
Molecular Profiling of Tumor Tissue in Mexican Patients with Colorectal Cancer


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Library Preparation and Targeted Sequencing
2.3. Data Analysis
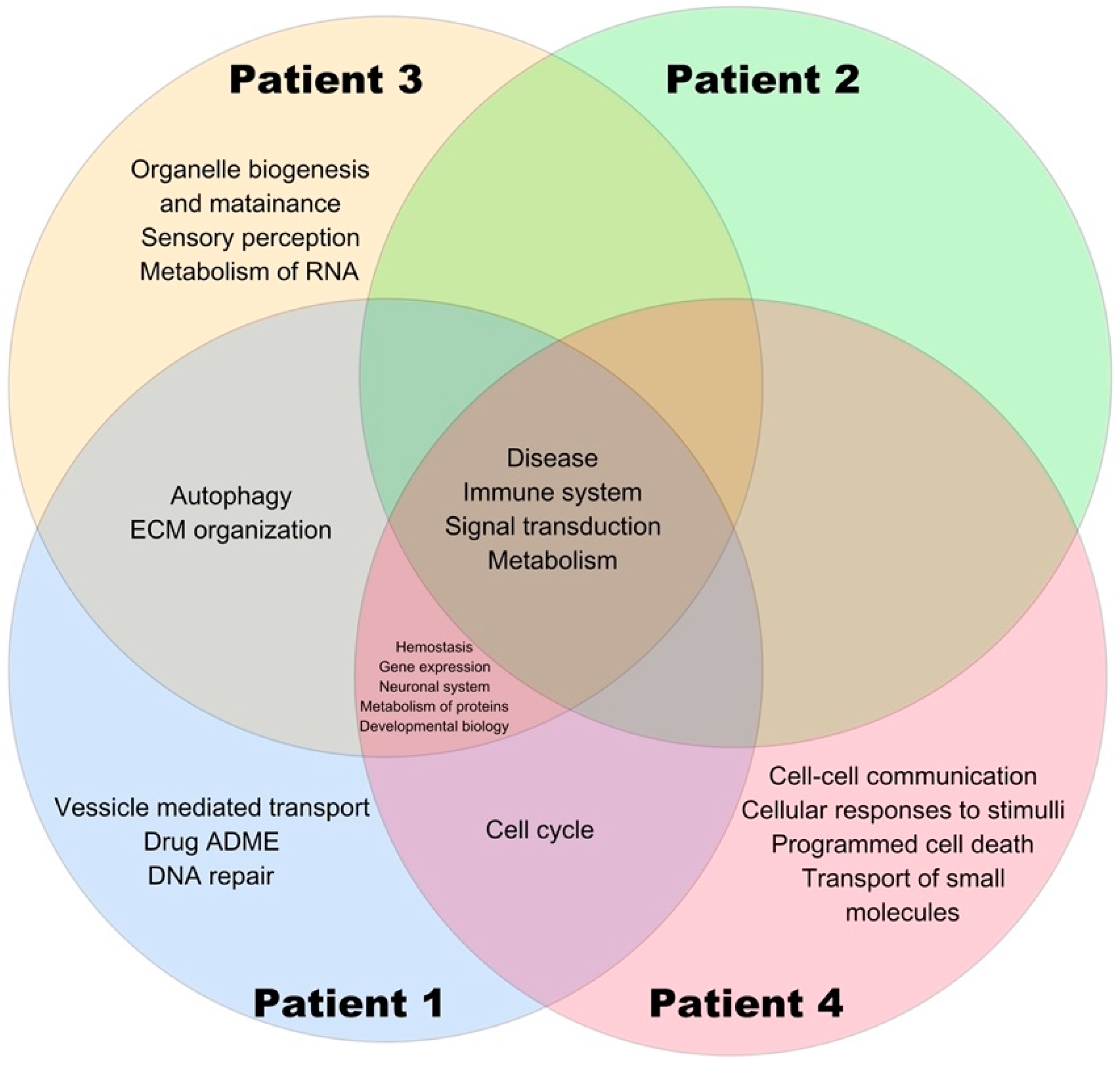
3. Results
3.1. Patient Characterization
3.2. Variant Identification in Tumor Samples
3.3. Data Processing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliore, L.; Migheli, F.; Spisni, R.; Copped, F. Genetics, cytogenetics, and epigenetics of colorectal cancer. J. Biomed. Biotechnol. 2011, 2011, 792362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, J.; Walker, L.C.; Robinson, B.A.; Frizelle, F.A.; Church, J.M.; Eglinton, T.W. Clinical implications of the genetics of sporadic colorectal cancer. ANZ J. Surg. 2019, 89, 1224–1229. [Google Scholar] [CrossRef]
- Del Vecchio, F.; Mastroiaco, V.; Di Marco, A.; Compagnoni, C.; Capece, D.; Zazzeroni, F.; Capalbo, C.; Alesse, E.; Tessitore, A. Next-generation sequencing: Recent applications to the analysis of colorectal cancer. J. Transl. Med. 2017, 15, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M. International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [Green Version]
- Win, A.K.; Jenkins, M.A.; Buchanan, D.D.; Clendenning, M.; Young, J.P.; Giles, G.G.; Goldblatt, J.; Leggett, B.A.; Hopper, J.L.; Thibodeau, S.N.; et al. Determining the frequency of de novo germline mutations in DNA mismatch repair genes. J. Med. Genet. 2011, 48, 530–534. [Google Scholar] [CrossRef]
- Barani, S.; Hosseini, S.V.; Ghaderi, A. Activating and inhibitory killer cell immunoglobulin like receptors (KIR) genes are involved in an increased susceptibility to colorectal adenocarcinoma and protection against invasion and metastasis. Immunobiology 2019, 224, 681–686. [Google Scholar] [CrossRef]
- Revel, M.; Daugan, M.V.; Sautés-Fridman, C.; Fridman, W.H.; Roumenina, L.T. Complement System: Promoter or Suppressor of Cancer Progression? Antibodies 2020, 9, 57. [Google Scholar] [CrossRef]
- Lang, M.; Gasche, C. Chemoprevention of colorectal cancer. Dig. Dis. 2015, 33, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Zhu, L.; Gu, Y.; Liu, P.; Tong, X.; Wu, G.; Zhu, W.; Shen, W.; Bao, H.; Ma, X.; et al. Genomic Profiling Reveals the Molecular Landscape of Gastrointestinal Tract Cancers in Chinese Patients. Front. Genet. 2021, 12, 608742. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Behera, B.P.; Mishra, S.R.; Bhutia, S.K. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Sem. Cancer Biol. 2020, 66, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, G.; Morris, K.V. All I’s on the RADAR: Role of ADAR in gene regulation. FEBS Lett. 2018, 592, 2860–2873. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Rabbie, R.; Ferguson, P.; Wong, K.; Couturier, D.L.; Moran, U.; Turner, C.; Emanuel, P.; Haas, K.; Saunus, J.M.; Davidson, M.R.; et al. The mutational landscape of melanoma brain metastases presenting as the first visceral site of recurrence. Br. J. Cancer. 2021, 124, 156–160. [Google Scholar] [CrossRef]
- Kung, C.P.; Maggi, L.B., Jr.; Weber, J.D. The Role of RNA Editing in Cancer Development and Metabolic Disorders. Front. Endocrinol. 2018, 9, 762. [Google Scholar] [CrossRef] [Green Version]
- Naccarati, A.; Polakova, V.; Pardini, B.; Vodickova, L.; Hemminki, K.; Kumar, R.; Vodicka, P. Mutations and polymorphisms in TP53 gene—An overview on the role in colorectal cancer. Mutagenesis 2012, 27, 211–218. [Google Scholar] [CrossRef]
- Michel, M.; Kaps, L.; Maderer, A.; Galle, P.R.; Moehler, M. The Role of p53 Dysfunction in Colorectal Cancer and Its Implication for Therapy. Cancers 2021, 13, 2296. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Xicola, R.M.; Clark, J.R.; Carroll, T.; Alvikas, J.; Marwaha, P.; Regan, M.R.; Lopez-Giraldez, F.; Choi, J.; Emmadi, R.; Alagiozian-Angelova, V.; et al. Implication of DNA repair genes in Lynch-like syndrome. Fam. Cancer 2019, 18, 331–342. [Google Scholar] [CrossRef]
- Paschke, S.; Jafarov, S.; Staib, L.; Kreuser, E.D.; Maulbecker-Armstrong, C.; Roitman, M.; Holm, T.; Harris, C.C.; Link, K.H.; Kornmann, M. Are Colon and Rectal Cancer Two Different Tumor Entities? A Proposal to Abandon the Term Colorectal Cancer. Int. J. Mol. Sci. 2018, 19, 2577. [Google Scholar] [CrossRef] [Green Version]
- Sagaert, X.; Vanstapel, A.; Verbeek, S. Tumor Heterogeneity in Colorectal Cancer: What Do We Know So Far? Pathobiology 2018, 85, 72–84. [Google Scholar] [CrossRef]
- Trabucco, S.E.; Gowen, K.; Maund, S.L.; Sanford, E.; Fabrizio, D.A.; Hall, M.J.; Yakirevich, E.; Gregg, J.P.; Stephens, P.J.; Frampton, G.M.; et al. A Novel Next-Generation Sequencing Approach to Detecting Microsatellite Instability and Pan-Tumor Characterization of 1000 Microsatellite Instability-High Cases in 67,000 Patient Samples. J. Mol. Diagn. 2019, 21, 1053–1066. [Google Scholar] [CrossRef]

{kind=link}
| Patient | Age | Sex | Tumor Localization | Histology Type | Metastasis | Stage | Diagnosis |
|---|---|---|---|---|---|---|---|
| 1 | 26 | Male | Left colon | Mucinous Adenocarcinoma † | No | II | LS? |
| 2 | 34 | Female | Rectum | Adenocarcinoma † | Node | III | FAP |
| 3 | 48 | Male | Rectum | Adenocarcinoma † | Node | III | Sporadic CRC |
| 4 | 66 | Male | Rectum | Adenocarcinoma ‡ | Node | III | Sporadic CRC |
| Patient Number | 1 | 2 | 3 | 4 | Total |
|---|---|---|---|---|---|
| Variant Type | |||||
| Pathogenic | 22 | 7 | 9 | 2 | 40 |
| Likely pathogenic | 124 | 9 | 10 | 9 | 152 |
| Total of variants | 146 (76%) | 16 (8%) | 19 (10%) | 11 (6%) | 192 (100%) |
| Indels | 101 | 5 | 6 | 3 | 115 |
| SNV | 45 | 11 | 13 | 8 | 77 |
| Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|
| ABCB4, ABCC4, ACOX1, ADAMTS18, ADAMTSL2 *, ADAMTSL4, AFP, ALDOA, AMER1, ANK2, ANO10, ANXA11, APC *, ARID1A, ATM, BCO1, BCORL1, BMPR2, CASP5, CASR, CCDC40, CEL *, CFI, CHD2, COL6A5, CPZ, CSNK2A2, CUL5, CYLD, CYP2D6, DCLRE1C, DHX16, DISP1, DLGAP3, DNAI2, DPM1, DTNA, EGR2, EPHA2, EPHA3 *, FBN2, FBN3, FBXW7, FCN3, FGG, FLCN, GJA8, GRK4, GSE1, HAX1, HMBS, HNF1A, HPS6, HTR3C, HTT, HYDIN, IQGAP1, ITPKC, ITPR1, KAT6B, KIR2DL4, KIR3DL1, KMT2E, KRAS, LARS2, LIG3, LMTK3, LTBP4, MAD1L1, MAP7D3, MASTL, MIA3, MLH3, MOGS, MSH3, MSH6 *, MST1, MTMR9, MTUS1 *, MUC5B, MYB, MYH14, MYL2, MYO15A, MYO9B, NAT1, NBAS, NLRP12, NOD2, NRXN1, OBSL1, PCARE, PCDH15, PHF2 *, PHKB, PIK3C2G, PLEC, PLEKHG4, PRRT2, PRSS12, PRX, PTCH1, PTEN, PTPN21, PZP, RBBP8, REV3L, RNASEH2B, ROR2, RSPH4A, SALL4, SCN9A *, SEC63 *, SERPINA6, SETX, SLC9A9, SPTB, STRA6, SUCLG1, TAP2, TBC1D23, TBX1, TCF7, TCF7L2, TGFBR2, TGM1, TMPO, TNXB, TOP1MT, TPP2, TRPM1, TUBB2B, CCN6, ZC3H3, ZFP90, ZNF469 | ABCC6 *, APC, C8B, DOCK4, ENO3, GALNS, HLA-DRB1, HYDIN, PPP2R2B *, SCN9A *, TMPRSS5 | A4GALT, ADAR, ALDOB, CNGB1, COL4A3, EYS, FBN1, HNF1B, KIR2DL4, KRAS, MC1R, PKHD1, PRF1, PRKN, RET, ROPN1L, SARDH, SCO2, TRPV4 | CD109, HYDIN, KIR2DL4, KRAS, MS4A2, MUC6, PAFAH1B3, PIK3CA, PTCD1, TP53, TRPV4 |
| 136 genes | 11 genes | 19 genes | 11 genes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-López, B.A.; Ayala-Madrigal, M.d.l.L.; Moreno-Ortiz, J.M.; Peregrina-Sandoval, J.; Trujillo-Rojas, M.Á.; Venegas-Rodríguez, J.L.; Hernández-Ramírez, R.; Fernández-Galindo, M.A.; Gutiérrez-Angulo, M. Molecular Profiling of Tumor Tissue in Mexican Patients with Colorectal Cancer. Curr. Issues Mol. Biol. 2022, 44, 3770-3778. https://doi.org/10.3390/cimb44080258
Flores-López BA, Ayala-Madrigal MdlL, Moreno-Ortiz JM, Peregrina-Sandoval J, Trujillo-Rojas MÁ, Venegas-Rodríguez JL, Hernández-Ramírez R, Fernández-Galindo MA, Gutiérrez-Angulo M. Molecular Profiling of Tumor Tissue in Mexican Patients with Colorectal Cancer. Current Issues in Molecular Biology. 2022; 44(8):3770-3778. https://doi.org/10.3390/cimb44080258
Chicago/Turabian StyleFlores-López, Beatriz Armida, María de la Luz Ayala-Madrigal, José Miguel Moreno-Ortiz, Jorge Peregrina-Sandoval, Miguel Ángel Trujillo-Rojas, José Luis Venegas-Rodríguez, Rosario Hernández-Ramírez, Martha Alejandra Fernández-Galindo, and Melva Gutiérrez-Angulo. 2022. "Molecular Profiling of Tumor Tissue in Mexican Patients with Colorectal Cancer" Current Issues in Molecular Biology 44, no. 8: 3770-3778. https://doi.org/10.3390/cimb44080258
APA StyleFlores-López, B. A., Ayala-Madrigal, M. d. l. L., Moreno-Ortiz, J. M., Peregrina-Sandoval, J., Trujillo-Rojas, M. Á., Venegas-Rodríguez, J. L., Hernández-Ramírez, R., Fernández-Galindo, M. A., & Gutiérrez-Angulo, M. (2022). Molecular Profiling of Tumor Tissue in Mexican Patients with Colorectal Cancer. Current Issues in Molecular Biology, 44(8), 3770-3778. https://doi.org/10.3390/cimb44080258