
Using AI-Based Evolutionary Algorithms to Elucidate Adult Brain Tumor (Glioma) Etiology Associated with IDH1 for Therapeutic Target Identification


Abstract
:1. Introduction
2. Materials and Methods
2.1. In Silico Exploration of IDH1 Gene Expression in GBM
2.2. Data Description and Biomarker Identification Using ACE Software
2.3. Investigation of Genes as Potential Biomarkers and Validation Using GlioVis
2.4. Investigation of Gene Pathways Using DAVID Bioinformatic Resources
2.5. Further Investigation of the Potential Biomarkers for Gene Alterations with cBioPortal
2.6. Further Investigations for Correlations between IDH1 mRNA Expression and Methylation, and with Protein Expression in GBM Using GlioVis
3. Results
3.1. IDH1 Expression Patterns in GBM
3.2. Overview of the Gliomas Analyzed by ACE
3.3. Genes Associated with IDH1 in All Gliomas (IDH-Wildtype)
3.4. Genes Associated with IDH1 in Primary GBM (IDH-Wildtype)
3.5. Genes Associated with IDH1 in Recurrent GBM (IDH-Wildtype)
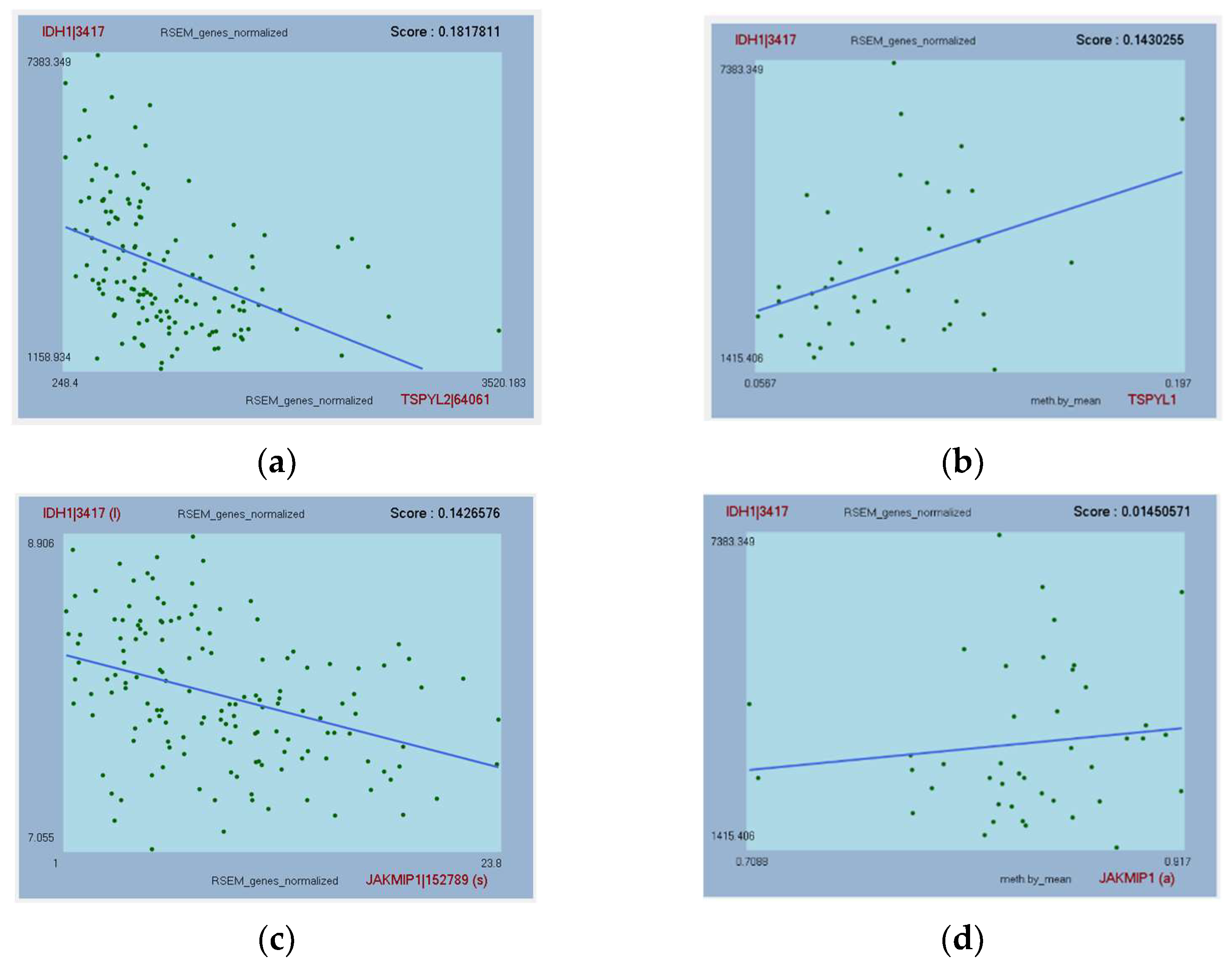
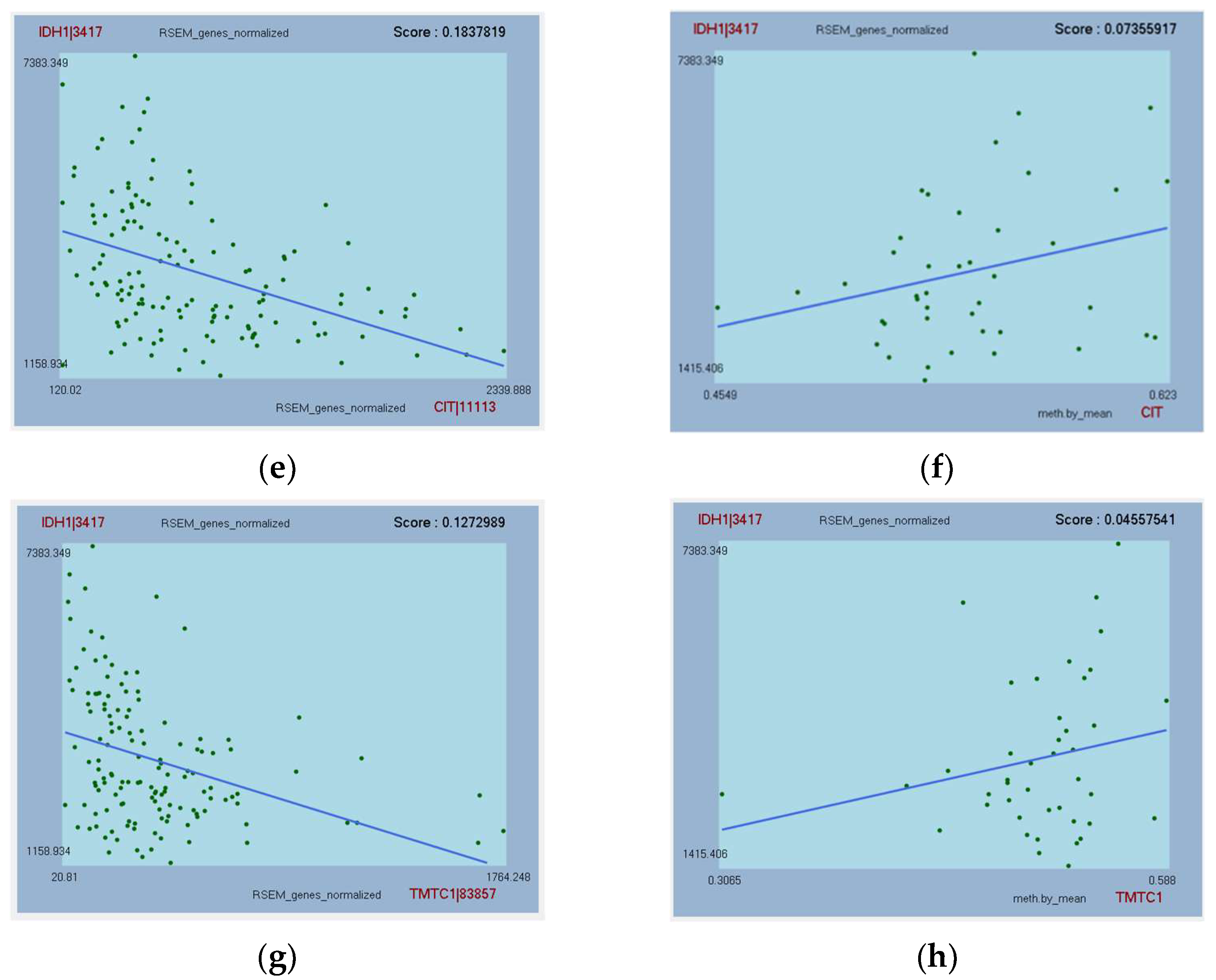
3.6. Further Analysis of Top-Hit Genes Associated with IDH1 as Potential Biomarkers
3.7. Genes Whose Protein Expression Is Associated with IDH1 Gene Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Office for National Statistics. Death Registration Summary Tables, England & Wales, Table 2. 2017. Available online: https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/datasets/deathregistrationssummarytablesenglandandwalesreferencetables (accessed on 8 November 2021).
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23 (Suppl. S3), iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Sun, R.; Cuthbert, H.; Watts, C. Fluorescence-Guided Surgery in the Surgical Treatment of Gliomas: Past, Present and Future. Cancers 2021, 13, 3508. [Google Scholar] [CrossRef] [PubMed]
- Heffron, T.P. Challenges of developing small-molecule kinase inhibitors for brain tumors and the need for emphasis on free drug levels. Neuro Oncol. 2018, 20, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, H.; Lee, C.K.; Tateishi, K.; Higuchi, F.; Subramanian, M.; Rafferty, S.; Melamed, L.; Miller, J.J.; Wakimoto, H.; Cahill, D.P. Poly (ADP-ribose) glycohydrolase inhibition sequesters NAD+ to potentiate the metabolic lethality of alkylating chemotherapy in IDH-mutant tumor cells. Cancer Discov. 2020, 10, 1672–1689. [Google Scholar] [CrossRef] [PubMed]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Wu, S.; Zhang, J.; Li, M.; Xu, F.; Wang, A.; Lei, Y.; Zhu, G. Wild-type IDH1 affects cell migration by modulating the PI3K/AKT/mTOR pathway in primary glioblastoma cells. Mol. Med. Rep. 2020, 22, 1949–1957. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Carey, B.W.; Finley, L.W.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Polewski, M.D.; Reveron-Thornton, R.F.; Cherryholmes, G.A.; Marinov, G.K.; Cassady, K.; Aboody, K.S. Increased expression of system xc− in glioblastoma confers an altered metabolic state and temozolomide resistance. Mol. Cancer Res. 2016, 14, 1229–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maus, A.; Peters, G.J. Glutamate and α-ketoglutarate: Key players in glioma metabolism. Amino Acids 2017, 49, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.B.; Dong, D.F.; Wang, M.D.; Gao, K. IDH1 overexpression induced chemotherapy resistance and IDH1 mutation enhanced chemotherapy sensitivity in Glioma cells in vitro and in vivo. Asian Pac. J. Cancer Prev. 2014, 15, 427–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swan, A.L.; Stekel, D.J.; Hodgman, C.; Allaway, D.; Alqahtani, M.H.; Mobasheri, A.; Bacardit, J. A machine learning heuristic to identify biologically relevant and minimal biomarker panels from omics data. BMC Genom. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Alderdice, M.; Craig, S.G.; Humphries, M.P.; Gilmore, A.; Johnston, N.; Bingham, V.; Coyle, V.; Senevirathne, S.; Longley, D.B.; Loughrey, M.B.; et al. Evolutionary genetic algorithm identifies IL2RB as a potential predictive biomarker for immune-checkpoint therapy in colorectal cancer. NAR Genom. Bioinform. 2021, 3, lqab016. [Google Scholar] [CrossRef]
- Gilmore, A.R.; Alderdice, M.; Savage, K.I.; O’Reilly, P.G.; Roddy, A.C.; Dunne, P.D.; Lawler, M.; McDade, S.S.; Waugh, D.J.; McArt, D.G. ACE: A workbench using evolutionary genetic algorithms for analyzing association in TCGA. Cancer Res. 2019, 79, 2072–2075. [Google Scholar] [CrossRef] [Green Version]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Gravendeel, L.A.; Kouwenhoven, M.C.; Gevaert, O.; de Rooi, J.J.; Stubbs, A.P.; Duijm, J.E.; French, P.J. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009, 69, 9065–9072. [Google Scholar] [CrossRef] [Green Version]
- Madhavan, S.; Zenklusen, J.C.; Kotliarov, Y.; Sahni, H.; Fine, H.A.; Buetow, K. Rembrandt: Helping personalized medicine become a reality through integrative translational research. Mol. Cancer Res. 2009, 7, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Kamoun, A.; Idbaih, A.; Dehais, C.; Elarouci, N.; Carpentier, C.; Letouzé, E.; Colin, C.; Mokhtari, K.; Jouvet, A.; Uro-Coste, E.; et al. Integrated multi-omics analysis of oligodendroglial tumours identifies three subgroups of 1p/19q co-deleted gliomas. Nat. Commun. 2016, 7, 11263. [Google Scholar] [CrossRef] [PubMed]
- Grzmil, M.; Morin, P., Jr.; Lino, M.M.; Merlo, A.; Frank, S.; Wang, Y.; Moncayo, G.; Hemmings, B.A. MAP kinase-interacting kinase 1 regulates SMAD2-dependent TGF-β signaling pathway in human glioblastoma. Cancer Res. 2011, 71, 2392–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murat, A.; Migliavacca, E.; Gorlia, T.; Lambiv, W.L.; Shay, T.; Hamou, M.F.; Hegi, M.E. Stem cell-related” self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J. Clin. Oncol. 2008, 26, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.G.; Foltz, G.D. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [Green Version]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. 2007–2015. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 12 January 2021).
- Zhao, Z.; Meng, F.; Wang, W.; Wang, Z.; Zhang, C.; Jiang, T. Comprehensive RNA-seq transcriptomic profiling in the malignant progression of gliomas. Sci. Data 2017, 4, 1–7. [Google Scholar] [CrossRef]
- Lee, Y.; Scheck, A.C.; Cloughesy, T.F.; Lai, A.; Dong, J.; Farooqi, H.K.; Nelson, S.F. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med. Genom. 2008, 1, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Wang, M.; Zhang, G.; Li, Y.; Wang, L.; Cui, H. POU2F2 regulates glycolytic reprogramming and glioblastoma progression via PDPK1-dependent activation of PI3K/AKT/mTOR pathway. Cell Death Dis. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Zhang, S. Selective autophagy and Huntingtin: Learning from disease. Cell Cycle 2015, 14, 1617–1618. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Xiao, J.; Ke, C.; Ni, X.; Xiu, R.; Tian, Q.; Pan, H.; Zou, L.; Wang, F.; Ma, T.; et al. TOPK inhibits autophagy by phosphorylating ULK1 and promotes glioma resistance to TMZ. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunez, F.; Garcia-Fabiani, M.; Kadiyala, P.; Hong, H.; Comba, A.; Lyssiotis, C.; Cheng, S.Y.; Lowenstein, P.; Castro, M. RDNA-03. Autophagy as a novel therapeutic target in mutant IDH1 gliomas. Neuro Oncol. 2019, 21 (Suppl. S6), vi207. [Google Scholar] [CrossRef]
- Tian, X.; Wang, Y.; Ding, X.; Cheng, W. High expression of GLO1 indicates unfavorable clinical outcomes in glioma patients. J. Neurosurg. Sci. 2019, 66(3), 228–233. [Google Scholar] [CrossRef] [PubMed]
- Jandial, R.; Neman, J.; Lim, P.P.; Tamae, D.; Kowolik, C.M.; Wuenschell, G.E.; Shuck, S.C.; Ciminera, A.K.; De Jesus, L.R.; Ouyang, C.; et al. Inhibition of GLO1 in glioblastoma multiforme increases DNA-AGEs, stimulates RAGE expression, and inhibits brain tumor growth in orthotopic mouse models. Int. J. Mol. Sci. 2018, 19, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sun, X.; Wang, J.; Zhang, K.; Yuan, Y.; Yao, L.; Li, X.; Shen, L. NDRG2 Inhibits Pyruvate Carboxylase-Mediated Anaplerosis and Combines with Glutamine Blockade To Inhibit The Growth of Glioma. ResearchSquare 2021. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yamaguchi, S.; Yonemura, S.; Kakiguchi, K.; Sato, Y.; Higashiyama, T.; Shimizu, T.; Hibi, M. Type IV collagen controls the axogenesis of cerebellar granule cells by regulating basement membrane integrity in zebrafish. PLoS Genet. 2015, 11, e1005587. [Google Scholar] [CrossRef] [Green Version]
- Le, M.T.; Shyh-Chang, N.; Khaw, S.L.; Chin, L.; The, C.; Tay, J.; O’Day, E.; Korzh, V.; Yang, H.; Lal, A.; et al. Conserved regulation of p53 network dosage by microRNA–125b occurs through evolving miRNA–target gene pairs. PLoS Genet. 2011, 7, e1002242. [Google Scholar] [CrossRef] [Green Version]
- Haemmig, S.; Baumgartner, U.; Glück, A.; Zbinden, S.; Tschan, M.P.; Kappeler, A.; Mariani, L.; Vajtai, I.; Vassella, E. miR-125b controls apoptosis and temozolomide resistance by targeting TNFAIP3 and NKIRAS2 in glioblastomas. Cell Death Dis. 2014, 5, e1279. [Google Scholar] [CrossRef] [Green Version]
- Qie, S.; Yoshida, A.; Parnham, S.; Oleinik, N.; Beeson, G.C.; Beeson, C.C.; Ogretmen, B.; Bass, A.J.; Wong, K.K.; Rustgi, A.K.; et al. Targeting glutamine-addiction and overcoming CDK4/6 inhibitor resistance in human esophageal squamous cell carcinoma. Nat. Commun. 2019, 10, 1296. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, L.M.; Smith, P.; Chen, Z.; Fouad, S.; D’Angiolella, V. The role of E3 ubiquitin ligases in the development and progression of glioblastoma. Cell Death Differ. 2021, 28, 522–537. [Google Scholar] [CrossRef]
- Zhao, W.; Huang, Y.; Zhang, J.; Liu, M.; Ji, H.; Wang, C.; Cao, N.; Li, C.; Xia, Y.; Jiang, Q.; et al. Polycomb group RING finger proteins 3/5 activate transcription via an interaction with the pluripotency factor Tex10 in embryonic stem cells. J. Biol. Chem. 2017, 292, 21527–21537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moey, C.; Hinze, S.J.; Brueton, L.; Morton, J.; McMullan, D.J.; Kamien, B.; Barnett, C.P.; Brunetti-Pierri, N.; Nicholl, J.; Gecz, J.; et al. Xp11. 2 microduplications including IQSEC2, TSPYL2 and KDM5C genes in patients with neurodevelopmental disorders. Eur. J. Hum. Genet. 2016, 24, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Peng, L.; So, J.; Tsang, K.H.; Chong, C.H.; Mak, P.H.; Chan, K.M.; Chan, S.Y. TSPYL2 regulates the expression of EZH2 target genes in neurons. Mol. Neurobiol. 2019, 56, 2640–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magni, M.; Buscemi, G.; Maita, L.; Peng, L.; Chan, S.Y.; Montecucco, A.; Delia, D.; Zannini, L. TSPYL2 is a novel regulator of SIRT1 and p300 activity in response to DNA damage. Cell Death Differ. 2019, 26, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Zhong, S.; Fields, C.R.; Kim, J.H.; Robertson, K.D. Epigenomic profiling reveals novel and frequent targets of aberrant DNA methylation-mediated silencing in malignant glioma. Cancer Res. 2006, 66, 7490–7501. [Google Scholar] [CrossRef] [Green Version]
- Epping, M.T.; Lunardi, A.; Nachmani, D.; Castillo-Martin, M.; Thin, T.H.; Cordon-Cardo, C.; Pandolfi, P.P. TSPYL2 is an essential component of the REST/NRSF transcriptional complex for TGF β signaling activation. Cell Death Differ. 2015, 22, 1353–1362. [Google Scholar] [CrossRef]
- Steindler, C.; Li, Z.; Algarté, M.; Alcover, A.; Libri, V.; Ragimbeau, J.; Pellegrini, S. Jakmip1 (marlin-1) defines a family of proteins interacting with janus kinases and microtubules. J. Biol. Chem. 2004, 279, 43168–43177. [Google Scholar] [CrossRef] [Green Version]
- Vidal, R.L.; Fuentes, P.; Valenzuela, J.I.; Alvarado-Diaz, C.P.; Ramírez, O.A.; Kukuljan, M.; Couve, A. RNA interference of Marlin-1/Jakmip1 results in abnormal morphogenesis and migration of cortical pyramidal neurons. Mol. Cell. Neurosci. 2012, 51, 1–11. [Google Scholar] [CrossRef]
- Lasser, M.; Tiber, J.; Lowery, L.A. The role of the microtubule cytoskeleton in neurodevelopmental disorders. Front. Cell. Neurosci. 2018, 12, 165. [Google Scholar] [CrossRef] [Green Version]
- Okai, I.; Wang, L.; Gong, L.; Arko-Boham, B.; Hao, L.; Zhou, X.; Qi, X.; Hu, J.; Shao, S. Overexpression of JAKMIP1 associates with Wnt/beta-catenin pathway activation and promotes cancer cell proliferation in vitro. Biomed. Pharmacother. 2013, 67, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Pallavicini, G.; Iegiani, G.; Berto, G.E.; Calamia, E.; Trevisiol, E.; Veltri, A.; Allis, S.; Di Cunto, F. CITK Loss inhibits growth of Group 3 and Group 4 medulloblastoma cells and sensitizes them to DNA-damaging agents. Cancers 2020, 12, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhaber, B.; Sinha, S.; Jadalanki, C.K.; Shitov, V.A.; Tan, Q.W.; Sirota, F.L.; Eisenhaber, F. Conserved sequence motifs in human TMTC1, TMTC2, TMTC3, and TMTC4, new O-mannosyltransferases from the GT-C/PMT clan, are rationalized as ligand binding sites. Biol. Direct 2021, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Larsen, I.S.; Narimatsu, Y.; Joshi, H.J.; Siukstaite, L.; Harrison, O.J.; Brasch, J.; Goodman, K.M.; Hansen, L.; Shapiro, L.; Honig, B.; et al. Discovery of an O-mannosylation pathway selectively serving cadherins and protocadherins. Proc. Natl. Acad. Sci. USA 2017, 114, 11163–11168. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, S.; Oliveira, T.; Bartels, M.F.; Miyoshi, E.; Pierce, M.; Taniguchi, N.; Carneiro, F.; Seruca, R.; Reis, C.A.; Strahl, S.; et al. O-mannosylation and N-glycosylation: Two coordinated mechanisms regulating the tumour suppressor functions of E-cadherin in cancer. Oncotarget 2016, 7, 65231. [Google Scholar] [CrossRef] [Green Version]
- Mealer, R.G.; Williams, S.E.; Daly, M.J.; Scolnick, E.M.; Cummings, R.D.; Smoller, J.W. Glycobiology and schizophrenia: A biological hypothesis emerging from genomic research. Mol. Psychiatry 2020, 25, 3129–3139. [Google Scholar] [CrossRef]
- Jerber, J.; Zaki, M.S.; Al-Aama, J.Y.; Rosti, R.O.; Ben-Omran, T.; Dikoglu, E.; Silhavy, J.L.; Caglar, C.; Musaev, D.; Albrecht, B.; et al. Biallelic mutations in TMTC3, encoding a transmembrane and TPR-containing protein, lead to cobblestone lissencephaly. Am. J. Hum. Genet. 2016, 99, 1181–1189. [Google Scholar] [CrossRef] [Green Version]
- Yun, E.J.; Vu, T.H. mSmile is necessary for bronchial smooth muscle and alveolar myofibroblast development. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2012, 295, 167–176. [Google Scholar] [CrossRef]
- Chiavari, M.; Ciotti, G.M.; Canonico, F.; Altieri, F.; Lacal, P.M.; Graziani, G.; Navarra, P.; Lisi, L. Pdia3 expression in glioblastoma modulates macrophage/microglia pro-tumor activation. Int. J. Mol. Sci. 2020, 21, 8214. [Google Scholar] [CrossRef]
- Li, Y.; Gan, S.; Ren, L.; Yuan, L.; Liu, J.; Wang, W.; Wang, X.; Zhang, Y.; Jiang, J.; Zhang, F.; et al. Multifaceted regulation and functions of replication factor C family in human cancers. Am. J. Cancer Res. 2018, 8, 1343–1355. [Google Scholar]
- Zhang, L.; Zhou, Q.; Qiu, Q.; Hou, L.; Wu, M.; Li, J.; Li, X.; Lu, B.; Cheng, X.; Liu, P.; et al. CircPLEKHM3 acts as a tumor suppressor through regulation of the miR-9/BRCA1/DNAJB6/KLF4/AKT1 axis in ovarian cancer. Mol. Cancer 2019, 18, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Semir, D.; Bezrookove, V.; Nosrati, M.; Scanlon, K.R.; Singer, E.; Judkins, J.; Kashani-Sabet, M. PHIP drives glioblastoma motility and invasion by regulating the focal adhesion complex. Proc. Natl. Acad. Sci. USA 2020, 117, 9064–9073. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Fang, H. Curcumin inhibits ovarian cancer progression by regulating circ-PLEKHM3/miR-320a/SMG1 axis. J. Ovarian Res. 2021, 14, 158. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.V.; Hao, X.; Aman, A.; Luchman, H.A.; Weiss, S. EGFR blockade in GBM brain tumor stem cells synergizes with JAK2/STAT3 pathway inhibition to abrogate compensatory mechanisms in vitro and in vivo. Neuro Oncol. Adv. 2020, 2, vdaa020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zhou, Y.; Chen, C.; Yang, T.; Zhou, S.; Chen, S.; Wu, Y.; Cui, Y. Efficacy and safety of mTOR inhibitors (rapamycin and its analogues) for tuberous sclerosis complex: A meta-analysis. Orphanet J. Rare Dis. 2019, 14, 39. [Google Scholar] [CrossRef]
- Gómez-Oliva, R.; Domínguez-García, S.; Carrascal, L.; Abalos-Martínez, J.; Pardillo-Díaz, R.; Verástegui, C.; Castro, C.; Nunez-Abades, P.; Geribaldi-Doldán, N. Evolution of experimental models in the study of glioblastoma: Toward finding efficient treatments. Front. Oncol. 2021, 10, 3245. [Google Scholar] [CrossRef]
- Pernik, M.N.; Bird, C.E.; Traylor, J.I.; Shi, D.D.; Richardson, T.E.; McBrayer, S.K.; Abdullah, K.G. Patient-Derived Cancer Organoids for Precision Oncology Treatment. J. Pers. Med. 2021, 11, 423. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IDH Status (Classifier) | |||||||
|---|---|---|---|---|---|---|---|
| Cancer Type | Primary/Recurrent | Grade | N | % | Wildtype | Mutant | Unknown |
| Oligoastrocytoma | Primary | II | 16 | 2.40 | 16 | 0 | 0 |
| Anaplastic Oligoastrocytoma | Primary | III | 10 | 1.50 | 10 | 0 | 0 |
| Oligodendroglioma | Primary | II | 10 | 1.50 | 10 | 1 | 0 |
| Oligodendroglioma | Recurrent | II | 1 | 0.15 | 1 | 0 | 0 |
| Astrocytoma | Primary | II | 9 | 1.35 | 9 | 0 | 0 |
| Astrocytoma | Recurrent | II | 1 | 0.15 | 1 | 0 | 0 |
| Anaplastic Astrocytoma | Primary | III | 47 | 7.04 | 47 | 0 | 0 |
| Glioblastoma | Primary | IV | 562 | 84.13 | 428 | 23 | 111 |
| Glioblastoma | Recurrent | IV | 12 | 1.80 | 9 | 3 | 0 |
| Total | 668 | 100 | 531 | 27 | 111 | ||
| Analysis | KEGG Term | KEGG Pathway | Count | Gene Name | Entrez Accession Numbers | p-Value (EASE Score) | p-Value Adjusted (Bonferroni) |
|---|---|---|---|---|---|---|---|
| 1. All | hsa00620 | Pyruvate metabolism | 2 | GLO1, PC | 5091, 2739 | 0.057 | 0.65 |
| 2. GBM NR | hsa04120 | Ubiquitin-mediated proteolysis | 3 | FBXO4, UBE2F, UBE3B | 26,272, 140,739, 89,910 | 0.023 | 0.69 |
| 2. GBM NR | hsa04510 | Focal adhesion | 3 | COL4A6, PPP1CA, PDPK1 | 1288, 5499, 5170 | 0.048 | 0.92 |
| 2. GBM NR | hsa04150 | mTOR signaling pathway | 2 | PDPK1, ULK1 | 5170, 8408 | 0.097 | 0.99 |
| 3. GBM R | hsa04150 | mTOR signaling pathway | 2 | PDPK1, ULK1 | 5170, 8408 | 0.089 | 0.99 |
| Genes Common Between: | Genes Exclusive To: | ||||
|---|---|---|---|---|---|
| All & GBM NR | All & GBM R | GBM NR & GBM R | All | GBM NR | GBM R |
| TSPYL2 C9orf45 | RFC2 PPIA | C20orf194 MECP2 PLEKHM3 C1orf198 MLLT6 HDAC5 ULK1 PDPK1 PSMA3 FKBP3 | MYH7B MINK1 EZH1 TOM1L2 GABPB1 NRG3 CRY2 GRAMD1 JAKMIP1 PC C5orf53 PLCXD3 SYNE1 TCEAL3 FNBP1 TBRG1 LLGL2 C10orf28 QRSL1 ZNF224 BZW1 DBF4 MED20 ILF2 C6orf153 ZNF410 EIF4A3 TIMP1 ZNF277 GLO1 | MPL LPAL2 FAM189A1 COL4A6 COLQ CIT UBE3B FBRS KCNQ5 PDZD8 FAM53C FAT3 IQCF1 MYOD1 DENND MFSD4 SNX6 C2orf80 NCRNA PPP1CA UBE2F FBXO4 ZEB1 | MYH15 HSFX2 ANKRD24 ZDHHC11 TMTC1 EDA FLYWCH1 TULP4 PER3 PCGF3 ABCC5 KIAA0355 MFNG FEZF1 TNFAIP6 PDGFA MEMO1 FAM3C COMMD1 FAM32A FAM98C FAM131B |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McInerney, C.E.; Lynn, J.A.; Gilmore, A.R.; Flannery, T.; Prise, K.M. Using AI-Based Evolutionary Algorithms to Elucidate Adult Brain Tumor (Glioma) Etiology Associated with IDH1 for Therapeutic Target Identification. Curr. Issues Mol. Biol. 2022, 44, 2982-3000. https://doi.org/10.3390/cimb44070206
McInerney CE, Lynn JA, Gilmore AR, Flannery T, Prise KM. Using AI-Based Evolutionary Algorithms to Elucidate Adult Brain Tumor (Glioma) Etiology Associated with IDH1 for Therapeutic Target Identification. Current Issues in Molecular Biology. 2022; 44(7):2982-3000. https://doi.org/10.3390/cimb44070206
Chicago/Turabian StyleMcInerney, Caitríona E., Joanna A. Lynn, Alan R. Gilmore, Tom Flannery, and Kevin M. Prise. 2022. "Using AI-Based Evolutionary Algorithms to Elucidate Adult Brain Tumor (Glioma) Etiology Associated with IDH1 for Therapeutic Target Identification" Current Issues in Molecular Biology 44, no. 7: 2982-3000. https://doi.org/10.3390/cimb44070206
APA StyleMcInerney, C. E., Lynn, J. A., Gilmore, A. R., Flannery, T., & Prise, K. M. (2022). Using AI-Based Evolutionary Algorithms to Elucidate Adult Brain Tumor (Glioma) Etiology Associated with IDH1 for Therapeutic Target Identification. Current Issues in Molecular Biology, 44(7), 2982-3000. https://doi.org/10.3390/cimb44070206