10. Whole-mtDNA Sequencing
11. Targeted-Exome Sequencing
12. RNA Sequencing
13. Case Examples
14. Challenges and Overcome of NGS Data Analysis Tools
15. Mitochondrial DNA and Mitochondrial Disease Databases
16. Future of the NGS in Mitochondrial Disorders
17. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tatsuta, T.; Model, K.; Langer, T. Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol. Biol. Cell 2005, 16, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Blerkom, J. Mitochondria in early mammalian development. Semin. Cell Dev. Biol. 2009, 20, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Kisilevsky, E.; Freund, P.; Margolin, E. Mitochondrial disorders and the eye. Surv. Ophthalmol. 2020, 65, 294–311. [Google Scholar] [CrossRef]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 297–348. [Google Scholar] [CrossRef] [Green Version]
- Duchen, M.R.; Szabadkai, G. Roles of mitochondria in human disease. Essays Biochem. 2010, 47, 115–137. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Bers, D.; Rizzuto, R. Mitochondria: From basic biology to cardiovascular disease. J. Mol. Cell. Cardiol. 2009, 46, 765–766. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.J.; Hartley, R.C.; Murphy, M.P. Mitochondria-targeted small molecule therapeutics and probes. Antioxidants Redox Signal. 2011, 15, 3021–3038. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Hartley, R.C.; Cochemé, H.M.; Murphy, M.P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef]
- Penta, J.S.; Johnson, F.M.; Wachsman, J.T.; Copeland, W.C. Mitochondrial DNA in human malignancy. Mutat. Res. Rev. Mutat. Res. 2001, 488, 119–133. [Google Scholar] [CrossRef]
- Kotrys, A.V.; Szczesny, R.J. Mitochondrial Gene Expression and Beyond-Novel Aspects of Cellular Physiology. Cells 2019, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Whitaker, R.M.; Corum, D.; Beeson, C.C.; Schnellmann, R.G. Mitochondrial Biogenesis as a Pharmacological Target: A New Approach to Acute and Chronic Diseases. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 229–249. [Google Scholar] [CrossRef]
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 2010, 11, 25–44. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; An, P.; Gu, Z.; Luo, Y.; Luo, J. Mitochondrial metal ion transport in cell metabolism and disease. Int. J. Mol. Sci. 2021, 22, 7525. [Google Scholar] [CrossRef]
- Mejia, E.M.; Hatch, G.M. Mitochondrial phospholipids: Role in mitochondrial function. J. Bioenerg. Biomembr. 2016, 48, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Zeviani, M. Disorders from perturbations of nuclear-mitochondrial intergenomic cross-talk. J. Intern. Med. 2009, 265, 174–192. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, Y.; Guan, M.X. A peep into mitochondrial disorder: Multifaceted from mitochondrial DNA mutations to nuclear gene modulation. Protein Cell 2015, 6, 862–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, K.; Collier, J.J.; Glasgow, R.I.C.; Robertson, F.M.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Oláhová, M.; McFarland, R.; Taylor, R.W. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J. Inherit. Metab. Dis. 2020, 43, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Dimauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef]
- Viscomi, C.; Zeviani, M. Strategies for fighting mitochondrial diseases. J. Intern. Med. 2020, 287, 665–684. [Google Scholar] [CrossRef]
- Maresca, A.; Del Dotto, V.; Romagnoli, M.; La Morgia, C.; Di Vito, L.; Capristo, M.; Valentino, M.L.; Carelli, V. Expanding and validating the biomarkers for mitochondrial diseases. J. Mol. Med. 2020, 98, 1467–1478. [Google Scholar] [CrossRef]
- DiMauro, S.; Hirano, M. Mitochondrial encephalomyopathies: An update. Neuromuscul. Disord. 2005, 15, 276–286. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [Green Version]
- Spinazzola, A. Mitochondrial DNA mutations and depletion in pediatric medicine. Semin. Fetal Neonatal Med. 2011, 16, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.M.; McFarland, R.; Blakely, E.L.; He, L.; Whittaker, R.G.; Taylor, R.W.; Chinnery, P.F.; Turnbull, D.M. Prevalence of mitochondrial DNA disease in adults. Ann. Neurol. 2008, 63, 35–39. [Google Scholar] [CrossRef]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [Green Version]
- DiMauro, S.; Hirano, M.; Schon, E.A. Approaches to the treatment of mitochondrial diseases. Muscle Nerve 2006, 34, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Viscomi, C.; Bottani, E.; Zeviani, M. Emerging concepts in the therapy of mitochondrial disease. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 544–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, Y.I.; Nonaka, I.; Horai, S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.S.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Petty, R.K.H.; Morgan-Hughes, J.A. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433. [Google Scholar]
- Wallace, D.C.; Singh, G.; Lotr, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.S.; Ii, L.J.E.; Nikoskelainen, E.K. Mitochondrial DNA. Science 1987, 729, 27–30. [Google Scholar]
- Wechsler, H.; Levine, S.; Idelson, R.K.; Rohman, M.; Taylor, J.O. The New England Journal of Medicine Downloaded from nejm.org at UNIVERSITY OF OTAGO on May 20, 2014. For personal use only. No other uses without permission. From the NEJM Archive. Copyright © 2022 Massachusetts Medical Society. All rights reserved. N. Engl. J. Med. 1983, 308, 97–100. [Google Scholar] [CrossRef]
- Rotig, A.; Colonna, M.; Bonnefont, J.P.; Blanche, S.; Fischer, A.; Saudubray, J.M.; Munnich, A. Mitochondrial Dna Deletion in Pearson’S Marrow/Pancreas Syndrome. Lancet 1989, 333, 902–903. [Google Scholar] [CrossRef]
- Chinnery, P.F. Primary Mitochondrial Disorders Overview 1. Clinical Characteristics of Mitochondrial Disorders; GeneReviews®: Seattle, DC, USA, 2021; pp. 1–16. [Google Scholar]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef] [Green Version]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Moyer, M.P.; Harrison, L.; Aw, T.Y. Contribution of glutathione status to oxidant-induced mitochondrial DNA damage in colonic epithelial cells. Free Radic. Biol. Med. 2009, 47, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Rachek, L.I.; Yuzefovych, L.V.; LeDoux, S.P.; Julie, N.L.; Wilson, G.L. Troglitazone, but not rosiglitazone, damages mitochondrial DNA and induces mitochondrial dysfunction and cell death in human hepatocytes. Toxicol. Appl. Pharmacol. 2009, 240, 348–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Reddy, T. Mitochondria as a Therapeutic Target for Aging and Neurodegenerative Diseases. Curr. Alzheimer Res. 2011, 8, 393–409. [Google Scholar] [CrossRef]
- Nicolson, G.L. Mitochondrial dysfunction and chronic disease: Treatment with natural supplements. Integr. Med. 2014, 13, 35–43. [Google Scholar]
- Karbowski, M.; Neutzner, A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. 2012, 123, 157–171. [Google Scholar] [CrossRef] [Green Version]
- Limongelli, G.; Masarone, D.; D’Alessandro, R.; Elliott, P.M. Mitochondrial diseases and the heart: An overview of molecular basis, diagnosis, treatment and clinical course. Future Cardiol. 2012, 8, 71–88. [Google Scholar] [CrossRef]
- Escribano-Lopez, I.; Diaz-Morales, N.; Rovira-Llopis, S.; Bañuls, C.; Lopez-Domenech, S.; Castelló, R.; Falcón, R.; Sola, E.; Rocha, M.; Hernández-Mijares, A.; et al. Atherosclerosis, Mitochondrial Dysfunction and Oxidative Stress: Mitochondria-Targeted Antioxidants as Potential Therapy. Front. Med. Chem. 2016, 96–135. [Google Scholar] [CrossRef]
- Fernandez, D.; Perl, A. Metabolic control of T cell activation and death in SLE. Autoimmun. Rev. 2009, 8, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, H.R.; Samuels, D.C.; Eden, J.A.; Relton, C.L.; Chinnery, P.F. Pathogenic Mitochondrial DNA Mutations Are Common in the General Population. Am. J. Hum. Genet. 2008, 83, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skladal, D.; Halliday, J.; Thorburn, D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Magner, M.; Kolářová, H.; Honzik, T.; Švandová, I.; Zeman, J. Clinical manifestation of mitochondrial diseases. Dev. period Med. 2015, 19, 441–449. [Google Scholar] [PubMed]
- Chow, J.; Rahman, J.; Achermann, J.C.; Dattani, M.T.; Rahman, S. Mitochondrial disease and endocrine dysfunction. Nat. Rev. Endocrinol. 2017, 13, 92–104. [Google Scholar] [CrossRef]
- Böhm, M.; Pronicka, E.; Karczmarewicz, E.; Pronicki, M.; Piekutowska-Abramczuk, D.; Sykut-Cegielska, J.; Mierzewska, H.; Hansikova, H.; Vesela, K.; Tesarova, M.; et al. Retrospective, multicentric study of 180 children with cytochrome c oxidase deficiency. Pediatr. Res. 2006, 59, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Dimauro, S. A history of mitochondrial diseases. J. Inherit. Metab. Dis. 2011, 34, 261–276. [Google Scholar] [CrossRef]
- Bannwarth, S.; Procaccio, V.; Lebre, A.S.; Jarde, C.; Chaussenot, A.; Hoarau, C.; Maoulida, H.; Charrier, N.; Gai, X.; Xie, H.M.; et al. Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J. Med. Genet. 2013, 50, 704–714. [Google Scholar] [CrossRef] [Green Version]
- Uriho, A.; Tang, X.; Le, G.; Yang, S.; Harimana, Y.; Ishimwe, S.P.; Yiping, L.; Zhang, K.; Ma, S.; Muhoza, B. Effects of resveratrol on mitochondrial biogenesis and physiological diseases. Adv. Tradit. Med. 2021, 21, 1–14. [Google Scholar] [CrossRef]
- DiMauro, S.; Emmanuele, V. Mitochondrial Disorders Due to Mutations in the Mitochondrial Genome. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease; Academic Press: Cambridge, MA, UK, 2020; ISBN 9780128139554. [Google Scholar]
- Kaufmann, P.; Engelstad, K.; Wei, Y.; Kulikova, R.; Oskoui, M.; Sproule, D.M.; Battista, V.; Koenigsberger, D.Y.; Pascual, J.M.; Shanske, S.; et al. Natural history of MELAS associated with mitochondrial DNA m.3243A > G genotype. Neurology 2011, 77, 1965–1971. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, A.; Minetti, C.; Moggio, M.; Mongini, T.; et al. The m.3243A > G mitochondrial DNA mutation and related phenotypes. A matter of gender? J. Neurol. 2014, 261, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.S.; DiMauro, S.; Turnbull, D.M. Mitochondrial medicine: A historical point of view. In Diagnosis and Management of Mitochondrial Disorders; Springer: Cham, Denmark, 2019; pp. 1–18. [Google Scholar]
- Mancuso, M.; Petrozzi, L.; Filosto, M.; Nesti, C.; Rocchi, A.; Choub, A.; Pistolesi, S.; Massetani, R.; Fontanini, G.; Siciliano, G. MERRF syndrome without ragged-red fibers: The need for molecular diagnosis. Biochem. Biophys. Res. Commun. 2007, 354, 1058–1060. [Google Scholar] [CrossRef] [PubMed]
- Huoponen, K. Leber hereditary optic neuropathy: Clinical and molecular genetic findings. Neurogenetics 2001, 3, 119–125. [Google Scholar] [CrossRef]
- Giordano, C.; Montopoli, M.; Perli, E.; Orlandi, M.; Fantin, M.; Ross-Cisneros, F.N.; Caparrotta, L.; Martinuzzi, A.; Ragazzi, E.; Ghelli, A.; et al. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain 2011, 134, 220–234. [Google Scholar] [CrossRef]
- Man, P.Y.W.; Griffiths, P.G.; Brown, D.T.; Howell, N.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of leber hereditary optic neuropathy in the North East of England. Am. J. Hum. Genet. 2003, 72, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, J.H.; Wolf, J.; Hoshitsuki, K.; Huddart, R.; Caudle, K.E.; Whirl-Carrillo, M.; Steyger, P.S.; Smith, R.J.H.; Cody, N.; Rodriguez-Antona, C.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for the Use of Aminoglycosides Based on MT-RNR1 Genotype. Clin. Pharmacol. Ther. 2022, 111, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.S.; Rahman, S.; Hanna, M.G. Single deletions in mitochondrial DNA—Molecular mechanisms and disease phenotypes in clinical practice. Neuromuscul. Disord. 2012, 22, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, M.A.; Federico, A.; Minetti, C.; Moggio, M.; et al. Redefining phenotypes associated with mitochondrial DNA single deletion. J. Neurol. 2015, 262, 1301–1309. [Google Scholar] [CrossRef]
- Heighton, J.N.; Brady, L.I.; Newman, M.C.; Tarnopolsky, M.A. Clinical and demographic features of chronic progressive external ophthalmoplegia in a large adult-onset cohort. Mitochondrion 2019, 44, 15–19. [Google Scholar] [CrossRef]
- Lee, H.-F.; Tsai, C.-R.; Chi, C.-S.; Lee, H.-J.; Chen, C.C.-C. Leigh Syndrome: Clinical and Neuroimaging. Pediatr. Neurol. 2009, 40, 88–93. [Google Scholar] [CrossRef]
- Leigh, D. Subacute necrotizing encephalomyelopathy in an infant. J. Neurol. Neurosurg. Psychiatry 1951, 14, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pincus, J.H. Subacute Necrotizing Encephalomyelopathy (Leigh’s Disease): A Consideration of Clinical Features and Etiology. Dev. Med. Child Neurol. 1912, 14, 87–101. [Google Scholar] [CrossRef]
- Ruitenbeek, W. Special article Leigh syndrome, a mitochondrial encephalo (myo) pathy. Clin. Neurol. Neurosurg. 1987, 89, 217–230. [Google Scholar]
- Sudo, A.; Honzawa, S.; Nonaka, I.; Goto, Y.-I. Leigh syndrome caused by mitochondrial DNA G13513A mutation: Frequency and clinical features in Japan. J. Hum. Genet. 2004, 49, 92–96. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Schon, E.A. Mitochondrial DNA mutations in human disease. Am. J. Med. Genet. 2001, 106, 18–26. [Google Scholar] [CrossRef]
- Johns, D.R. Mitochondrial DNA and disease. N. Engl. J. Med. 1995, 333, 638–644. [Google Scholar] [CrossRef]
- Pitceathly, R.D.S.; Rahman, S. Mitochondrial DNA-Related Diseases Associated with Single Large-Scale Deletions and Point Mutations. In The Human Mitochondrial Genome; Academic Press: Cambridge, MA, USA, 2020; pp. 353–374. ISBN 9780128196564. [Google Scholar]
- Liang, C.; Ahmad, K.; Sue, C.M. The broadening spectrum of mitochondrial disease: Shifts in the diagnostic paradigm. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1360–1367. [Google Scholar] [CrossRef] [Green Version]
- Lnnqvist, T.; Paetau, A.; Valanne, L.; Pihko, H. Recessive twinkle mutations cause severe epileptic encephalopathy. Brain 2009, 132, 1553–1562. [Google Scholar] [CrossRef]
- Zsurka, G.; Hampel, K.G.; Nelson, I.; Jardel, C.; Mirandola, S.R.; Sassen, R.; Kornblum, C.; Marcorelles, P.; Lavoué, S.; Lombès, A.; et al. Severe epilepsy as the major symptom of new mutations in the mitochondrial tRNAPhe gene. Neurology 2010, 74, 507–512. [Google Scholar] [CrossRef]
- Salomão, K.B.; Ayo, C.M.; Della-Rosa, V.A. Investigation of the A1555G mutation in mitochondrial DNA (MT-RNR1) in groups of Brazilian individuals with nonsyndromic deafness and normal-hearing. Indian J. Hum. Genet. 2013, 19, 54–57. [Google Scholar] [CrossRef] [Green Version]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Collins, A.; Cohen, B.H.; DeBrosse, S.D.; et al. Practice patterns of mitochondrial disease physicians in North America. Part 1: Diagnostic and clinical challenges. Mitochondrion 2014, 14, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, D.P.; Lawlor, M.W. Presentation and Diagnostic Evaluation of Mitochondrial Disease. Pediatr. Clin. North Am. 2017, 64, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wortmann, S.B.; Kluijtmans, L.A.J.; Rodenburg, R.J.; Sass, J.O.; Nouws, J.; Van Kaauwen, E.P.; Kleefstra, T.; Tranebjaerg, L.; De Vries, M.C.; Isohanni, P.; et al. 3-Methylglutaconic aciduria—Lessons from 50 genes and 977 patients. J. Inherit. Metab. Dis. 2013, 36, 913–921. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, T.; Mashimo, T. Creating knockout and knockin rodents using engineered endonucleases via direct embryo injection. Methods Mol. Biol. 2015, 1239, 307–315. [Google Scholar] [CrossRef]
- Watson, E.; Davis, R.; Sue, C.M. New diagnostic pathways for mitochondrial disease. J. Transl. Genet. Genomics 2020, 4, 188–202. [Google Scholar] [CrossRef]
- Phadke, R. Myopathology of Adult and Paediatric Mitochondrial Diseases. J. Clin. Med. 2017, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Rodenburg, R.J.T.; Schoonderwoerd, G.C.; Tiranti, V.; Taylor, R.W.; Rötig, A.; Valente, L.; Invernizzi, F.; Chretien, D.; He, L.; Backx, G.P.B.M.; et al. A multi-center comparison of diagnostic methods for the biochemical evaluation of suspected mitochondrial disorders. Mitochondrion 2013, 13, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DaRe, J.T.; Vasta, V.; Penn, J.; Tran, N.T.B.; Hahn, S.H. Targeted exome sequencing for mitochondrial disorders reveals high genetic heterogeneity. BMC Med. Genet. 2013, 14, 118. [Google Scholar] [CrossRef] [Green Version]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Koenig, M.K. Presentation and Diagnosis of Mitochondrial Disorders in Children. Pediatr. Neurol. 2008, 38, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Molnar, M.J.; Kovacs, G.G. Mitochondrial Diseases, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 145, ISBN 9780128023952. [Google Scholar]
- Wortmann, S.B.; Mayr, J.A.; Nuoffer, J.M.; Prokisch, H.; Sperl, W. A Guideline for the Diagnosis of Pediatric Mitochondrial Disease: The Value of Muscle and Skin Biopsies in the Genetics Era. Neuropediatrics 2017, 48, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Neveling, K.; Feenstra, I.; Gilissen, C.; Hoefsloot, L.H.; Kamsteeg, E.J.; Mensenkamp, A.R.; Rodenburg, R.J.T.; Yntema, H.G.; Spruijt, L.; Vermeer, S.; et al. A Post-Hoc Comparison of the Utility of Sanger Sequencing and Exome Sequencing for the Diagnosis of Heterogeneous Diseases. Hum. Mutat. 2013, 34, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Vasta, V.; Merritt, J.L.; Saneto, R.P.; Hahn, S.H. Next-generation sequencing for mitochondrial diseases: A wide diagnostic spectrum. Pediatr. Int. 2012, 54, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Young, L.T. Is bipolar disorder a mitochondrial disease? J. Psychiatry Neurosci. 2007, 32, 160. [Google Scholar]
- Kendall, F.D. Mitochondrial disorders: Overview of diagnostic tools and new diagnostic trends. J. Pediatr. Biochem. 2012, 2, 193–203. [Google Scholar] [CrossRef]
- ElBeheiry, A.A.; Abougabal, A.M.; Omar, T.I.; Etaby, A.N. Role of brain magnetic resonance spectroscopy in the evaluation of suspected mitochondrial diseases in children: Experience in 30 pediatric cases. Egypt. J. Radiol. Nucl. Med. 2014, 45, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Thorburn, D.R.; Sugiana, C.; Salemi, R.; Kirby, D.M.; Worgan, L.; Ohtake, A.; Ryan, M.T. Biochemical and molecular diagnosis of mitochondrial respiratory chain disorders. Biochim. Biophys. Acta Bioenerg. 2004, 1659, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Meyers, D.E.; Basha, H.I.; Koenig, M.K. Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Texas Hear. Inst. J. 2013, 40, 385–394. [Google Scholar]
- Wong, L.J.C.; Scaglia, F.; Graham, B.H.; Craigen, W.J. Current molecular diagnostic algorithm for mitochondrial disorders. Mol. Genet. Metab. 2010, 100, 111–117. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, X. Significance of mitochondria DNA mutations in diseases. Adv. Exp. Med. Biol. 2017, 1038, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Epidemiology and treatment of mitochondrial disorders. Am. J. Med. Genet. Semin. Med. Genet. 2001, 106, 94–101. [Google Scholar] [CrossRef]
- Davis, R.L.; Liang, C.; Sue, C.M. Mitochondrial Diseases, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 147, ISBN 9780444632333. [Google Scholar]
- McCormick, E.M.; Zolkipli-Cunningham, Z.; Falk, M.J. Mitochondrial disease genetics update: Recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr. Opin. Pediatr. 2018, 30, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, T.E.J.; Nguyen, M.; Kamps, R.; Hendrickx, A.T.; Sallevelt, S.C.E.H.; Gottschalk, R.W.H.; Calis, C.M.; Stassen, A.P.M.; De Koning, B.; Mulder-Den Hartog, E.N.M.; et al. Whole exome sequencing is the preferred strategy to identify the genetic defect in patients with a probable or possible mitochondrial cause. Front. Genet. 2018, 9, 400. [Google Scholar] [CrossRef]
- Wang, J.; Venegas, V.; Li, F.; Wong, L.J. Analysis of mitochondrial DNA point mutation heteroplasmy by ARMS quantitative PCR. Curr. Protoc. Hum. Genet. 2011, 68, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.M.; Perin, J.C.; Schurr, T.G.; Dulik, M.C.; Zhadanov, S.I.; Baur, J.A.; King, M.P.; Place, E.; Clarke, C.; Grauer, M.; et al. Mitochondrial genome sequence analysis: A custom bioinformatics pipeline substantially improves Affymetrix MitoChip v2.0 call rate and accuracy. BMC Bioinform. 2011, 12, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, E.; Place, E.; Falk, M.J. Molecular Genetic Testing for Mitochondrial Disease: From One Generation to the Next. Neurotherapeutics 2013, 10, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Pfundt, R.; Del Rosario, M.; Vissers, L.E.L.M.; Kwint, M.P.; Janssen, I.M.; De Leeuw, N.; Yntema, H.G.; Nelen, M.R.; Lugtenberg, D.; Kamsteeg, E.J.; et al. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genet. Med. 2017, 19, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Huang, T. Next generation sequencing to characterize mitochondrial genomic DNA heteroplasmy. Curr. Protoc. Hum. Genet. 2011, 71, 19.8.1–19.8.12. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Schönberg, A.; Schaefer, M.; Schroeder, R.; Nasidze, I.; Stoneking, M. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am. J. Hum. Genet. 2010, 87, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Alemi, M.; Prigione, A.; Wong, A.; Schoenfeld, R.; DiMauro, S.; Hirano, M.; Cortopassi, G. Mitochondrial DNA deletions inhibit proteasomal activity and stimulate an autophagic transcript. Free Radic. Biol. Med. 2007, 42, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Poulton, J.; Marchington, D.; Suomalainen, A. Decrease of 3243 A→G mtDNA mutation from blood in MELAS syndrome: A longitudinal study. Am. J. Hum. Genet. 2001, 68, 238–240. [Google Scholar] [CrossRef] [Green Version]
- Macken, W.L.; Vandrovcova, J.; Hanna, M.G.; Pitceathly, R.D.S. Applying genomic and transcriptomic advances to mitochondrial medicine. Nat. Rev. Neurol. 2021, 17, 215–230. [Google Scholar] [CrossRef]
- Sikkema-Raddatz, B.; Johansson, L.F.; de Boer, E.N.; Almomani, R.; Boven, L.G.; van den Berg, M.P.; van Spaendonck-Zwarts, K.Y.; van Tintelen, J.P.; Sijmons, R.H.; Jongbloed, J.D.H.; et al. Targeted Next-Generation Sequencing can Replace Sanger Sequencing in Clinical Diagnostics. Hum. Mutat. 2013, 34, 1035–1042. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Li, F.; Chen, D.; Wang, G.; Truong, C.K.; Enns, G.M.; Graham, B.; Milone, M.; Landsverk, M.L.; Wang, J.; et al. Comprehensive next-generation sequence analyses of the entire mitochondrial genome reveal new insights into the molecular diagnosis of mitochondrial DNA disorders. Genet. Med. 2013, 15, 388–394. [Google Scholar] [CrossRef] [Green Version]
- McMillan, H.J.; Schwartzentruber, J.; Smith, A.; Lee, S.; Chakraborty, P.; Bulman, D.E.; Beaulieu, C.L.; Majewski, J.; Boycott, K.M.; Geraghty, M.T. Compound heterozygous mutations in glycyl-tRNA synthetase are a proposed cause of systemic mitochondrial disease. BMC Med. Genet. 2014, 15, 36. [Google Scholar] [CrossRef] [Green Version]
- Solomon, D.A. 5—Integrating Molecular Diagnostics with Surgical Neuropathology. In Practical Surgical Neuropathology: A Diagnostic Approach A Volume in the Pattern Recognition Series; Elsevier: Amsterdam, The Netherlands, 2018; ISBN 9780323449410. [Google Scholar]
- Dai, Y.; Wang, C.; Nie, Z.; Han, J.; Chen, T.; Zhao, X.; Ai, C.; Ji, Y.; Gao, T.; Jiang, P. Mutation analysis of Leber’s hereditary optic neuropathy using a multi-gene panel. Biomed. Rep. 2018, 8, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Xing, R.; Cui, J.; Li, W.; Lu, Y. Investigation of frequent somatic mutations of MTND5 gene in gastric cancer cell lines and tissues. Mitochondrial DNA Part B Resour. 2018, 3, 1004–1010. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, A.; Tétreault, M.; Dyment, D.A.; Zou, R.; Kernohan, K.; Geraghty, M.T.; Hartley, T.; Boycott, K.M. Concordance between whole-exome sequencing and clinical sanger sequencing: Implications for patient care. Mol. Genet. Genomic Med. 2016, 4, 504–512. [Google Scholar] [CrossRef] [Green Version]
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef]
- Jongbloed, J.D.H.; Psafalvi, A.; Kerstjens-Frederikse, W.S.; Sinke, R.J.; Van Tintelen, J.P. New clinical molecular diagnostic methods for congenital and inherited heart disease. Expert Opin. Med. Diagn. 2011, 5, 9–24. [Google Scholar] [CrossRef]
- Norton, N.; Li, D.; Hershberger, R.E. Next-generation sequencing to identify genetic causes of cardiomyopathies. Curr. Opin. Cardiol. 2012, 27, 214–220. [Google Scholar] [CrossRef]
- Vasta, V.; Ng, S.B.; Turner, E.H.; Shendure, J.; Hahn, S.H. Next generation sequence analysis for mitochondrial disorders. Genome Med. 2009, 1, 100. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S. Mitochondrial Disease in Children. J. Intern. Med. 2020, 287, 609–633. [Google Scholar] [CrossRef]
- Carroll, C.J.; Brilhante, V.; Suomalainen, A. Next-generation sequencing for mitochondrial disorders. Br. J. Pharmacol. 2014, 171, 1837–1853. [Google Scholar] [CrossRef] [Green Version]
- Shendure, J.; Aiden, E.L. The expanding scope of DNA sequencing. Nat. Biotechnol. 2012, 30, 1084–1094. [Google Scholar] [CrossRef] [Green Version]
- Shendure, J.; Mitra, R.D.; Varma, C.; Church, G.M. Advanced sequencing technologies: Methods and goals. Nat. Rev. Genet. 2004, 5, 335–344. [Google Scholar] [CrossRef]
- Ahmad, R.; Hasan, M.Y. Next-generation sequencing technology in the diagnosis of mitochondrial disorders. Int. J. Health Sci. 2021, 15, 1–2. [Google Scholar]
- Chin, J.; Marotta, R.; Chiotis, M.; Allan, E.H.; Collins, S.J. Detection rates and phenotypic spectrum of m.3243A > G in the MT-TL1 gene: A molecular diagnostic laboratory perspective. Mitochondrion 2014, 17, 34–41. [Google Scholar] [CrossRef]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; Van De Vorst, M.; Van Bon, B.W.M.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef]
- Ma, M.; Ru, Y.; Chuang, L.S.; Hsu, N.Y.; Shi, L.S.; Hakenberg, J.; Cheng, W.Y.; Uzilov, A.; Ding, W.; Glicksberg, B.S.; et al. Disease-associated variants in different categories of disease located in distinct regulatory elements. BMC Genomics 2015, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cummings, B.; Marshall, J.; Tukiainen, T.; Lek, M.; Donkervoort, S.; Foley, A.R.; Bolduc, V.; Waddell, L.; Sandaradura, S.; O’Grady, G.; et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 2016, 5209, 074153. [Google Scholar] [CrossRef] [Green Version]
- Frésard, L.; Smail, C.; Ferraro, N.M.; Teran, N.A.; Li, X.; Smith, K.S.; Bonner, D.; Kernohan, K.D.; Marwaha, S.; Zappala, Z.; et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat. Med. 2019, 25, 911–919. [Google Scholar] [CrossRef]
- Lee, H.; Huang, A.Y.; Wang, L.-k.; Yoon, A.J.; Renteria, G.; Eskin, A.; Signer, R.H.; Dorrani, N.; Nieves-Rodriguez, S.; Wan, J.; et al. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genet. Med. 2020, 22, 490–499. [Google Scholar] [CrossRef]
- Bick, D.; Dimmock, D. Whole exome and whole genome sequencing. Curr. Opin. Pediatr. 2011, 23, 594–600. [Google Scholar] [CrossRef]
- Rius, R.; Compton, A.G.; Baker, N.L.; Welch, A.E.; Coman, D.; Kava, M.P.; Minoche, A.E.; Cowley, M.J.; Thorburn, D.R.; Christodoulou, J. Application of genome sequencing from blood to diagnose mitochondrial diseases. Genes 2021, 12, 607. [Google Scholar] [CrossRef]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Tamiy, G.; Makino, S.; Hayashi, M.; Abe, A.; Numakura, C.; Ueki, M.; Tanaka, A.; Ito, C.; Toshimori, K.; Ogawa, N.; et al. A Mutation of COX6A1 causes a recessive axonal or mixed form of charcot-marie-tooth disease. Am. J. Hum. Genet. 2014, 95, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Malicdan, M.C.V.; Vilboux, T.; Ben-Zeev, B.; Guo, J.; Eliyahu, A.; Pode-Shakked, B.; Dori, A.; Kakani, S.; Chandrasekharappa, S.C.; Ferreira, C.R.; et al. A novel inborn error of the coenzyme Q10 biosynthesis pathway: Cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Hum. Mutat. 2018, 39, 69–79. [Google Scholar] [CrossRef]
- Schon, K.R.; Ratnaike, T.; van den Ameele, J.; Horvath, R.; Chinnery, P.F. Mitochondrial Diseases: A Diagnostic Revolution. Trends Genet. 2020, 36, 702–717. [Google Scholar] [CrossRef]
- Dewey, F.E.; Grove, M.E.; Pan, C.; Goldstein, B.A.; Bernstein, J.A.; Chaib, H.; Merker, J.D.; Goldfeder, R.L.; Enns, G.M.; David, S.P.; et al. Clinical interpretation and implications of whole-genome sequencing. JAMA J. Am. Med. Assoc. 2014, 311, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.M.; Hildreth, A.; Batalov, S.; Ding, Y.; Chowdhury, S.; Watkins, K.; Ellsworth, K.; Camp, B.; Kint, C.I.; Yacoubian, C.; et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci. Transl. Med. 2019, 11, eaat6177. [Google Scholar] [CrossRef]
- Wright, C.F.; McRae, J.F.; Clayton, S.; Gallone, G.; Aitken, S.; FitzGerald, T.W.; Jones, P.; Prigmore, E.; Rajan, D.; Lord, J.; et al. Making new genetic diagnoses with old data: Iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet. Med. 2018, 20, 1216–1223. [Google Scholar] [CrossRef] [Green Version]
- Stark, Z.; Schofield, D.; Martyn, M.; Rynehart, L.; Shrestha, R.; Alam, K.; Lunke, S.; Tan, T.Y.; Gaff, C.L.; White, S.M. Does genomic sequencing early in the diagnostic trajectory make a difference? A follow-up study of clinical outcomes and cost-effectiveness. Genet. Med. 2019, 21, 173–180. [Google Scholar] [CrossRef]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar] [CrossRef]
- Gambin, T.; Akdemir, Z.C.; Yuan, B.; Gu, S.; Chiang, T.; Carvalho, C.M.B.; Shaw, C.; Jhangiani, S.; Boone, P.M.; Eldomery, M.K.; et al. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 2017, 45, 1633–1648. [Google Scholar] [CrossRef]
- Gorski, M.M.; Blighe, K.; Lotta, L.A.; Pappalardo, E.; Garagiola, I.; Mancini, I.; Mancuso, M.E.; Fasulo, M.R.; Santagostino, E.; Peyvandi, F. Whole-exome sequencing to identify genetic risk variants underlying inhibitor development in severe hemophilia A patients. Blood 2016, 127, 2924–2933. [Google Scholar] [CrossRef] [Green Version]
- Legati, A.; Reyes, A.; Nasca, A.; Invernizzi, F.; Lamantea, E.; Tiranti, V.; Garavaglia, B.; Lamperti, C.; Ardissone, A.; Moroni, I.; et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1326–1335. [Google Scholar] [CrossRef]
- Wagner, M.; Berutti, R.; Lorenz-Depiereux, B.; Graf, E.; Eckstein, G.; Mayr, J.A.; Meitinger, T.; Ahting, U.; Prokisch, H.; Strom, T.M.; et al. Mitochondrial DNA mutation analysis from exome sequencing—A more holistic approach in diagnostics of suspected mitochondrial disease. J. Inherit. Metab. Dis. 2019, 42, 909–917. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Koolen, D.A.; Smeitink, J.A.; van den Heuvel, L.; Rodenburg, R.J. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J. Inherit. Metab. Dis. 2015, 38, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Griffin, H.R.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Duff, J.; Hudson, G.; Horvath, R.; Wilson, I.J.; Santibanez-Koref, M.; Taylor, R.W.; et al. Accurate mitochondrial DNA sequencing using off-target reads provides a single test to identify pathogenic point mutations. Genet. Med. 2014, 16, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucinska-Wieckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosinska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 2016, 14, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Picardi, E.; Pesole, G. Mitochondrial genomes gleaned from human whole-exome sequencing. Nat. Methods 2012, 9, 523–524. [Google Scholar] [CrossRef]
- Weigl, S.; Paradiso, A.; Tommasi, S. Mitochondria and Familial Predisposition to Breast Cancer. Curr. Genomics 2013, 14, 195–203. [Google Scholar] [CrossRef]
- Neiman, M.; Taylor, D.R. The causes of mutation accumulation in mitochondrial genomes. Proc. R. Soc. B Biol. Sci. 2009, 276, 1201–1209. [Google Scholar] [CrossRef] [Green Version]
- Prates Mori, M.; de Souza-Pinto, N.C. Role of mitochondrial dysfunction in the pathophysiology of DNA repair disorders. Cell Biol. Int. 2018, 42, 643–650. [Google Scholar] [CrossRef]
- Fendt, L.; Zimmermann, B.; Daniaux, M.; Parson, W. Sequencing strategy for the whole mitochondrial genome resulting in high quality sequences. BMC Genomics 2009, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Herbers, E.; Kekäläinen, N.J.; Hangas, A.; Pohjoismäki, J.L.; Goffart, S. Tissue specific differences in mitochondrial DNA maintenance and expression. Mitochondrion 2019, 44, 85–92. [Google Scholar] [CrossRef]
- Lieber, D.S.; Calvo, S.E.; Shanahan, K.; Slate, N.G.; Liu, S.; Hershman, S.G.; Gold, N.B.; Chapman, B.A.; Thorburn, D.R.; Berry, G.T.; et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology 2013, 80, 1762–1770. [Google Scholar] [CrossRef] [Green Version]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Koopman, W.J.H.; Willems, P.H.G.M.; Smeitink, J.A.M. Monogenic Mitochondrial Disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Bewicke-Copley, F.; Arjun Kumar, E.; Palladino, G.; Korfi, K.; Wang, J. Applications and analysis of targeted genomic sequencing in cancer studies. Comput. Struct. Biotechnol. J. 2019, 17, 1348–1359. [Google Scholar] [CrossRef]
- Ma, E.S.K.; Wan, T.S.K.; Au, C.H.; Ho, D.N.; Ma, S.Y.; Ng, M.H.L.; Chan, T.L. Next-generation sequencing and molecular cytogenetic characterization of ETV6-LYN fusion due to chromosomes 1, 8 and 12 rearrangement in acute myeloid leukemia. Cancer Genet. 2017, 218–219, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Zhang, P.; Samuels, D.C.; Lehmann, B.; Stricker, T.; Pietenpol, J.; Shyr, Y.; Guo, Y. Mitochondria sequence mapping strategies and practicability of mitochondria variant detection from exome and RNA sequencing data. Brief. Bioinform. 2016, 17, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Horvath, R.; Chinnery, P.F. Diagnosis and Management of Mitochondrial Disorders. Diagnosis Manag. Mitochondrial Disord. 2019, 281–287. [Google Scholar] [CrossRef]
- Schlieben, L.D.; Prokisch, H. The Dimensions of Primary Mitochondrial Disorders. Front. Cell Dev. Biol. 2020, 8, 1–11. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612. [Google Scholar] [CrossRef]
- Dinwiddie, D.L.; Smith, L.D.; Miller, N.A.; Atherton, A.M.; Farrow, E.G.; Strenk, M.E.; Soden, S.E.; Saunders, C.J.; Kingsmore, S.F. Diagnosis of mitochondrial disorders by concomitant next-generation sequencing of the exome and mitochondrial genome. Genomics 2013, 102, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Puusepp, S.; Reinson, K.; Pajusalu, S.; Murumets, Ü.; Õiglane-Shlik, E.; Rein, R.; Talvik, I.; Rodenburg, R.J.; Õunap, K. Effectiveness of whole exome sequencing in unsolved patients with a clinical suspicion of a mitochondrial disorder in Estonia. Mol. Genet. Metab. Rep. 2018, 15, 80–89. [Google Scholar] [CrossRef]
- Kerr, M.; Hume, S.; Omar, F.; Koo, D.; Barnes, H.; Khan, M.; Aman, S.; Wei, X.C.; Alfuhaid, H.; McDonald, R.; et al. MITO-FIND: A study in 390 patients to determine a diagnostic strategy for mitochondrial disease. Mol. Genet. Metab. 2020, 131, 66–82. [Google Scholar] [CrossRef]
- Kathiresan, N.; Temanni, R.; Almabrazi, H.; Syed, N.; Jithesh, P.; Al-Ali, R. Accelerating next generation sequencing data analysis with system level optimizations. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Kulkarni, P.; Frommolt, P. Challenges in the Setup of Large-scale Next-Generation Sequencing Analysis Workflows. Comput. Struct. Biotechnol. J. 2017, 15, 471–477. [Google Scholar] [CrossRef]
- Dillies, M.A.; Rau, A.; Aubert, J.; Hennequet-Antier, C.; Jeanmougin, M.; Servant, N.; Keime, C.; Marot, N.S.; Castel, D.; Estelle, J.; et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief. Bioinform. 2013, 14, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Han, T.; Lin, X.; Song, X.; Xie, H.; He, J.; Chen, W. A novel computational tool for copy number variation detection in targeted circulating tumor DNA. J. Clin. Oncol. 2019, 37, e13051. [Google Scholar] [CrossRef]
- Zare, F.; Dow, M.; Monteleone, N.; Hosny, A.; Nabavi, S. An evaluation of copy number variation detection tools for cancer using whole exome sequencing data. BMC Bioinform. 2017, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Crispatzu, G.; Kulkarni, P.; Toliat, M.R.; Nürnberg, P.; Herling, M.; Herling, C.D.; Frommolt, P. Semi-automated cancer genome analysis using high-performance computing. Hum. Mutat. 2017, 38, 1325–1335. [Google Scholar] [CrossRef]
- Peters, D.; Luo, X.; Qiu, K.; Liang, P. Speeding Up Large-Scale Next Generation Sequencing Data Analysis with pBWA. J. Appl. Bioinforma. Comput. Biol. 2017, 1. [Google Scholar] [CrossRef]
- Diroma, M.A.; Calabrese, C.; Simone, D.; Santorsola, M.; Calabrese, F.M.; Gasparre, G.; Attimonelli, M. Extraction and annotation of human mitochondrial genomes from 1000 Genomes Whole Exome Sequencing data. BMC Genom. 2014, 15, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Manuscript, A. A map of human genome variation from population-scale sequencing. Nature 2011, 473, 544. [Google Scholar] [CrossRef]
- Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; Gabriel, S.B.; et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Clima, R.; Preste, R.; Calabrese, C.; Diroma, M.A.; Santorsola, M.; Scioscia, G.; Simone, D.; Shen, L.; Gasparre, G.; Attimonelli, M. HmtDB 2016: Data update, a better performing query system and human mitochondrial DNA haplogroup predictor. Nucleic Acids Res. 2017, 45, D698–D706. [Google Scholar] [CrossRef]
- Ruiz-Pesini, E.; Lott, M.T.; Procaccio, V.; Poole, J.C.; Brandon, M.C.; Mishmar, D.; Yi, C.; Kreuziger, J.; Baldi, P.; Wallace, D.C. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007, 35, 823–828. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C.; Stugard, C.; Murdock, D.; Schurr, T.; Brown, M.D. Ancient mtDNA sequences in the human nuclear genome: A potential source of errors in identifying pathogenic mutations. Proc. Natl. Acad. Sci. USA 1997, 94, 14900–14905. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.E.; Miller, S.; Herrnstadt, C.; Ghosh, S.S.; Fahy, E.; Shinobu, L.A.; Galasko, D.; Thal, L.J.; Beal, M.F.; Howell, N.; et al. Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 4526–4531. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Diroma, M.A.; Gonzalez, M.; Navarro-Gomez, D.; Leipzig, J.; Lott, M.T.; van Oven, M.; Wallace, D.C.; Muraresku, C.C.; Zolkipli-Cunningham, Z.; et al. MSeqDR: A Centralized Knowledge Repository and Bioinformatics Web Resource to Facilitate Genomic Investigations in Mitochondrial Disease. Hum. Mutat. 2016, 37, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.J.; Shen, L.; Gonzalez, M.; Leipzig, J.; Lott, M.T.; Stassen, A.P.M.; Diroma, M.A.; Navarro-Gomez, D.; Yeske, P.; Bai, R.; et al. Mitochondrial Disease Sequence Data Resource (MSeqDR): A global grass-roots consortium to facilitate deposition, curation, annotation, and integrated analysis of genomic data for the mitochondrial disease clinical and research communities. Mol. Genet. Metab. 2015, 114, 388–396. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; McCormick, E.M.; Muraresku, C.C.; Falk, M.J.; Gai, X. Clinical Bioinformatics in Precise Diagnosis of Mitochondrial Disease. Clin. Lab. Med. 2020, 40, 149–161. [Google Scholar] [CrossRef]
- Shen, L.; Attimonelli, M.; Bai, R.; Lott, M.T.; Wallace, D.C.; Falk, M.J.; Gai, X. MSeqDR mvTool: A mitochondrial DNA Web and API resource for comprehensive variant annotation, universal nomenclature collation, and reference genome conversion. Hum. Mutat. 2018, 39, 806–810. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, 980–985. [Google Scholar] [CrossRef] [Green Version]
- Hakeem, K.R.; Tombuloğlu, H.; Tombuloğlu, G. Plant Omics: Trends and Applications; Springer: Cham, Switzerland, 2016; ISBN 9783319317038. [Google Scholar]
- Dames, S.; Chou, L.S.; Xiao, Y.; Wayman, T.; Stocks, J.; Singleton, M.; Eilbeck, K.; Mao, R. The development of next-generation sequencing assays for the mitochondrial genome and 108 nuclear genes associated with mitochondrial disorders. J. Mol. Diagn. 2013, 15, 526–534. [Google Scholar] [CrossRef]
- Seneca, S.; Vancampenhout, K.; Van Coster, R.; Smet, J.; Lissens, W.; Vanlander, A.; De Paepe, B.; Jonckheere, A.; Stouffs, K.; De Meirleir, L. Analysis of the whole mitochondrial genome: Translation of the Ion Torrent Personal Genome Machine system to the diagnostic bench? Eur. J. Hum. Genet. 2015, 23, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Broomfield, A.; Sweeney, M.G.; Woodward, C.E.; Fratter, C.; Morris, A.M.; Leonard, J.V.; Abulhoul, L.; Grunewald, S.; Clayton, P.T.; Hanna, M.G.; et al. Paediatric single mitochondrial DNA deletion disorders: An overlapping spectrum of disease. J. Inherit. Metab. Dis. 2015, 38, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, K.; Klein, C. Next Generation Sequencing and the Future of Genetic Diagnosis. Neurotherapeutics 2014, 11, 699–707. [Google Scholar] [CrossRef] [Green Version]


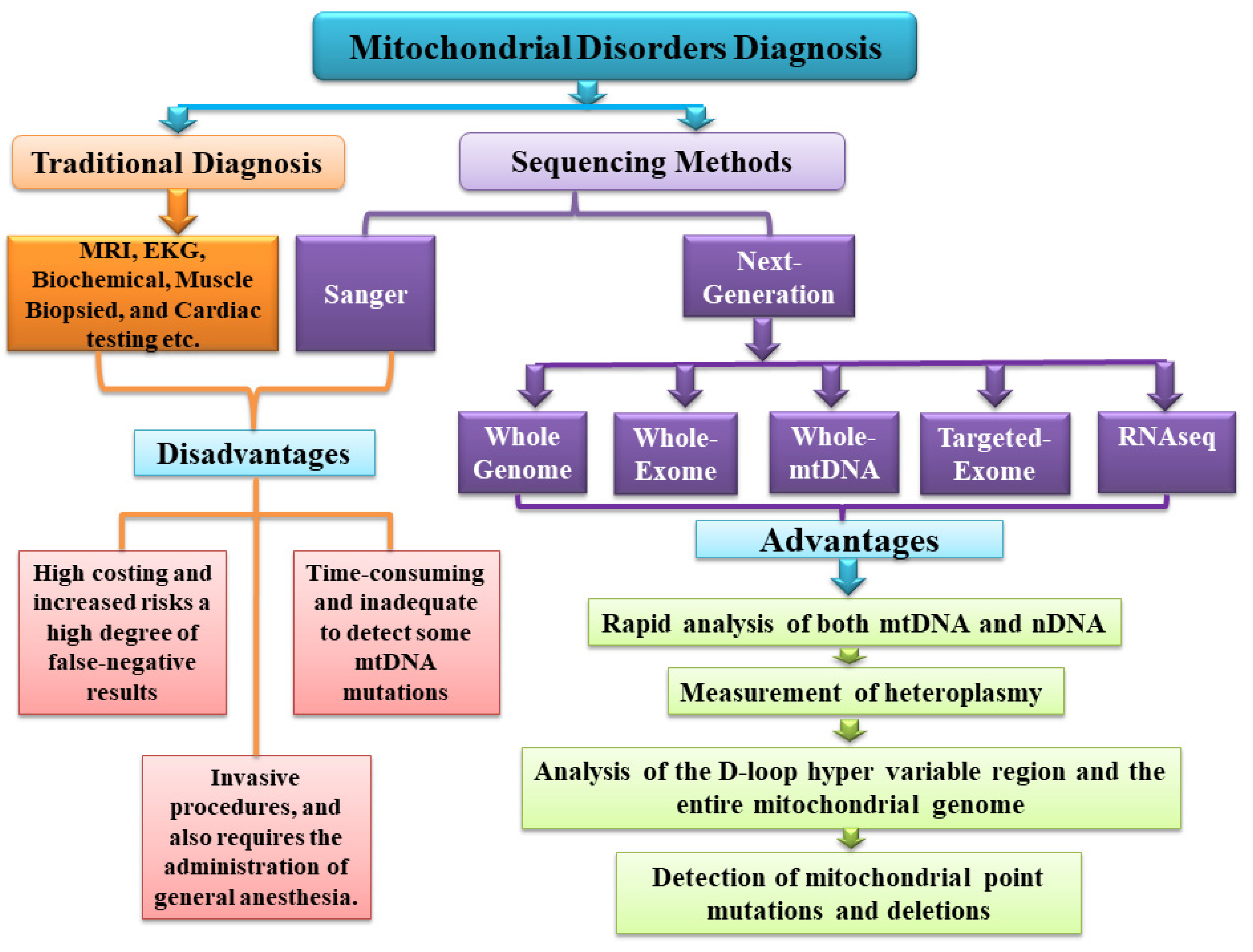{kind=link}
| Clinical Phenotype | Genetic Mutation | Most Common Clinical Features |
|---|---|---|
| MELAS Syndrome [60] | Mostly linked with the mutation designated m.3243A > G in the MTTL gene. Another notable point mutation is m.13513G > A in (MTND5) gene | Migraine, hearing loss, exercise intolerance, growth failure, diabetes, gastrointestinal disturbances, cardiopathy, and ophthalmoparesis |
| MERRF Syndrome [60] | Typical m.8344A > G mutation in the gene denominated as MTTK | Fatigue, ataxia, myoclonus, seizure, weakness in the muscle, ptosis, numerous lipomas, and hearing damage |
| NARP Syndrome [60] | Caused by 70% mutation in the gene entitled MT-ATP6 | Seizures, dementia, ataxia |
| MILS Syndrome [60] | Caused by 90% mutation in the gene recognized as MT-ATP6 | Basal ganglia and brainstem lesion |
| LHON Syndrome [60] | Mutations in three genes m.3460G > A in the gene identified as MTND1, m.14484T > C in the gene acknowledged as MTND6 and m.11778G > A in MTND4 of complex I (ND genes) | Dyschromatopsia, pseudoedema |
| Kearns Sayre syndrome (KSS) [78] | Single large-scale deletion | Pigmentary retinopathy, progressive external ophthalmoplegia with ptosis, and cardiac conduction flaws |
| Progressive external ophthalmoplegia (PEO)/(CPEO) [78,79] | Single large-scale deletion; mutations known as m.3243A > G, m.3243A > T, m.4298G > A, m.4308G > A, m.5690A > G, m.5703G > A, m.12276G > A, m.12294G > A, m.12315G > A, m.12316G > A involving MT-TL1, MT-TI, MT-TN, MT-TL2 genes | Impaired eye movements, ptosis |
| Leigh’s disease [80] | Point mutation occurs in the subunit of protein | Lesions in basal ganglia, psychomotor interruption, problems in movement, lactic acidosis |
| Pearson syndrome [78] | Single large-scale deletion | Sideroblastic anemia, short stature, exocrine pancreatic insufficiency, and failure to thrive |
| Epilepsy [81,82] | Recessive POLG mutations; mutations recognized as m.8344A > G in MT-TK, m.3243A > G in MT-TL1, m.611G > A, and m.583G > A in MT-TF gene | Refractory status epilepticus, migraine, psychiatric |
| Nonsyndromic sensorineural hearing loss [83] | Point mutations at m.1555A > G, m.7445A > G, m.1494C > T, and m.7511T > C in MT-RNR1, MT-TS1 genes | Moderate improving hearing loss, acute deafness |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmud, S.; Biswas, S.; Afrose, S.; Mita, M.A.; Hasan, M.R.; Shimu, M.S.S.; Paul, G.K.; Chung, S.; Saleh, M.A.; Alshehri, S.; et al. Use of Next-Generation Sequencing for Identifying Mitochondrial Disorders. Curr. Issues Mol. Biol. 2022, 44, 1127-1148. https://doi.org/10.3390/cimb44030074
Mahmud S, Biswas S, Afrose S, Mita MA, Hasan MR, Shimu MSS, Paul GK, Chung S, Saleh MA, Alshehri S, et al. Use of Next-Generation Sequencing for Identifying Mitochondrial Disorders. Current Issues in Molecular Biology. 2022; 44(3):1127-1148. https://doi.org/10.3390/cimb44030074
Chicago/Turabian StyleMahmud, Shafi, Suvro Biswas, Shamima Afrose, Mohasana Akter Mita, Md. Robiul Hasan, Mst. Sharmin Sultana Shimu, Gobindo Kumar Paul, Sanghyun Chung, Md. Abu Saleh, Sultan Alshehri, and et al. 2022. "Use of Next-Generation Sequencing for Identifying Mitochondrial Disorders" Current Issues in Molecular Biology 44, no. 3: 1127-1148. https://doi.org/10.3390/cimb44030074
APA StyleMahmud, S., Biswas, S., Afrose, S., Mita, M. A., Hasan, M. R., Shimu, M. S. S., Paul, G. K., Chung, S., Saleh, M. A., Alshehri, S., Ghoneim, M. M., Alruwaily, M., & Kim, B. (2022). Use of Next-Generation Sequencing for Identifying Mitochondrial Disorders. Current Issues in Molecular Biology, 44(3), 1127-1148. https://doi.org/10.3390/cimb44030074