

Optic Neuritis in Multiple Sclerosis—A Review of Molecular Mechanisms Involved in the Degenerative Process


 ,
, {kind=link}
{kind=link}
Abstract
:1. Background
2. Pathophysiology of Optic Neuritis, a Projection of MS Pathomechanism
2.1. Inflammatory Phase
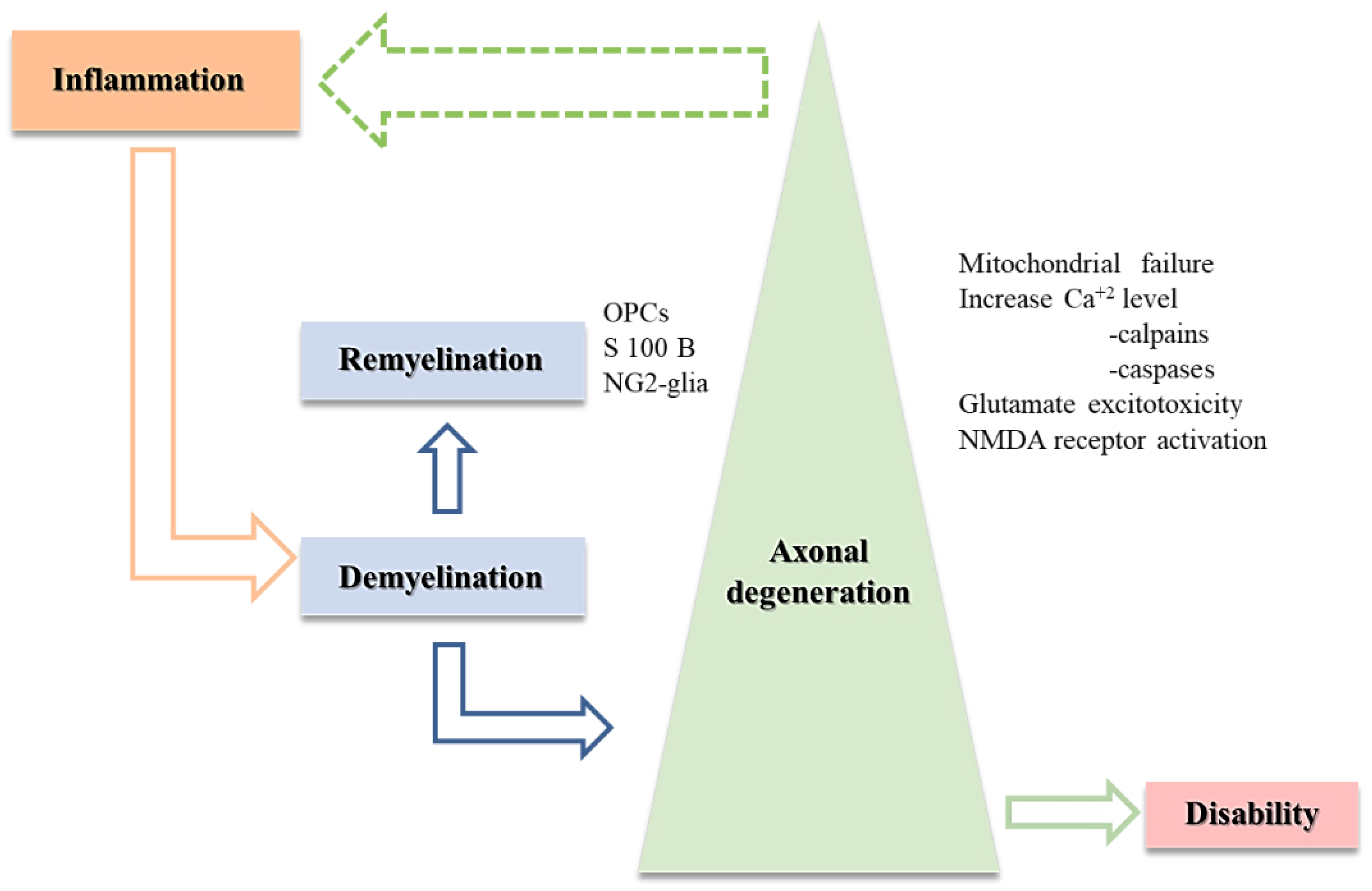
2.2. MS Triad: Demyelination, Axonal Degeneration, and Remyelination
2.2.1. Demyelination and Axonal Loss
2.2.2. Animal Models Used to Study Remyelination
Experimental Autoimmune Encephalomyelitis
Demyelinating Illness Caused by the Virus Theiler’ s Murine Encephalitis
The Role of Cuprizone or Other Toxins
The Role of Lysophospholipid Lysophosphatidylcholine (LPC, Lysolecithin)
2.2.3. Axonal and Neuronal Degeneration
2.2.4. Remyelination in Optic Neuritis
3. The “Big” Picture behind the MS Triad
3.1. MRI
3.2. Visual Evoked Potentials Analysis
3.3. Cerebrospinal Fluid Examination
3.4. Optical Coherence Tomography
3.5. Transorbital B-Mode Ultrasonography
4. New Therapeutic Targets
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data availability statement
Conflicts of Interest
References
- Handel, A.E.; Giovannoni, G.; Ebers, G.C.; Ramagopalan, S.V. Environmental Factors and Their Timing in Adult-Onset Multiple Sclerosis. Nat. Rev. Neurol. 2010, 6, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Gourraud, P.-A.; Harbo, H.F.; Hauser, S.L.; Baranzini, S.E. The Genetics of Multiple Sclerosis: An up-to-Date Review: The Genetics of Multiple Sclerosis. Immunol. Rev. 2012, 248, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Parker Harp, C.; Shindler, K. Optic Neuritis: A Model for the Immuno-Pathogenesis of Central Nervous System Inflammatory Demyelinating Diseases. Curr. Immunol. Rev. 2015, 11, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Redler, Y.; Levy, M. Rodent Models of Optic Neuritis. Front. Neurol. 2020, 11, 580951. [Google Scholar] [CrossRef] [PubMed]
- Baumann, N.; Pham-Dinh, D. Biology of Oligodendrocyte and Myelin in the Mammalian Central Nervous System. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef]
- Bando, Y. Roads to Formation of Normal Myelin Structure and Pathological Myelin Structure. In Myelin: Advances in Experimental Medicine and Biology; Sango, K., Yamauchi, J., Ogata, T., Susuki, K., Eds.; Springer: Singapore, 2019; Volume 1190, pp. 257–264. ISBN 978-981-329-635-0. [Google Scholar]
- Denic, A.; Johnson, A.J.; Bieber, A.J.; Warrington, A.E.; Rodriguez, M.; Pirko, I. The Relevance of Animal Models in Multiple Sclerosis Research. Pathophysiology 2011, 18, 21–29. [Google Scholar] [CrossRef]
- Frohman, E.M.; Dwyer, M.G.; Frohman, T.; Cox, J.L.; Salter, A.; Greenberg, B.M.; Hussein, S.; Conger, A.; Calabresi, P.; Balcer, L.J.; et al. Relationship of Optic Nerve and Brain Conventional and Non-Conventional MRI Measures and Retinal Nerve Fiber Layer Thickness, as Assessed by OCT and GDx: A Pilot Study. J. Neurol. Sci. 2009, 282, 96–105. [Google Scholar] [CrossRef]
- Costello, F.; Hodge, W.; Pan, Y.I.; Eggenberger, E.; Coupland, S.; Kardon, R.H. Tracking Retinal Nerve Fiber Layer Loss after Optic Neuritis: A Prospective Study Using Optical Coherence Tomography. Mult. Scler. Houndmills Basingstoke Engl. 2008, 14, 893–905. [Google Scholar] [CrossRef]
- Henderson, A.P.D.; Altmann, D.R.; Trip, A.S.; Kallis, C.; Jones, S.J.; Schlottmann, P.G.; Garway-Heath, D.F.; Plant, G.T.; Miller, D.H. A Serial Study of Retinal Changes Following Optic Neuritis with Sample Size Estimates for Acute Neuroprotection Trials. Brain J. Neurol. 2010, 133, 2592–2602. [Google Scholar] [CrossRef]
- Calabia, J.; Torguet, P.; Garcia, I.; Martin, N.; Mate, G.; Marin, A.; Molina, C.; Valles, M. The Relationship between Renal Resistive Index, Arterial Stiffness, and Atherosclerotic Burden: The Link between Macrocirculation and Microcirculation. J. Clin. Hypertens. Greenwich Conn 2014, 16, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Preiningerova, J.L.; Grishko, A.; Sobisek, L.; Andelova, M.; Benova, B.; Kucerova, K.; Havrdova, E.K. Do Eyes with and without Optic Neuritis in Multiple Sclerosis Age Equally? Neuropsychiatr. Dis. Treat. 2018, 14, 2281–2285. [Google Scholar] [CrossRef]
- Matthews, P.M.; Roncaroli, F.; Waldman, A.; Sormani, M.P.; De Stefano, N.; Giovannoni, G.; Reynolds, R. A Practical Review of the Neuropathology and Neuroimaging of Multiple Sclerosis. Pract. Neurol. 2016, 16, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Khalilpour, S.; Latifi, S.; Behnammanesh, G.; Majid, A.M.S.A.; Majid, A.S.A.; Tamayol, A. Ischemic Optic Neuropathy as a Model of Neurodegenerative Disorder: A Review of Pathogenic Mechanism of Axonal Degeneration and the Role of Neuroprotection. J. Neurol. Sci. 2017, 375, 430–441. [Google Scholar] [CrossRef]
- Herold, S.; Kumar, P.; Wichert, S.P.; Kretzschmar, B.; Bähr, M.; Rossner, M.J.; Hein, K. Neurodegeneration in Autoimmune Optic Neuritis Is Associated with Altered APP Cleavage in Neurons and Up-Regulation of P53. PLoS ONE 2015, 10, e0138852. [Google Scholar] [CrossRef]
- Bando, Y. Mechanism of Demyelination and Remyelination in Multiple Sclerosis. Clin. Exp. Neuroimmunol. 2020, 11, 14–21. [Google Scholar] [CrossRef]
- Wagner, C.A.; Roqué, P.J.; Goverman, J.M. Pathogenic T Cell Cytokines in Multiple Sclerosis. J. Exp. Med. 2020, 217, e20190460. [Google Scholar] [CrossRef]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple Sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef]
- Britze, J.; Frederiksen, J.L. Optical Coherence Tomography in Multiple Sclerosis. Eye Lond. Engl. 2018, 32, 884–888. [Google Scholar] [CrossRef]
- Lightman, S.; McDonald, W.I.; Bird, A.C.; Francis, D.A.; Hoskins, A.; Batchelor, J.R.; Halliday, A.M. Retinal Venous Sheathing in Optic Neuritis. Its Significance for the Pathogenesis of Multiple Sclerosis. Brain J. Neurol. 1987, 110 Pt 2, 405–414. [Google Scholar] [CrossRef]
- Barnett, M.H.; Henderson, A.P.; Prineas, J.W. The Macrophage in MS: Just a Scavenger after All? Pathology and Pathogenesis of the Acute MS Lesion. Mult. Scler. J. 2006, 12, 121–132. [Google Scholar] [CrossRef]
- Duffy, S.S.; Lees, J.G.; Moalem-Taylor, G. The Contribution of Immune and Glial Cell Types in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Mult. Scler. Int. 2014, 2014, 285245. [Google Scholar] [CrossRef]
- Adam, C.A.; Șalaru, D.L.; Prisacariu, C.; Marcu, D.T.M.; Sascău, R.A.; Stătescu, C. Novel Biomarkers of Atherosclerotic Vascular Disease—Latest Insights in the Research Field. Int. J. Mol. Sci. 2022, 23, 4998. [Google Scholar] [CrossRef]
- Frontiers|Editorial: “Inside-Out”s, vs. “Outside-In” Paradigms in Multiple Sclerosis Etiopathogenesis. Available online: https://www.frontiersin.org/articles/10.3389/fncel.2021.666529/full (accessed on 14 July 2022).
- Morgan, B.P.; Gommerman, J.L.; Ramaglia, V. An “Outside-In” and “Inside-Out” Consideration of Complement in the Multiple Sclerosis Brain: Lessons From Development and Neurodegenerative Diseases. Front. Cell. Neurosci. 2021, 14, 600656. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Malpas, S.C. Sympathetic Nervous System Overactivity and Its Role in the Development of Cardiovascular Disease. Physiol. Rev. 2010, 90, 513–557. [Google Scholar] [CrossRef]
- Hemmer, B.; Pinilla, C.; Gran, B.; Vergelli, M.; Ling, N.; Conlon, P.; McFarland, H.F.; Houghten, R.; Martin, R. Contribution of Individual Amino Acids Within MHC Molecule or Antigenic Peptide to TCR Ligand Potency. J. Immunol. 2000, 164, 861–871. [Google Scholar] [CrossRef]
- Tada, T.; Takemori, T.; Okumura, K.; Nonaka, M.; Tokuhisa, T. Two Distinct Types of Helper T Cells Involved in the Secondary Antibody Response: Independent and Synergistic Effects of Ia− and Ia+ Helper T Cells. J. Exp. Med. 1978, 147, 446–458. [Google Scholar] [CrossRef]
- Tian, A.-Y.; Zhang, R.-W.; Shi, X.-G.; Yu, H.-M. Alteration of T Helper Cell Subsets in the Optic Nerve of Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Med. 2010, 25, 869–874. [Google Scholar] [CrossRef]
- Simsek, I.; Erdem, H.; Pay, S.; Sobaci, G.; Dinc, A. Optic Neuritis Occurring with Anti-tumour Necrosis Factor α Therapy. Ann. Rheum. Dis. 2007, 66, 1255–1258. [Google Scholar] [CrossRef]
- Traugott, U. Characterization and Distribution of Lymphocyte Subpopulations in Multiple Sclerosis Plaques versus Autoimmune Demyelinating Lesions. Semin. Immunopathol. 1985, 8, 71–95. [Google Scholar] [CrossRef]
- Hauser, T.H.; Pinto, D.S.; Josephson, M.E.; Zimetbaum, P. Safety and Feasibility of a Clinical Pathway for the Outpatient Initiation of Antiarrhythmic Medications in Patients with Atrial Fibrillation or Atrial Flutter. Am. J. Cardiol. 2003, 91, 1437–1441. [Google Scholar] [CrossRef]
- Losy, J. Is MS an Inflammatory or Primary Degenerative Disease? J. Neural Transm. 2013, 120, 1459–1462. [Google Scholar] [CrossRef]
- Jacobsen, M.; Cepok, S.; Quak, E.; Happel, M.; Gaber, R.; Ziegler, A.; Schock, S.; Oertel, W.H.; Sommer, N.; Hemmer, B. Oligoclonal Expansion of Memory CD8+ T Cells in Cerebrospinal Fluid from Multiple Sclerosis Patients. Brain 2002, 125, 538–550. [Google Scholar] [CrossRef]
- Gobin, S.J.P.; Montagne, L.; Van Zutphen, M.; Van Der Valk, P.; Van Den Elsen, P.J.; De Groot, C.J.A. Upregulation of Transcription Factors Controlling MHC Expression in Multiple Sclerosis Lesions. Glia 2001, 36, 68–77. [Google Scholar] [CrossRef]
- Zang, Y.C.Q.; Li, S.; Rivera, V.M.; Hong, J.; Robinson, R.R.; Breitbach, W.T.; Killian, J.; Zhang, J.Z. Increased CD8+ Cytotoxic T Cell Responses to Myelin Basic Protein in Multiple Sclerosis. J. Immunol. 2004, 172, 5120–5127. [Google Scholar] [CrossRef]
- Melzer, N.; Meuth, S.G.; Wiendl, H. CD8+ T Cells and Neuronal Damage: Direct and Collateral Mechanisms of Cytotoxicity and Impaired Electrical Excitability. FASEB J. 2009, 23, 3659–3673. [Google Scholar] [CrossRef]
- Lu, L.-F.; Rudensky, A. Molecular Orchestration of Differentiation and Function of Regulatory T Cells. Genes Dev. 2009, 23, 1270–1282. [Google Scholar] [CrossRef]
- Gregori, S.; Goudy, K.S.; Roncarolo, M.G. The Cellular and Molecular Mechanisms of Immuno-Suppression by Human Type 1 Regulatory T Cells. Front. Immunol. 2012, 3, 30. [Google Scholar] [CrossRef]
- Schneider, A.; Long, S.A.; Cerosaletti, K.; Ni, C.T.; Samuels, P.; Kita, M.; Buckner, J.H. In Active Relapsing-Remitting Multiple Sclerosis, Effector T Cell Resistance to Adaptive Tregs Involves IL-6–Mediated Signaling. Sci. Transl. Med. 2013, 5, 170ra15. [Google Scholar] [CrossRef]
- Chiarini, M.; Serana, F.; Zanotti, C.; Capra, R.; Rasia, S.; Rottoli, M.; Rovaris, M.; Caputo, D.; Cavaletti, G.; Frigo, M.; et al. Modulation of the Central Memory and Tr1-like Regulatory T Cells in Multiple Sclerosis Patients Responsive to Interferon-Beta Therapy. Mult. Scler. J. 2012, 18, 788–798. [Google Scholar] [CrossRef]
- Lee, H.-M.; Bautista, J.L.; Scott-Browne, J.; Mohan, J.F.; Hsieh, C.-S. A Broad Range of Self-Reactivity Drives Thymic Regulatory T Cell Selection to Limit Responses to Self. Immunity 2012, 37, 475–486. [Google Scholar] [CrossRef]
- Bielekova, B.; Catalfamo, M.; Reichert-Scrivner, S.; Packer, A.; Cerna, M.; Waldmann, T.A.; McFarland, H.; Henkart, P.A.; Martin, R. Regulatory CD56 bright Natural Killer Cells Mediate Immunomodulatory Effects of IL-2Rα-Targeted Therapy (Daclizumab) in Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5941–5946. [Google Scholar] [CrossRef]
- Morandi, B.; Bramanti, P.; Bonaccorsi, I.; Montalto, E.; Oliveri, D.; Pezzino, G.; Navarra, M.; Ferlazzo, G. Role of Natural Killer Cells in the Pathogenesis and Progression of Multiple Sclerosis. Pharmacol. Res. 2008, 57, 1–5. [Google Scholar] [CrossRef]
- Laroni, A.; Armentani, E.; Kerlero de Rosbo, N.; Ivaldi, F.; Marcenaro, E.; Sivori, S.; Gandhi, R.; Weiner, H.L.; Moretta, A.; Mancardi, G.L.; et al. Dysregulation of Regulatory CD56 Bright NK Cells/T Cells Interactions in Multiple Sclerosis. J. Autoimmun. 2016, 72, 8–18. [Google Scholar] [CrossRef]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Rünzi, A.; Kuhlmann, T.; Posevitz-Fejfár, A.; Schwab, N.; Schneider-Hohendorf, T.; Herich, S.; Held, K.; Konjević, M.; et al. Impaired NK-Mediated Regulation of T-Cell Activity in Multiple Sclerosis Is Reconstituted by IL-2 Receptor Modulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2973–E2982. [Google Scholar] [CrossRef]
- Heidelberger, M.; Kabat, E.A.; Mayer, M. A Further Study of the Cross Reaction between the Specific Polysaccharides of Types III and VIII Pneumococci in Horse Antisera. J. Exp. Med. 1942, 75, 35–47. [Google Scholar] [CrossRef]
- Henderson, V.W.; Guthrie, J.R.; Dennerstein, L. Serum Lipids and Memory in a Population Based Cohort of Middle Age Women. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1530–1535. [Google Scholar] [CrossRef]
- Lovato, L.; Willis, S.N.; Rodig, S.J.; Caron, T.; Almendinger, S.E.; Howell, O.W.; Reynolds, R.; O’ Connor, K.C.; Hafler, D.A. Related B Cell Clones Populate the Meninges and Parenchyma of Patients with Multiple Sclerosis. Brain 2011, 134, 534–541. [Google Scholar] [CrossRef]
- Cepok, S. Patterns of Cerebrospinal Fluid Pathology Correlate with Disease Progression in Multiple Sclerosis. Brain 2001, 124, 2169–2176. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of Ectopic B-Cell Follicles with Germinal Centers in the Meninges of Patients with Secondary Progressive Multiple Sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef]
- Naismith, R.T.; Piccio, L.; Lyons, J.A.; Lauber, J.; Tutlam, N.T.; Parks, B.J.; Trinkaus, K.; Song, S.K.; Cross, A.H. Rituximab Add-on Therapy for Breakthrough Relapsing Multiple Sclerosis: A 52-Week Phase II Trial. Neurology 2010, 74, 1860–1867. [Google Scholar] [CrossRef]
- Rai, N.K.; Singh, V.; Li, L.; Willard, B.; Tripathi, A.; Dutta, R. Comparative Proteomic Profiling Identifies Reciprocal Expression of Mitochondrial Proteins Between White and Gray Matter Lesions From Multiple Sclerosis Brains. Front. Neurol. 2021, 12, 779003. [Google Scholar]
- Cunniffe, N.; Coles, A. Promoting Remyelination in Multiple Sclerosis. J. Neurol. 2021, 268, 30–44. [Google Scholar] [CrossRef]
- Sampaio-Baptista, C.; Johansen-Berg, H. White Matter Plasticity in the Adult Brain. Neuron 2017, 96, 1239–1251. [Google Scholar] [CrossRef]
- Ffrench-Constant, C.; Raff, M.C. Proliferating Bipotential Glial Progenitor Cells in Adult Rat Optic Nerve. Nature 1986, 319, 499–502. [Google Scholar] [CrossRef]
- Psachoulia, K.; Jamen, F.; Young, K.M.; Richardson, W.D. Cell Cycle Dynamics of NG2 Cells in the Postnatal and Ageing Mouse Brain. Neuron Glia Biol. 2009, 5, 57–67. [Google Scholar] [CrossRef]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.J.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-Resident Glial Progenitor/Stem Cells Produce Schwann Cells as Well as Oligodendrocytes during Repair of CNS Demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar] [CrossRef]
- Blakemore, W.F. Pattern of Remyelination in the CNS. Nature 1974, 249, 577–578. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS Myelin—From Mechanisms to Experimental Medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef]
- Baxter, A.G. The Origin and Application of Experimental Autoimmune Encephalomyelitis. Nat. Rev. Immunol. 2007, 7, 904–912. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’ Brien, K.; Gran, B. Experimental Autoimmune Encephalomyelitis (EAE) as a Model for Multiple Sclerosis (MS): EAE as Model for MS. Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [Green Version]
- Krishnamoorthy, G.; Wekerle, H. EAE: An Immunologist’ s Magic Eye: FORUM. Eur. J. Immunol. 2009, 39, 2031–2035. [Google Scholar] [CrossRef]
- Wekerle, H. Lessons from Multiple Sclerosis: Models, Concepts, Observations. Ann. Rheum. Dis. 2008, 67, iii56–iii60. [Google Scholar] [CrossRef]
- Drescher, K.M. Being a Mouse in a Man’ s World: What TMEV Has Taught Us about Human Disease. Front. Biosci. 2008, 13, 3775–3785. [Google Scholar] [CrossRef]
- McCarthy, D.P.; Richards, M.H.; Miller, S.D. Mouse Models of Multiple Sclerosis: Experimental Autoimmune Encephalomyelitis and Theiler’ s Virus-Induced Demyelinating Disease. In Autoimmunity: Methods in Molecular Biology; Perl, A., Ed.; Humana Press: Totowa, NJ, USA, 2012; Volume 900, pp. 381–401. ISBN 978-1-60761-719-8. [Google Scholar]
- Lipton, H.L. Human Vilyuisk Encephalitis. Rev. Med. Virol. 2008, 18, 347–352. [Google Scholar] [CrossRef]
- Pachner, A.R. Experimental Models of Multiple Sclerosis. Curr. Opin. Neurol. 2011, 24, 291–299. [Google Scholar] [CrossRef]
- Lane, T.; Buchmeier, M. Murine Coronavirus Infection: A Paradigm for Virus-Induced Demyelinating Disease. Trends Microbiol. 1997, 5, 9–14. [Google Scholar] [CrossRef]
- Matthews, A.; Weiss, S.; Paterson, Y. Murine Hepatitis Virus--A Model for Virus-Induced CNS Demyelination. J. Neurovirol. 2002, 8, 76–85. [Google Scholar] [CrossRef]
- van der Star, B.J.; Vogel, D.Y.S.; Kipp, M.; Puentes, F.; Baker, D.; Amor, S. In Vitro and In Vivo Models of Multiple Sclerosis. CNS Neurol. Disord.-Drug Targets 2012, 11, 570–588. [Google Scholar] [CrossRef]
- Hesse, A.; Wagner, M.; Held, J.; Brück, W.; Salinas-Riester, G.; Hao, Z.; Waisman, A.; Kuhlmann, T. In Toxic Demyelination Oligodendroglial Cell Death Occurs Early and Is FAS Independent. Neurobiol. Dis. 2010, 37, 362–369. [Google Scholar] [CrossRef]
- Torkildsen, Ø.; Brunborg, L.A.; Myhr, K.-M.; Bø, L. The Cuprizone Model for Demyelination. Acta Neurol. Scand. 2008, 117, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Plemel, J.R.; Michaels, N.J.; Weishaupt, N.; Caprariello, A.V.; Keough, M.B.; Rogers, J.A.; Yukseloglu, A.; Lim, J.; Patel, V.V.; Rawji, K.S.; et al. Mechanisms of Lysophosphatidylcholine-Induced Demyelination: A Primary Lipid Disrupting Myelinopathy. Glia 2018, 66, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, S.; Aref, E.; Raoufy, M.R.; Javan, M. An Optimized Animal Model of Lysolecithin Induced Demyelination in Optic Nerve; More Feasible, More Reproducible, Promising for Studying the Progressive Forms of Multiple Sclerosis. J. Neurosci. Methods 2021, 352, 109088. [Google Scholar] [CrossRef] [PubMed]
- GrandPré, T.; Nakamura, F.; Vartanian, T.; Strittmatter, S.M. Identification of the Nogo Inhibitor of Axon Regeneration as a Reticulon Protein. Nature 2000, 403, 439–444. [Google Scholar] [CrossRef] [PubMed]
- McKerracher, L.; David, S.; Jackson, D.L.; Kottis, V.; Dunn, R.J.; Braun, P.E. Identification of Myelin-Associated Glycoprotein as a Major Myelin-Derived Inhibitor of Neurite Growth. Neuron 1994, 13, 805–811. [Google Scholar] [CrossRef]
- Wang, K.C.; Kim, J.A.; Sivasankaran, R.; Segal, R.; He, Z. P75 Interacts with the Nogo Receptor as a Co-Receptor for Nogo, MAG and OMgp. Nature 2002, 420, 74–78. [Google Scholar] [CrossRef]
- Fournier, A.E.; GrandPre, T.; Strittmatter, S.M. Identification of a Receptor Mediating Nogo-66 Inhibition of Axonal Regeneration. Nature 2001, 409, 341–346. [Google Scholar] [CrossRef]
- Satoh, J.-I.; Onoue, H.; Arima, K.; Yamamura, T. Nogo-A and Nogo Receptor Expression in Demyelinating Lesions of Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2005, 64, 129–138. [Google Scholar] [CrossRef]
- Huber, G.; Schuster, F.; Raasch, W. Brain Renin-Angiotensin System in the Pathophysiology of Cardiovascular Diseases. Pharmacol. Res. 2017, 125, 72–90. [Google Scholar] [CrossRef]
- Larochelle, C.; Lécuyer, M.-A.; Alvarez, J.I.; Charabati, M.; Saint-Laurent, O.; Ghannam, S.; Kebir, H.; Flanagan, K.; Yednock, T.; Duquette, P.; et al. Melanoma Cell Adhesion Molecule-Positive CD8 T Lymphocytes Mediate Central Nervous System Inflammation: MCAM + CD8 in Neuroinflammation. Ann. Neurol. 2015, 78, 39–53. [Google Scholar] [CrossRef]
- Stirling, D.P.; Cummins, K.; Wayne Chen, S.R.; Stys, P. Axoplasmic Reticulum Ca2+ Release Causes Secondary Degeneration of Spinal Axons: Intra-Axonal Ca2+ Stores. Ann. Neurol. 2014, 75, 220–229. [Google Scholar] [CrossRef]
- Bojcevski, J.; Stojic, A.; Hoffmann, D.B.; Williams, S.K.; Müller, A.; Diem, R.; Fairless, R. Influence of Retinal NMDA Receptor Activity during Autoimmune Optic Neuritis. J. Neurochem. 2020, 153, 693–709. [Google Scholar] [CrossRef]
- Aizenman, E.; Frosch, M.P.; Lipton, S.A. Responses Mediated by Excitatory Amino Acid Receptors in Solitary Retinal Ganglion Cells from Rat. J. Physiol. 1988, 396, 75–91. [Google Scholar] [CrossRef]
- Choi, H.J.; Byun, M.S.; Yi, D.; Choe, Y.M.; Sohn, B.K.; Baek, H.W.; Lee, J.H.; Kim, H.J.; Han, J.Y.; Yoon, E.J.; et al. Association Between Serum Triglycerides and Cerebral Amyloidosis in Cognitively Normal Elderly. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2016, 24, 604–612. [Google Scholar] [CrossRef]
- Sühs, K.-W.; Wegner, F.; Skripuletz, T.; Trebst, C.; Tayeb, S.B.; Raab, P.; Stangel, M. Heterogeneity of Clinical Features and Corresponding Antibodies in Seven Patients with Anti-NMDA Receptor Encephalitis. Exp. Ther. Med. 2015, 10, 1283–1292. [Google Scholar] [CrossRef]
- Hoffmann, D.B.; Williams, S.K.; Jovana, B.; Müller, A.; Stadelmann, C.; Naidoo, V.; Bahr, B.A.; Diem, R.; Fairless, R. Calcium Influx and Calpain Activation Mediate Preclinical Retinal Neurodegeneration in Autoimmune Optic Neuritis. J. Neuropathol. Exp. Neurol. 2013, 72, 745–757. [Google Scholar] [CrossRef]
- Halpain, S.; Hipolito, A.; Saffer, L. Regulation of F-Actin Stability in Dendritic Spines by Glutamate Receptors and Calcineurin. J. Neurosci. 1998, 18, 9835–9844. [Google Scholar] [CrossRef]
- Yin, H.L.; Stossel, T.P. Control of Cytoplasmic Actin Gel–Sol Transformation by Gelsolin, a Calcium-Dependent Regulatory Protein. Nature 1979, 281, 583–586. [Google Scholar] [CrossRef]
- Kothakota, S.; Azuma, T.; Reinhard, C.; Klippel, A.; Tang, J.; Chu, K.; McGarry, T.J.; Kirschner, M.W.; Koths, K.; Kwiatkowski, D.J.; et al. Caspase-3-Generated Fragment of Gelsolin: Effector of Morphological Change in Apoptosis. Science 1997, 278, 294–298. [Google Scholar] [CrossRef]
- Harms, C.; Bösel, J.; Lautenschlager, M.; Harms, U.; Braun, J.S.; Hörtnagl, H.; Dirnagl, U.; Kwiatkowski, D.J.; Fink, K.; Endres, M. Neuronal Gelsolin Prevents Apoptosis by Enhancing Actin Depolymerization. Mol. Cell. Neurosci. 2004, 25, 69–82. [Google Scholar] [CrossRef]
- Brown, S.B.; Bailey, K.; Savill, J. Actin Is Cleaved during Constitutive Apoptosis. Biochem. J. 1997, 323, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, E.C.K. Cellular and Molecular Mechanisms in the Pathogenesis of Multiple Sclerosis. Cells 2020, 9, 2223. [Google Scholar] [CrossRef] [PubMed]
- Carroll, W.M.; Jennings, A.R.; Mastaglia, F.L. The Origin of Remyelinating Oligodendrocytes in Antiserum-Mediated Demyelinative Optic Neuropathy. Brain 1990, 113, 953–973. [Google Scholar] [CrossRef] [PubMed]
- Scolding, N. Oligodendrocyte Progenitors Are Present in the Normal Adult Human CNS and in the Lesions of Multiple Sclerosis. Brain 1998, 121, 2221–2228. [Google Scholar] [CrossRef]
- Al-Louzi, O.; Saidha, S. Pathophysiology of Optic Neuritis. Mult. Scler. Mech. View 2016, 281–309. [Google Scholar] [CrossRef]
- Rodriguez, M.; Scheithauer, B.W.; Forbes, G.; Kelly, P.J. Oligodendrocyte Injury Is an Early Event in Lesions of Multiple Sclerosis. Mayo Clin. Proc. 1993, 68, 627–636. [Google Scholar] [CrossRef]
- Prineas, J.W.; Barnard, R.O.; Kwon, E.E.; Sharer, L.R.; Cho, E.-S. Multiple Sclerosis: Remyelination of Nascent Lesions: Remyelination of Nascent Lesions. Ann. Neurol. 1993, 33, 137–151. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Brück, W.; Rodriguez, M.; Lassmann, H. Distinct Patterns of Multiple Sclerosis Pathology Indicates Heterogeneity in Pathogenesis. Brain Pathol. 1996, 6, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, K.A.; Sloane, J.A. Regulation of Remyelination in Multiple Sclerosis. FEBS Lett. 2011, 585, 3821–3828. [Google Scholar] [CrossRef]
- Wu, L.M.N.; Williams, A.; Delaney, A.; Sherman, D.L.; Brophy, P.J. Increasing Internodal Distance in Myelinated Nerves Accelerates Nerve Conduction to a Flat Maximum. Curr. Biol. 2012, 22, 1957–1961. [Google Scholar] [CrossRef]
- Jäkel, S.; Agirre, E.; Mendanha Falcão, A.; van Bruggen, D.; Lee, K.W.; Knuesel, I.; Malhotra, D.; ffrench-Constant, C.; Williams, A.; Castelo-Branco, G. Altered Human Oligodendrocyte Heterogeneity in Multiple Sclerosis. Nature 2019, 566, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Bodini, B.; Veronese, M.; García-Lorenzo, D.; Battaglini, M.; Poirion, E.; Chardain, A.; Freeman, L.; Louapre, C.; Tchikviladze, M.; Papeix, C.; et al. Dynamic Imaging of Individual Remyelination Profiles in Multiple Sclerosis. Ann. Neurol. 2016, 79, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Brück, W.; Lucchinetti, C.; Lassmann, H. Remyelination Is Extensive in a Subset of Multiple Sclerosis Patients. Brain J. Neurol. 2006, 129, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Sutiwisesak, R.; Burns, T.C.; Rodriguez, M.; Warrington, A.E. Remyelination Therapies for Multiple Sclerosis: Optimizing Translation from Animal Models into Clinical Trials. Expert Opin. Investig. Drugs 2021, 30, 857–876. [Google Scholar] [CrossRef] [PubMed]
- Hooijmans, C.R.; Hlavica, M.; Schuler, F.A.F.; Good, N.; Good, A.; Baumgartner, L.; Galeno, G.; Schneider, M.P.; Jung, T.; de Vries, R.; et al. Remyelination Promoting Therapies in Multiple Sclerosis Animal Models: A Systematic Review and Meta-Analysis. Sci. Rep. 2019, 9, 822. [Google Scholar] [CrossRef]
- Zorina, Y.; Stricker, J.; Caggiano, A.O.; Button, D.C. Human IgM Antibody RHIgM22 Promotes Phagocytic Clearance of Myelin Debris by Microglia. Sci. Rep. 2018, 8, 9392. [Google Scholar] [CrossRef]
- Eisen, A.; Greenberg, B.M.; Bowen, J.D.; Arnold, D.L.; Caggiano, A.O. A Double-Blind, Placebo-Controlled, Single Ascending-Dose Study of Remyelinating Antibody RHIgM22 in People with Multiple Sclerosis. Mult. Scler. J.-Exp. Transl. Clin. 2017, 3, 2055217317743097. [Google Scholar] [CrossRef]
- Green, A.J.; Gelfand, J.M.; A Cree, B.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef]
- Pelletier, J.; Ranjeva, J.-P.; Tourbah, A.; Edan, G.; Barillot, C.; Le Lamer, S.; Audoin, B.; Rico, A.; Crespy, L.; Rid-ley, B.; et al. Results of a Phase 1b Study to Confirm Safety and Tolerability of Olesoxime in Multiple Sclerosis Patients; American Academy of Neurology: Minneapolis, MN, USA, 2015. [Google Scholar]
- Magalon, K.; Zimmer, C.; Cayre, M.; Khaldi, J.; Bourbon, C.; Robles, I.; Tardif, G.; Viola, A.; Pruss, R.M.; Bordet, T.; et al. Olesoxime accelerates myelination and promotes repair in models of demyelination. Ann. Neurol. 2011, 71, 213–226. [Google Scholar] [CrossRef]
- Bordet, T.; Berna, P.; Abitbol, J.-L.; Pruss, R.M. Olesoxime (TRO19622): A Novel Mitochondrial-Targeted Neuroprotective Compound. Pharmaceuticals 2010, 3, 345–368. [Google Scholar] [CrossRef]
- Chiang, A.C.A.; Seua, A.V.; Singhmar, P.; Arroyo, L.D.; Mahalingam, R.; Hu, J.; Kavelaars, A.; Heijnen, C.J. Bexarotene normalizes chemotherapy-induced myelin decompaction and reverses cognitive and sensorimotor deficits in mice. Acta Neuropathol. Commun. 2020, 8, 193. [Google Scholar] [CrossRef] [PubMed]
- A Randomised Placebo-Controlled Study of the Safety and Tolerability of a Retinoid-X Receptor Agonist’ s Ability to Promote Remyelination in People with Relapsing-Remitting Multiple Sclerosis Already on Interferon-Beta Therapy: A Phase 2a Trial-AdisInsight. Available online: https://adisinsight.springer.com/trials/700259921 (accessed on 29 August 2022).
- Shen, X.; Liu, W.; Gao, X.; Lu, Z.; Wu, X.; Gao, X. Mechanisms of Oxidase and Superoxide Dismutation-like Activities of Gold, Silver, Platinum, and Palladium, and Their Alloys: A General Way to the Activation of Molecular Oxygen. J. Am. Chem. Soc. 2015, 137, 15882–15891. [Google Scholar] [CrossRef] [PubMed]
- Clene Nanomedicine VISIONARY-MS LTE: A Multi-Center, Open-Label Long-Term Extension Study Assessing the Safety, Efficacy, Tolerability, and Pharmacokinetics of CNM-Au8 In Patients With Stable Relapsing Multiple Sclerosis. 2022. Available online: https://clinicaltrials.gov/ (accessed on 20 August 2022).
- Koch, M.W.; Liu, W.-Q.; Camara-Lemarroy, C.; Zhang, Y.; Pike, G.B.; Metz, L.; Yong, V.W. Domperidone-induced elevation of serum prolactin levels and immune response in multiple sclerosis. J. Neuroimmunol. 2019, 334, 576974. [Google Scholar] [CrossRef]
- Zhornitsky, S.; Yong, V.W.; Weiss, S.; Metz, L.M. Prolactin in multiple sclerosis. Mult. Scler. J. 2012, 19, 15–23. [Google Scholar] [CrossRef]
- Jepson, S.; Vought, B.; Gross, C.H.; Gan, L.; Austen, D.; Frantz, J.D.; Zwahlen, J.; Lowe, D.; Markland, W.; Krauss, R. LINGO-1, a Transmembrane Signaling Protein, Inhibits Oligodendrocyte Differentiation and Myelination through Intercellular Self-interactions. J. Biol. Chem. 2012, 287, 22184–22195. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.Q.; Rana, J.; Barkhof, F.; Melamed, I.; Gevorkyan, H.; Wattjes, M.P.; de Jong, R.; Brosofsky, K.; Ray, S.; Xu, L.; et al. Randomized phase I trials of the safety/tolerability of anti-LINGO-1 monoclonal antibody BIIB033. Neurol.-Neuroimmunol. Neuroinflammation 2014, 1, e18. [Google Scholar] [CrossRef]
- Cadavid, D.; Balcer, L.; Galetta, S.; Aktas, O.; Ziemssen, T.; Vanopdenbosch, L.; Frederiksen, J.; Skeen, M.; Jaffe, G.J.; Butzkueven, H.; et al. Safety and efficacy of opicinumab in acute optic neuritis (RENEW): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2017, 16, 189–199. [Google Scholar] [CrossRef]
- Cadavid, D.; Mellion, M.; Hupperts, R.; Edwards, K.R.; A Calabresi, P.; Drulović, J.; Giovannoni, G.; Hartung, H.-P.; Arnold, D.L.; Fisher, E.; et al. Safety and efficacy of opicinumab in patients with relapsing multiple sclerosis (SYNERGY): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2019, 18, 845–856. [Google Scholar] [CrossRef]
- Kale, N. Optic Neuritis as an Early Sign of Multiple Sclerosis. Eye Brain 2016, 8, 195–202. [Google Scholar] [CrossRef]
- Mamarabadi, M.; Razjouyan, H.; Mohammadi, F.; Moghaddasi, M. Assessment of Outcome Predictors after First Attack of Optic Neuritis. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 2011, 38, 887–895. [Google Scholar] [CrossRef]
- Frohman, E.; Costello, F.; Zivadinov, R.; Stuve, O.; Conger, A.; Winslow, H.; Trip, A.; Frohman, T.; Balcer, L. Optical Coherence Tomography in Multiple Sclerosis. Lancet Neurol. 2006, 5, 853–863. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of Multiple Sclerosis: 2017 Revisions of the McDonald Criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Bove, R.M.; Hauser, S.L. Diagnosing Multiple Sclerosis: Art and Science. Lancet Neurol. 2018, 17, 109–111. [Google Scholar] [CrossRef]
- Kupersmith, M.J.; Alban, T.; Zeiffer, B.; Lefton, D. Contrast-Enhanced MRI in Acute Optic Neuritis: Relationship to Visual Performance. Brain J. Neurol. 2002, 125, 812–822. [Google Scholar] [CrossRef]
- Bennett, J.L. Optic Neuritis. Contin. Minneap. Minn 2019, 25, 1236–1264. [Google Scholar] [CrossRef]
- Swanton, J.K.; Fernando, K.T.; Dalton, C.M.; Miszkiel, K.A.; Altmann, D.R.; Plant, G.T.; Thompson, A.J.; Miller, D.H. Early MRI in Optic Neuritis: The Risk for Disability. Neurology 2009, 72, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Hoorbakht, H.; Bagherkashi, F. Optic Neuritis, Its Differential Diagnosis and Management. Open Ophthalmol. J. 2012, 6, 65–72. [Google Scholar] [CrossRef]
- Optic Neuritis Study Group Multiple Sclerosis Risk after Optic Neuritis: Final Optic Neuritis Treatment Trial Follow-Up. Arch. Neurol. 2008, 65, 727–732. [CrossRef]
- Lebrun, C.; Bensa, C.; Debouverie, M.; Wiertlevski, S.; Brassat, D.; de Seze, J.; Rumbach, L.; Pelletier, J.; Labauge, P.; Brochet, B.; et al. Association between Clinical Conversion to Multiple Sclerosis in Radiologically Isolated Syndrome and Magnetic Resonance Imaging, Cerebrospinal Fluid, and Visual Evoked Potential: Follow-up of 70 Patients. Arch. Neurol. 2009, 66, 841–846. [Google Scholar] [CrossRef]
- Brownlee, W.J.; Miszkiel, K.A.; Tur, C.; Barkhof, F.; Miller, D.H.; Ciccarelli, O. Inclusion of Optic Nerve Involvement in Dissemination in Space Criteria for Multiple Sclerosis. Neurology 2018, 91, e1130–e1134. [Google Scholar] [CrossRef]
- Tintoré, M.; Rovira, A.; Río, J.; Nos, C.; Grivé, E.; Sastre-Garriga, J.; Pericot, I.; Sánchez, E.; Comabella, M.; Montalban, X. New Diagnostic Criteria for Multiple Sclerosis: Application in First Demyelinating Episode. Neurology 2003, 60, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Wattjes, M.P.; Costello, F.; Flores-Rivera, J.; Fraser, C.L.; Fujihara, K.; Leavitt, J.; Marignier, R.; Paul, F.; Schippling, S.; et al. The Investigation of Acute Optic Neuritis: A Review and Proposed Protocol. Nat. Rev. Neurol. 2014, 10, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.W. Recent Advances in Optic Neuritis Related to Multiple Sclerosis. Acta Ophthalmol. 2012, 90, 203–209. [Google Scholar] [CrossRef]
- Zafeiropoulos, P.; Katsanos, A.; Kitsos, G.; Stefaniotou, M.; Asproudis, I. The Contribution of Multifocal Visual Evoked Potentials in Patients with Optic Neuritis and Multiple Sclerosis: A Review. Doc. Ophthalmol. Adv. Ophthalmol. 2021, 142, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.C.; Odel, J.G.; Winn, B.J. The Multifocal Visual Evoked Potential. J. Neuro-Ophthalmol. Off. J. N. Am. Neuro-Ophthalmol. Soc. 2003, 23, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C.; Klistorner, A.; Graham, S.; Garrick, R.; Billson, F.; Grigg, J. Multifocal Visual Evoked Potential Latency Analysis: Predicting Progression to Multiple Sclerosis. Arch. Neurol. 2006, 63, 847–850. [Google Scholar] [CrossRef]
- Klistorner, A.; Arvind, H.; Nguyen, T.; Garrick, R.; Paine, M.; Graham, S.; Yiannikas, C. Fellow Eye Changes in Optic Neuritis Correlate with the Risk of Multiple Sclerosis. Mult. Scler. Houndmills Basingstoke Engl. 2009, 15, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Pihl-Jensen, G.; Schmidt, M.F.; Frederiksen, J.L. Multifocal Visual Evoked Potentials in Optic Neuritis and Multiple Sclerosis: A Review. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2017, 128, 1234–1245. [Google Scholar] [CrossRef]
- Klistorner, A.; Fraser, C.; Garrick, R.; Graham, S.; Arvind, H. Correlation between Full-Field and Multifocal VEPs in Optic Neuritis. Doc. Ophthalmol. Adv. Ophthalmol. 2008, 116, 19–27. [Google Scholar] [CrossRef]
- De Santiago, L.; Ortiz Del Castillo, M.; Blanco, R.; Barea, R.; Rodríguez-Ascariz, J.M.; Miguel-Jiménez, J.M.; Sánchez-Morla, E.M.; Boquete, L. A Signal-to-Noise-Ratio-Based Analysis of Multifocal Visual-Evoked Potentials in Multiple Sclerosis Risk Assessment. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2016, 127, 1574–1580. [Google Scholar] [CrossRef]
- Petrillo, J.; Balcer, L.; Galetta, S.; Chai, Y.; Xu, L.; Cadavid, D. Initial Impairment and Recovery of Vision-Related Functioning in Participants With Acute Optic Neuritis From the RENEW Trial of Opicinumab. J. Neuro-Ophthalmol. Off. J. N. Am. Neuro-Ophthalmol. Soc. 2019, 39, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Grover, L.K.; Hood, D.C.; Ghadiali, Q.; Grippo, T.M.; Wenick, A.S.; Greenstein, V.C.; Behrens, M.M.; Odel, J.G. A Comparison of Multifocal and Conventional Visual Evoked Potential Techniques in Patients with Optic Neuritis/Multiple Sclerosis. Doc. Ophthalmol. Adv. Ophthalmol. 2008, 117, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Klistorner, A.; Arvind, H.; Nguyen, T.; Garrick, R.; Paine, M.; Graham, S.; O’ Day, J.; Yiannikas, C. Multifocal VEP and OCT in Optic Neuritis: A Topographical Study of the Structure-Function Relationship. Doc. Ophthalmol. Adv. Ophthalmol. 2009, 118, 129–137. [Google Scholar] [CrossRef]
- Laron, M.; Cheng, H.; Zhang, B.; Schiffman, J.S.; Tang, R.A.; Frishman, L.J. Comparison of Multifocal Visual Evoked Potential, Standard Automated Perimetry and Optical Coherence Tomography in Assessing Visual Pathway in Multiple Sclerosis Patients. Mult. Scler. Houndmills Basingstoke Engl. 2010, 16, 412–426. [Google Scholar] [CrossRef]
- Olesen, M.N.; Soelberg, K.; Debrabant, B.; Nilsson, A.C.; Lillevang, S.T.; Grauslund, J.; Brandslund, I.; Madsen, J.S.; Paul, F.; Smith, T.J.; et al. Cerebrospinal Fluid Biomarkers for Predicting Development of Multiple Sclerosis in Acute Optic Neuritis: A Population-Based Prospective Cohort Study. J. Neuroinflamm. 2019, 16, 59. [Google Scholar] [CrossRef]
- Ottervald, J.; Franzén, B.; Nilsson, K.; Andersson, L.I.; Khademi, M.; Eriksson, B.; Kjellström, S.; Marko-Varga, G.; Végvári, Á.; Harris, R.A.; et al. Multiple Sclerosis: Identification and Clinical Evaluation of Novel CSF Biomarkers. J. Proteom. 2010, 73, 1117–1132. [Google Scholar] [CrossRef]
- Weinshenker, B.G.; Wingerchuk, D.M. Neuromyelitis Spectrum Disorders. Mayo Clin. Proc. 2017, 92, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, J.L.; Larsson, H.B.; Olesen, J. Correlation of Magnetic Resonance Imaging and CSF Findings in Patients with Acute Monosymptomatic Optic Neuritis. Acta Neurol. Scand. 1992, 86, 317–322. [Google Scholar] [CrossRef]
- Tumani, H.; Hartung, H.-P.; Hemmer, B.; Teunissen, C.; Deisenhammer, F.; Giovannoni, G.; Zettl, U.K. Cerebrospinal Fluid Biomarkers in Multiple Sclerosis. Neurobiol. Dis. 2009, 35, 117–127. [Google Scholar] [CrossRef]
- Uhlig, H.H. Monogenic Diseases Associated with Intestinal Inflammation: Implications for the Understanding of Inflammatory Bowel Disease. Gut 2013, 62, 1795–1805. [Google Scholar] [CrossRef]
- Nakashima, I.; Fujihara, K.; Misu, T.; Okita, N.; Takase, S.; Itoyama, Y. Significant Correlation between IL-10 Levels and IgG Indices in the Cerebrospinal Fluid of Patients with Multiple Sclerosis. J. Neuroimmunol. 2000, 111, 64–67. [Google Scholar] [CrossRef]
- Ries, S.; Hilgenberg, E.; Lampropoulou, V.; Shen, P.; Dang, V.D.; Wilantri, S.; Sakwa, I.; Fillatreau, S. B-Type Suppression: A Role Played by “Regulatory B Cells” or “Regulatory Plasma Cells”? Eur. J. Immunol. 2014, 44, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.-A.; Hümmert, M.W.; et al. MOG-IgG in NMO and Related Disorders: A Multicenter Study of 50 Patients. Part 2: Epidemiology, Clinical Presentation, Radiological and Laboratory Features, Treatment Responses, and Long-Term Outcome. J. Neuroinflamm. 2016, 13, 280. [Google Scholar] [CrossRef]
- Jarius, S.; Paul, F.; Franciotta, D.; Ruprecht, K.; Ringelstein, M.; Bergamaschi, R.; Rommer, P.; Kleiter, I.; Stich, O.; Reuss, R.; et al. Cerebrospinal Fluid Findings in Aquaporin-4 Antibody Positive Neuromyelitis Optica: Results from 211 Lumbar Punctures. J. Neurol. Sci. 2011, 306, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.L.; Briggs, F.B.S.; Winnike, J.H.; Natanzon, Y.; Maichle, S.; Knagge, K.J.; Newby, L.K.; Gregory, S.G. Metabolome-Based Signature of Disease Pathology in MS. Mult. Scler. Relat. Disord. 2019, 31, 12–21. [Google Scholar] [CrossRef]
- Liu, Z.; Waters, J.; Rui, B. Metabolomics as a Promising Tool for Improving Understanding of Multiple Sclerosis: A Review of Recent Advances. Biomed. J. 2022, in press. [CrossRef]
- Housley, W.J.; Pitt, D.; Hafler, D.A. Biomarkers in Multiple Sclerosis. Clin. Immunol. 2015, 161, 51–58. [Google Scholar] [CrossRef]
- Wishart, D.S. Emerging Applications of Metabolomics in Drug Discovery and Precision Medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef]
- Reinke, S.; Broadhurst, D.; Sykes, B.; Baker, G.; Catz, I.; Warren, K.; Power, C. Metabolomic Profiling in Multiple Sclerosis: Insights into Biomarkers and Pathogenesis. Mult. Scler. J. 2014, 20, 1396–1400. [Google Scholar] [CrossRef]
- Lutz, N.W.; Viola, A.; Malikova, I.; Confort-Gouny, S.; Audoin, B.; Ranjeva, J.-P.; Pelletier, J.; Cozzone, P.J. Inflammatory Multiple-Sclerosis Plaques Generate Characteristic Metabolic Profiles in Cerebrospinal Fluid. PLoS ONE 2007, 2, e595. [Google Scholar] [CrossRef]
- Jenkins, T.M.; Toosy, A.T. Optic Neuritis: The Eye as a Window to the Brain. Curr. Opin. Neurol. 2017, 30, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorga, R.E.; Moraru, A.; Ozturk, M.R.; Costin, D. The Role of Optical Coherence Tomography in Optic Neuropathies. Rom. J. Ophthalmol. 2018, 62, 3–14. [Google Scholar] [CrossRef]
- Trip, S.A.; Schlottmann, P.G.; Jones, S.J.; Altmann, D.R.; Garway-Heath, D.F.; Thompson, A.J.; Plant, G.T.; Miller, D.H. Retinal Nerve Fiber Layer Axonal Loss and Visual Dysfunction in Optic Neuritis. Ann. Neurol. 2005, 58, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Costello, F.; Coupland, S.; Hodge, W.; Lorello, G.R.; Koroluk, J.; Pan, Y.I.; Freedman, M.S.; Zackon, D.H.; Kardon, R.H. Quantifying Axonal Loss after Optic Neuritis with Optical Coherence Tomography. Ann. Neurol. 2006, 59, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Saidha, S.; Syc, S.B.; Ibrahim, M.A.; Eckstein, C.; Warner, C.V.; Farrell, S.K.; Oakley, J.D.; Durbin, M.K.; Meyer, S.A.; Balcer, L.J.; et al. Primary Retinal Pathology in Multiple Sclerosis as Detected by Optical Coherence Tomography. Brain 2011, 134, 518–533. [Google Scholar] [CrossRef]
- Burkholder, B.M.; Osborne, B.; Loguidice, M.J.; Bisker, E.; Frohman, T.C.; Conger, A.; Ratchford, J.N.; Warner, C.; Markowitz, C.E.; Jacobs, D.A.; et al. Macular Volume Determined by Optical Coherence Tomography as a Measure of Neuronal Loss in Multiple Sclerosis. Arch. Neurol. 2009, 66, 1366–1372. [Google Scholar] [CrossRef]
- Kupersmith, M.J.; Mandel, G.; Anderson, S.; Meltzer, D.E.; Kardon, R. Baseline, One and Three Month Changes in the Peripapillary Retinal Nerve Fiber Layer in Acute Optic Neuritis: Relation to Baseline Vision and MRI. J. Neurol. Sci. 2011, 308, 117–123. [Google Scholar] [CrossRef]
- Chen, J.J.; Sotirchos, E.S.; Henderson, A.D.; Vasileiou, E.S.; Flanagan, E.P.; Bhatti, M.T.; Jamali, S.; Eggenberger, E.R.; Dinome, M.; Frohman, L.P.; et al. OCT Retinal Nerve Fiber Layer Thickness Differentiates Acute Optic Neuritis from MOG Antibody-Associated Disease and Multiple Sclerosis. Mult. Scler. Relat. Disord. 2022, 58, 103525. [Google Scholar] [CrossRef]
- Lochner, P.; Leone, M.A.; Coppo, L.; Nardone, R.; Zedde, M.L.; Cantello, R.; Brigo, F. B-Mode Transorbital Ultrasononography for the Diagnosis of Acute Optic Neuritis. A Systematic Review. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2016, 127, 803–809. [Google Scholar] [CrossRef]
- Phuljhele, S.; Kedar, S.; Saxena, R. Approach to Optic Neuritis: An Update. Indian J. Ophthalmol. 2021, 69, 2266–2276. [Google Scholar] [CrossRef]
- Wilhelm, H.; Schabet, M. The Diagnosis and Treatment of Optic Neuritis. Dtsch. Ärztebl. Int. 2015, 112, 616–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcu, D.T.M.; Adam, C.A.; Dorobanțu, D.-M.; Șalaru, D.L.; Sascău, R.A.; Balasanian, M.O.; Macovei, L.; Arsenescu-Georgescu, C.; Stătescu, C. Beta-Blocker-Related Atrioventricular Conduction Disorders—A Single Tertiary Referral Center Experience. Medicina 2022, 58, 320. [Google Scholar] [CrossRef] [PubMed]
- Marcu, D.T.M.; Arsenescu-Georgescu, C. Adverse Drug Reactions and Atrioventricular Conduction Disorders—A Female Gender Related Aproach. Intern. Med. 2021, 18, 15–29. [Google Scholar] [CrossRef]
- Shan, F.; Long, Y.; Qiu, W. Autoimmune Glial Fibrillary Acidic Protein Astrocytopathy: A Review of the Literature. Front. Immunol. 2018, 9, 2802. [Google Scholar] [CrossRef]
- Flanagan, E.P.; Hinson, S.R.; Lennon, V.A.; Fang, B.; Aksamit, A.J.; Morris, P.P.; Basal, E.; Honorat, J.A.; Alfugham, N.B.; Linnoila, J.J.; et al. Glial Fibrillary Acidic Protein Immunoglobulin G as Biomarker of Autoimmune Astrocytopathy: Analysis of 102 Patients. Ann. Neurol. 2017, 81, 298–309. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and Neuromyelitis Optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG Marker of Optic-Spinal Multiple Sclerosis Binds to the Aquaporin-4 Water Channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef]
- Collongues, N.; de Seze, J. Current and Future Treatment Approaches for Neuromyelitis Optica. Ther. Adv. Neurol. Disord. 2011, 4, 111–121. [Google Scholar] [CrossRef]
- Tradtrantip, L.; Zhang, H.; Saadoun, S.; Phuan, P.-W.; Lam, C.; Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Anti-Aquaporin-4 Monoclonal Antibody Blocker Therapy for Neuromyelitis Optica. Ann. Neurol. 2012, 71, 314–322. [Google Scholar] [CrossRef]
- Tradtrantip, L.; Zhang, H.; Anderson, M.O.; Saadoun, S.; Phuan, P.-W.; Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Small-Molecule Inhibitors of NMO-IgG Binding to Aquaporin-4 Reduce Astrocyte Cytotoxicity in Neuromyelitis Optica. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 2197–2208. [Google Scholar] [CrossRef]
- Saadoun, S.; Waters, P.; MacDonald, C.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Neutrophil Protease Inhibition Reduces Neuromyelitis Optica-Immunoglobulin G-Induced Damage in Mouse Brain. Ann. Neurol. 2012, 71, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Herges, K.; de Jong, B.A.; Kolkowitz, I.; Dunn, C.; Mandelbaum, G.; Ko, R.M.; Maini, A.; Han, M.H.; Killestein, J.; Polman, C.; et al. Protective Effect of an Elastase Inhibitor in a Neuromyelitis Optica-like Disease Driven by a Peptide of Myelin Oligodendroglial Glycoprotein. Mult. Scler. Houndmills Basingstoke Engl. 2012, 18, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bennett, J.L.; Verkman, A.S. Ex Vivo Spinal Cord Slice Model of Neuromyelitis Optica Reveals Novel Immunopathogenic Mechanisms. Ann. Neurol. 2011, 70, 943–954. [Google Scholar] [CrossRef]
- Smith, K.J. Sodium Channels and Multiple Sclerosis: Roles in Symptom Production, Damage and Therapy. Brain Pathol. 2007, 17, 230–242. [Google Scholar] [CrossRef] [PubMed]
- da Silva-Junior, A.J.; Mesentier-Louro, L.A.; Nascimento-dos-Santos, G.; Teixeira-Pinheiro, L.C.; Vasques, J.F.; Chimeli-Ormonde, L.; Bodart-Santos, V.; de Carvalho, L.R.P.; Santiago, M.F.; Mendez-Otero, R. Human Mesenchymal Stem Cell Therapy Promotes Retinal Ganglion Cell Survival and Target Reconnection after Optic Nerve Crush in Adult Rats. Stem Cell Res. Ther. 2021, 12, 69. [Google Scholar] [CrossRef] [PubMed]
- Labrador-Velandia, S.; Alonso-Alonso, M.L.; Alvarez-Sanchez, S.; González-Zamora, J.; Carretero-Barrio, I.; Pastor, J.C.; Fernandez-Bueno, I.; Srivastava, G.K. Mesenchymal Stem Cell Therapy in Retinal and Optic Nerve Diseases: An Update of Clinical Trials. World J. Stem Cells 2016, 8, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Nicoară, S.D.; Brie, I.; Jurj, A.; Sorițău, O. The Future of Stem Cells and Their Derivates in the Treatment of Glaucoma. A Critical Point of View. Int. J. Mol. Sci. 2021, 22, 11077. [Google Scholar] [CrossRef]
- Aneesh, A.; Liu, A.; Moss, H.E.; Feinstein, D.; Ravindran, S.; Mathew, B.; Roth, S. Emerging Concepts in the Treatment of Optic Neuritis: Mesenchymal Stem Cell-Derived Extracellular Vesicles. Stem Cell Res. Ther. 2021, 12, 594. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciapă, M.A.; Șalaru, D.L.; Stătescu, C.; Sascău, R.A.; Bogdănici, C.M. Optic Neuritis in Multiple Sclerosis—A Review of Molecular Mechanisms Involved in the Degenerative Process. Curr. Issues Mol. Biol. 2022, 44, 3959-3979. https://doi.org/10.3390/cimb44090272
Ciapă MA, Șalaru DL, Stătescu C, Sascău RA, Bogdănici CM. Optic Neuritis in Multiple Sclerosis—A Review of Molecular Mechanisms Involved in the Degenerative Process. Current Issues in Molecular Biology. 2022; 44(9):3959-3979. https://doi.org/10.3390/cimb44090272
Chicago/Turabian StyleCiapă, Manuela Andreea, Delia Lidia Șalaru, Cristian Stătescu, Radu Andy Sascău, and Camelia Margareta Bogdănici. 2022. "Optic Neuritis in Multiple Sclerosis—A Review of Molecular Mechanisms Involved in the Degenerative Process" Current Issues in Molecular Biology 44, no. 9: 3959-3979. https://doi.org/10.3390/cimb44090272
APA StyleCiapă, M. A., Șalaru, D. L., Stătescu, C., Sascău, R. A., & Bogdănici, C. M. (2022). Optic Neuritis in Multiple Sclerosis—A Review of Molecular Mechanisms Involved in the Degenerative Process. Current Issues in Molecular Biology, 44(9), 3959-3979. https://doi.org/10.3390/cimb44090272