
Therapeutic Plasma Exchange and Multiple Sclerosis Dysregulations: Focus on the Removal of Pathogenic Circulatory Factors and Altering Nerve Growth Factor and Sphingosine-1-Phosphate Plasma Levels


Abstract
:1. Introduction
2. Methodology
3. Pathogenesis of MS
3.1. Between Space and Time Axes
3.2. Between Detrimental and Beneficial Neuro-Inflammatory Responses
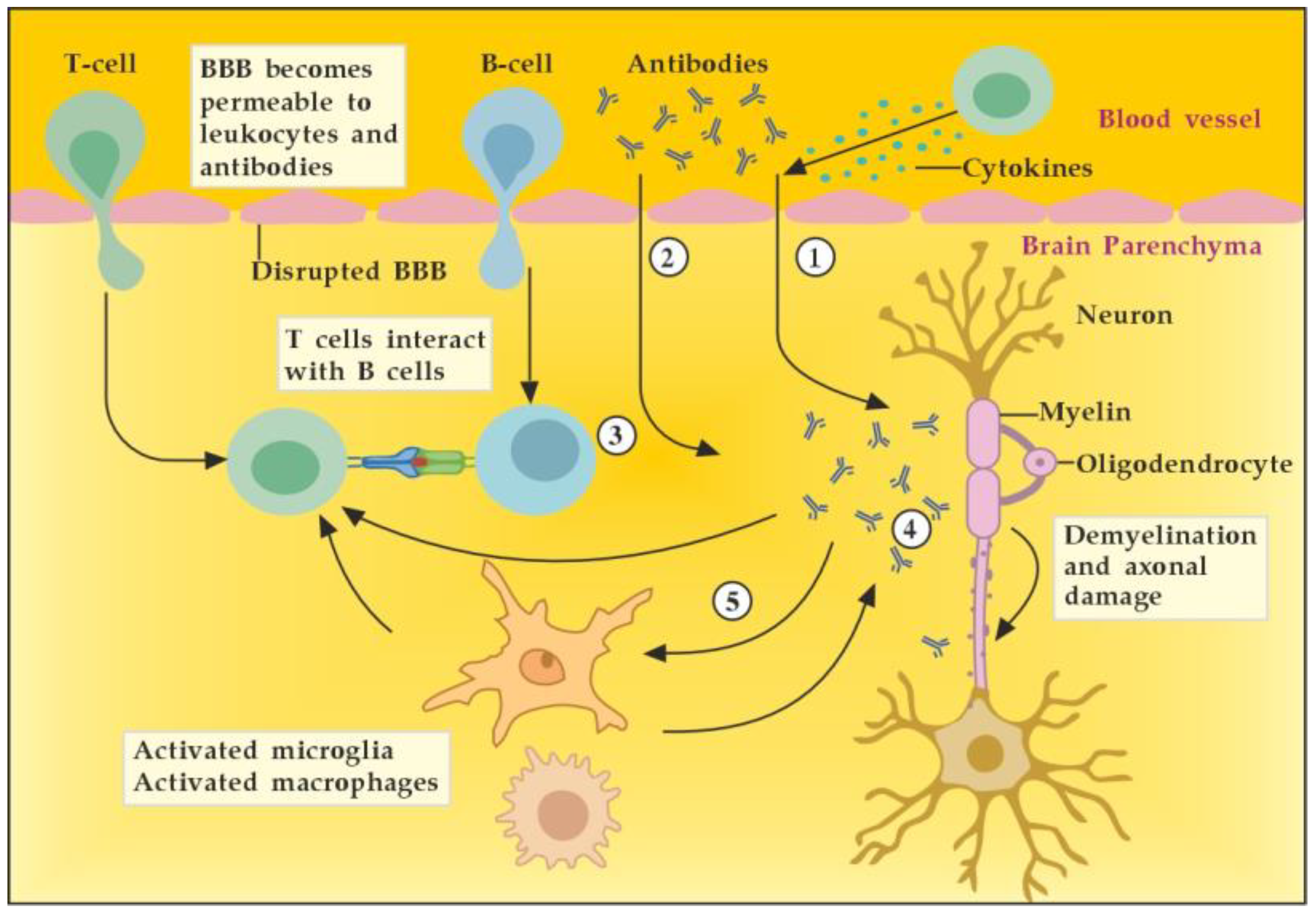
3.2.1. The Role of Peripheral Immune Cells
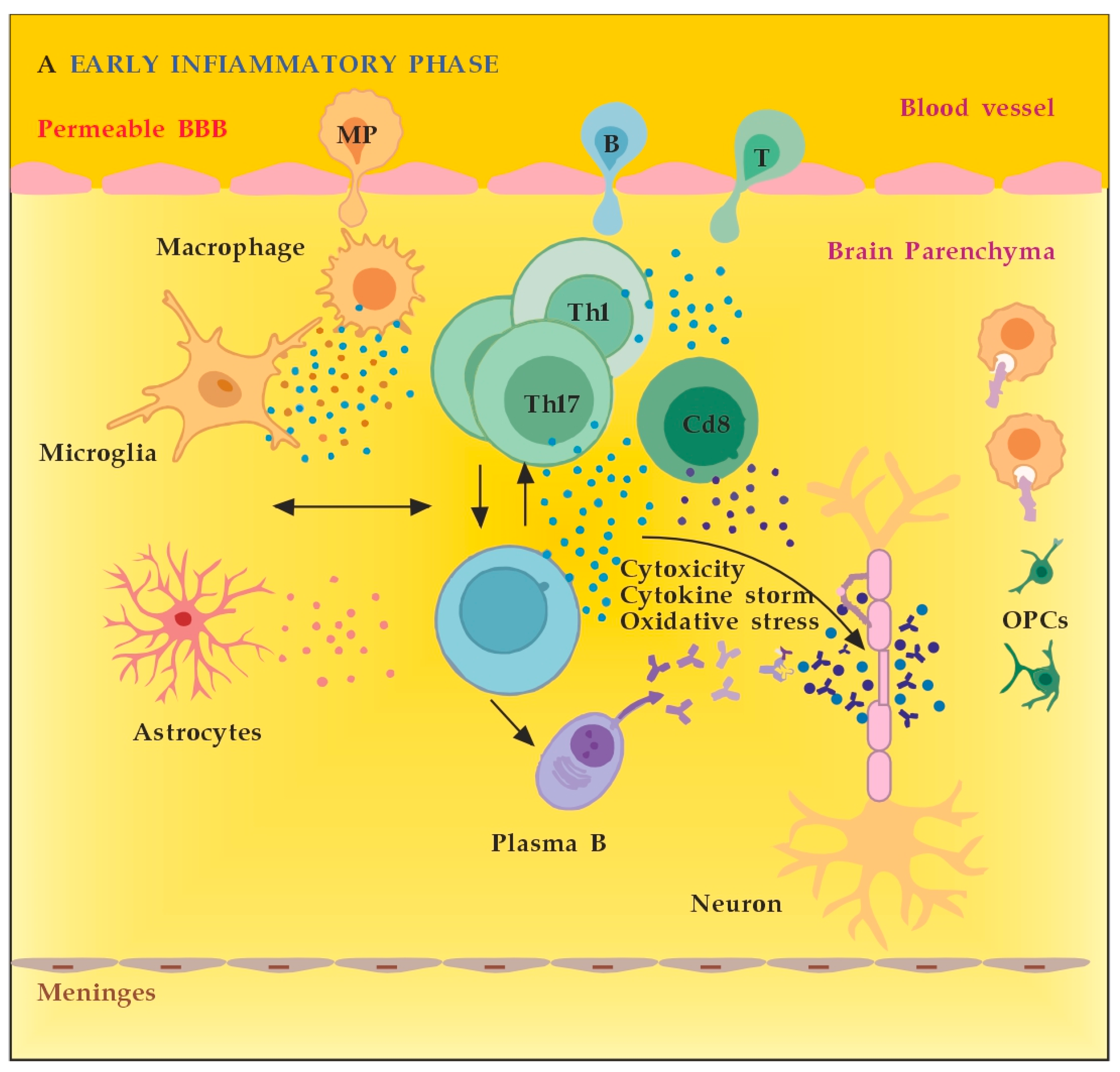
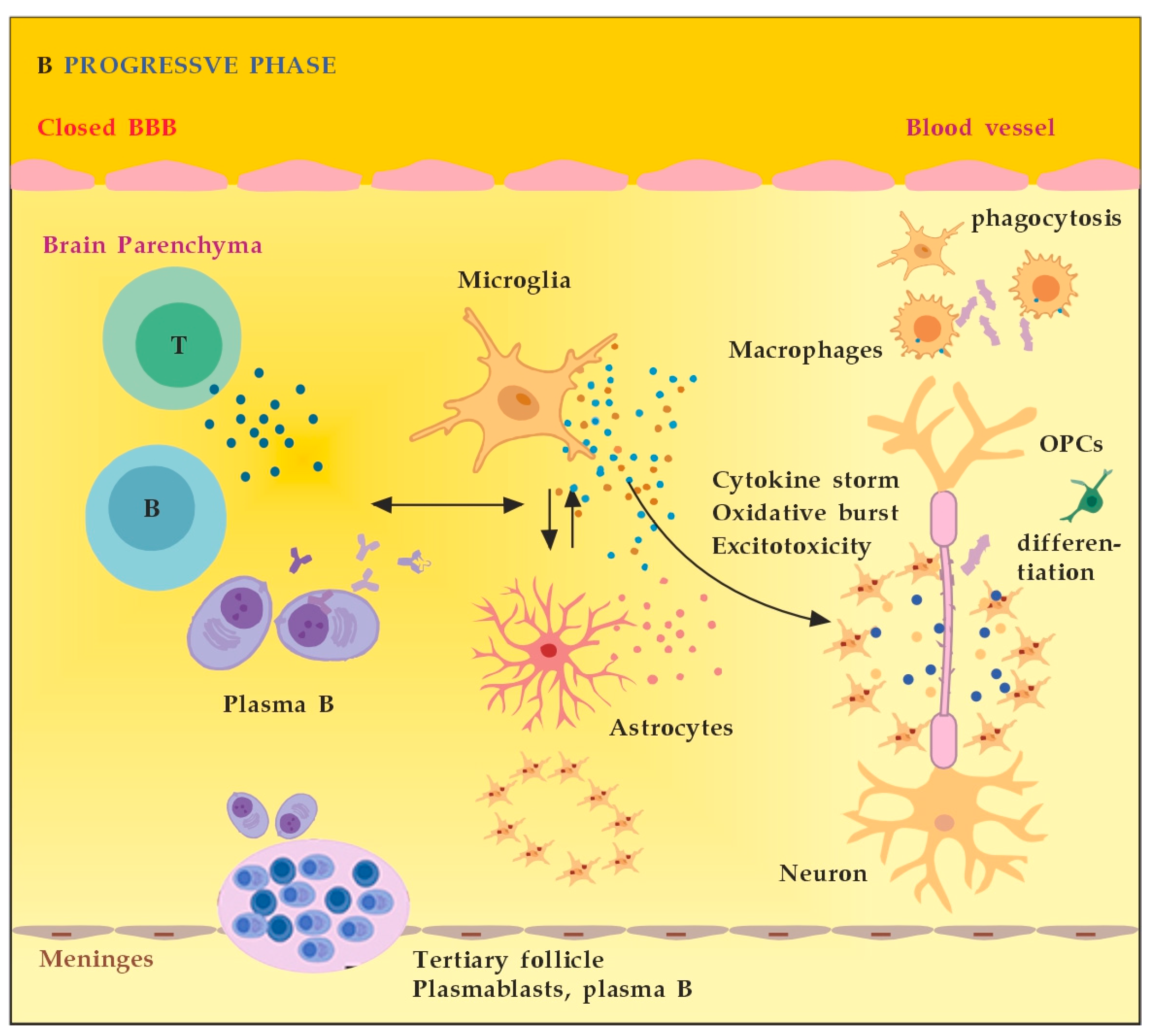
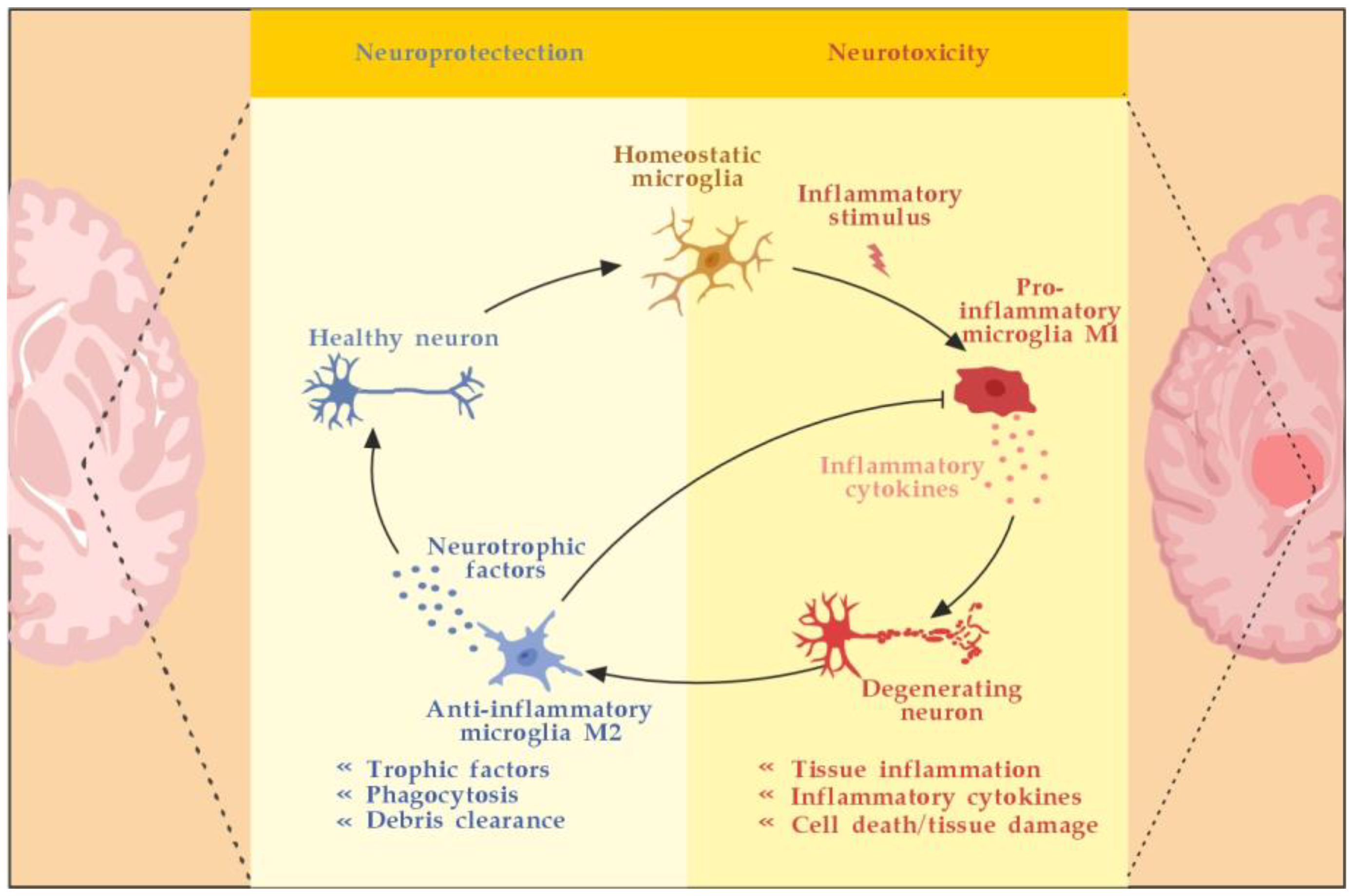
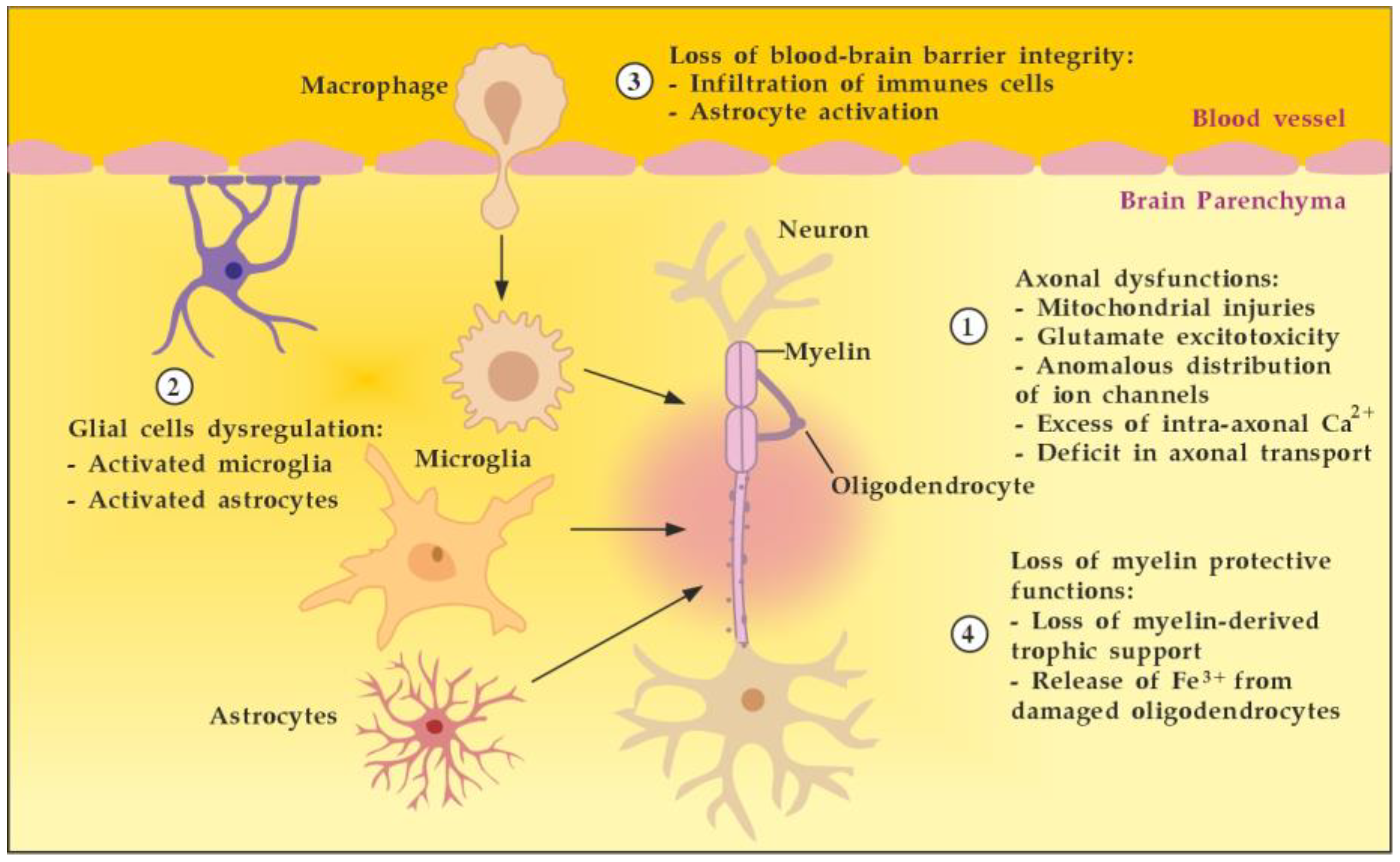
3.2.2. The Role of the Innate Resident CNS Cells
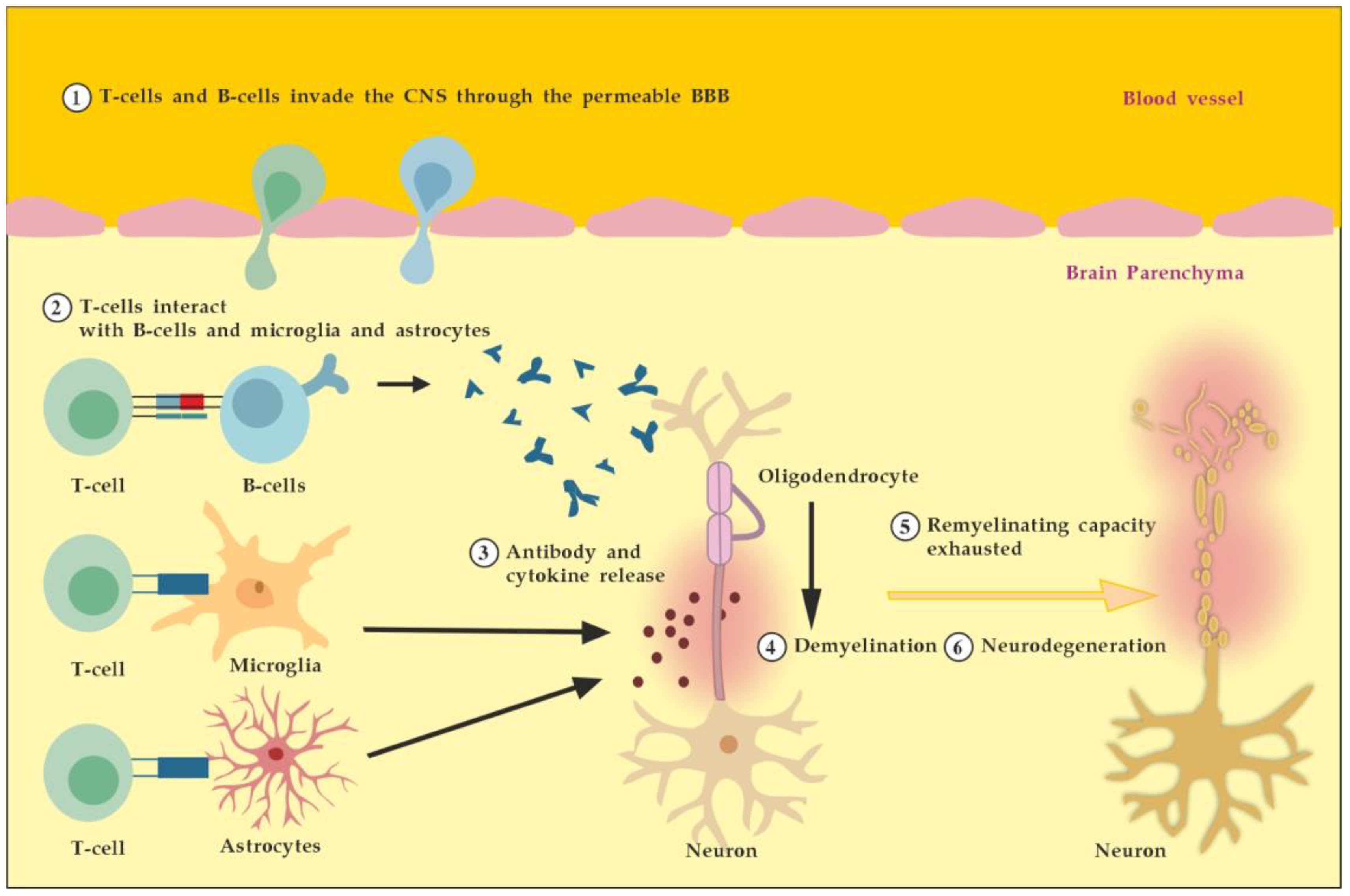
4. The Role of Autoantibodies
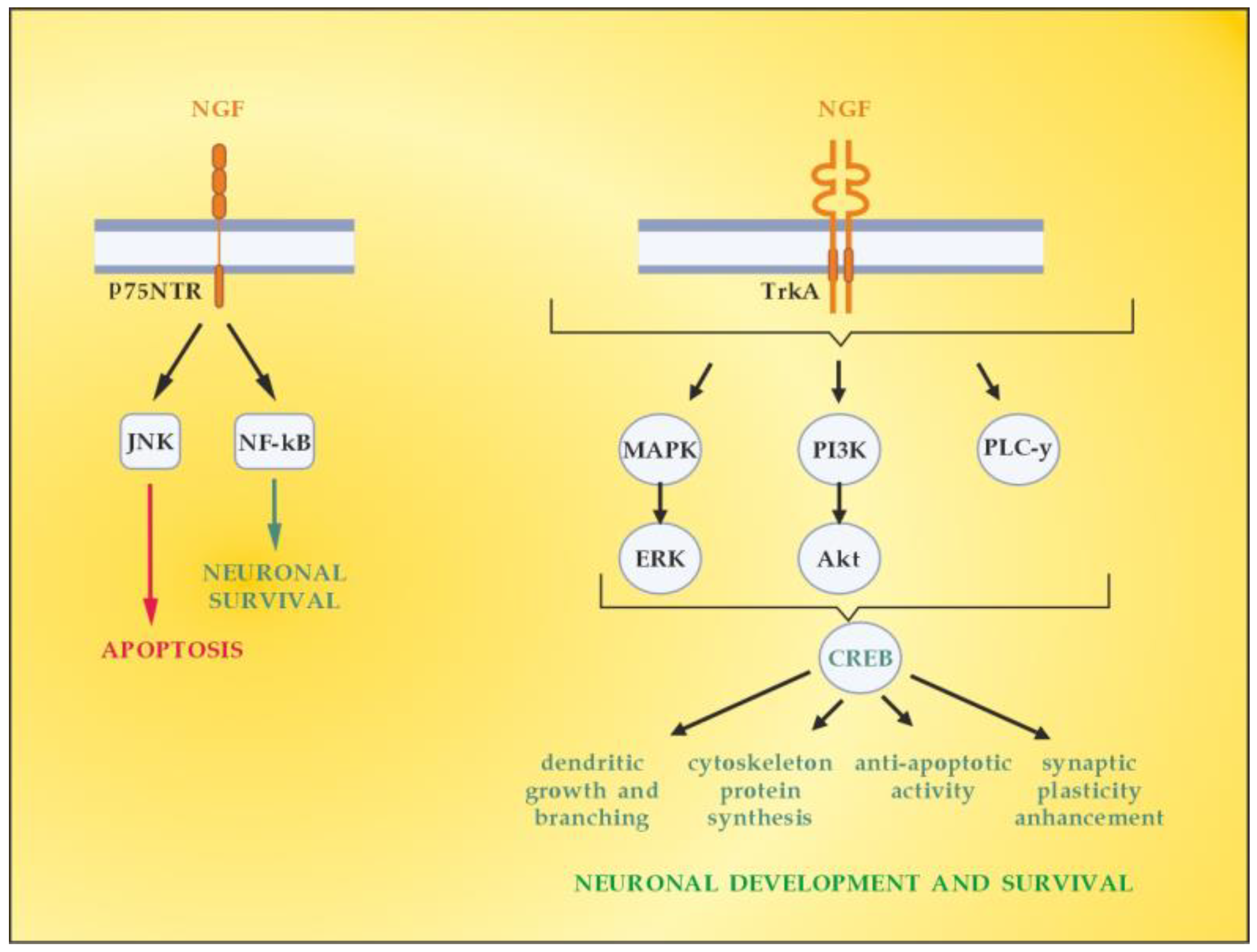
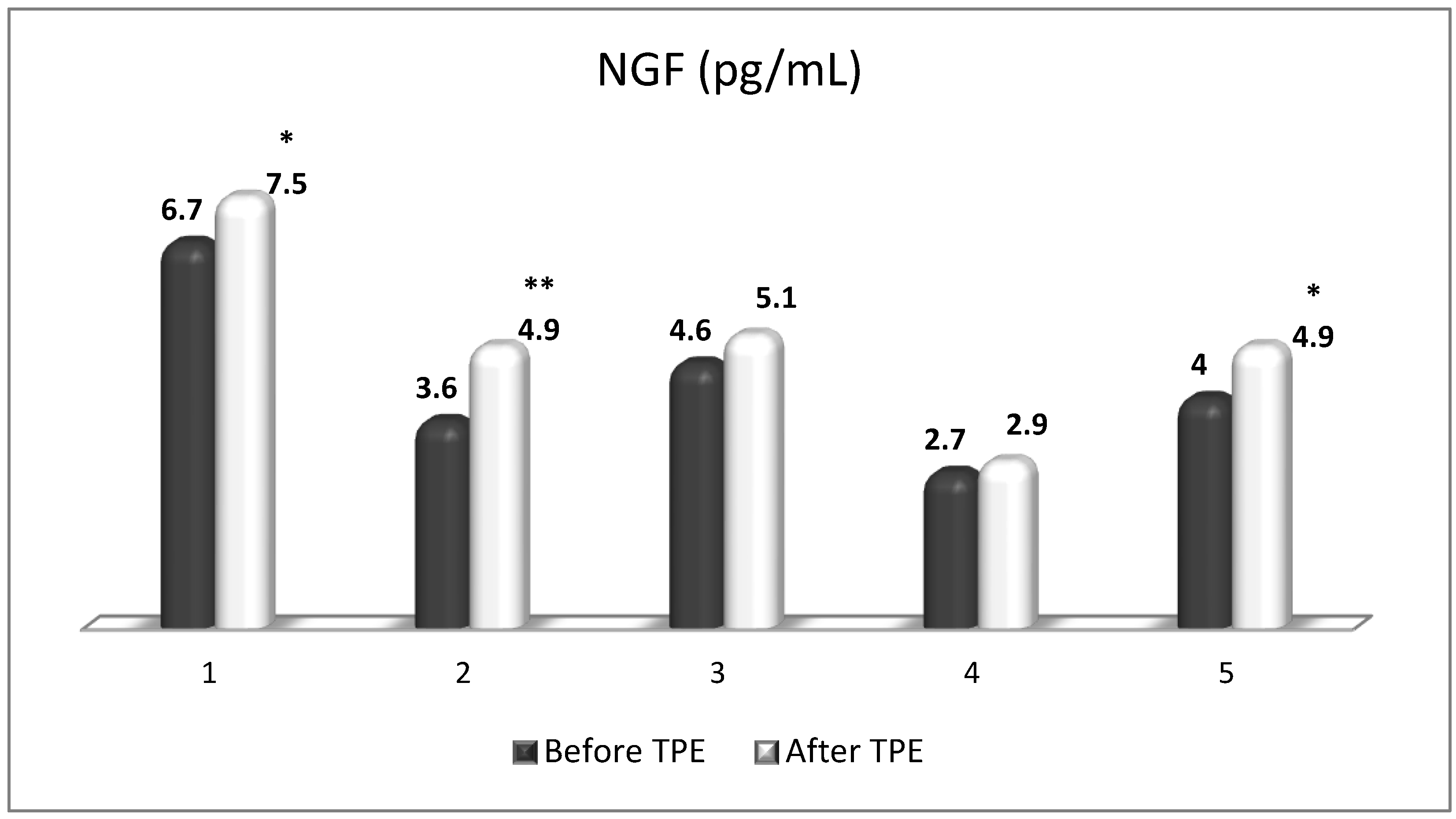
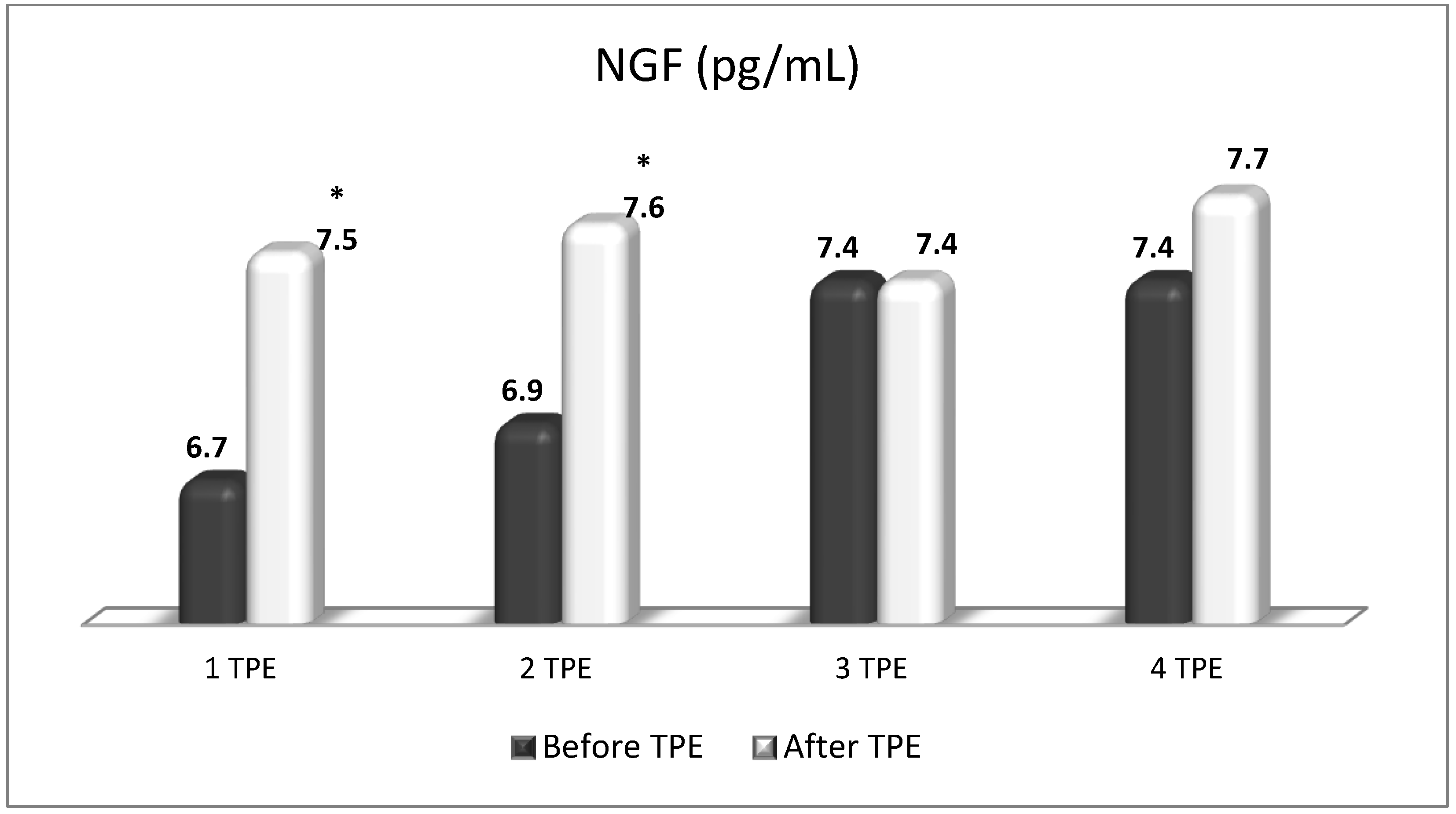
5. The Role of NGF
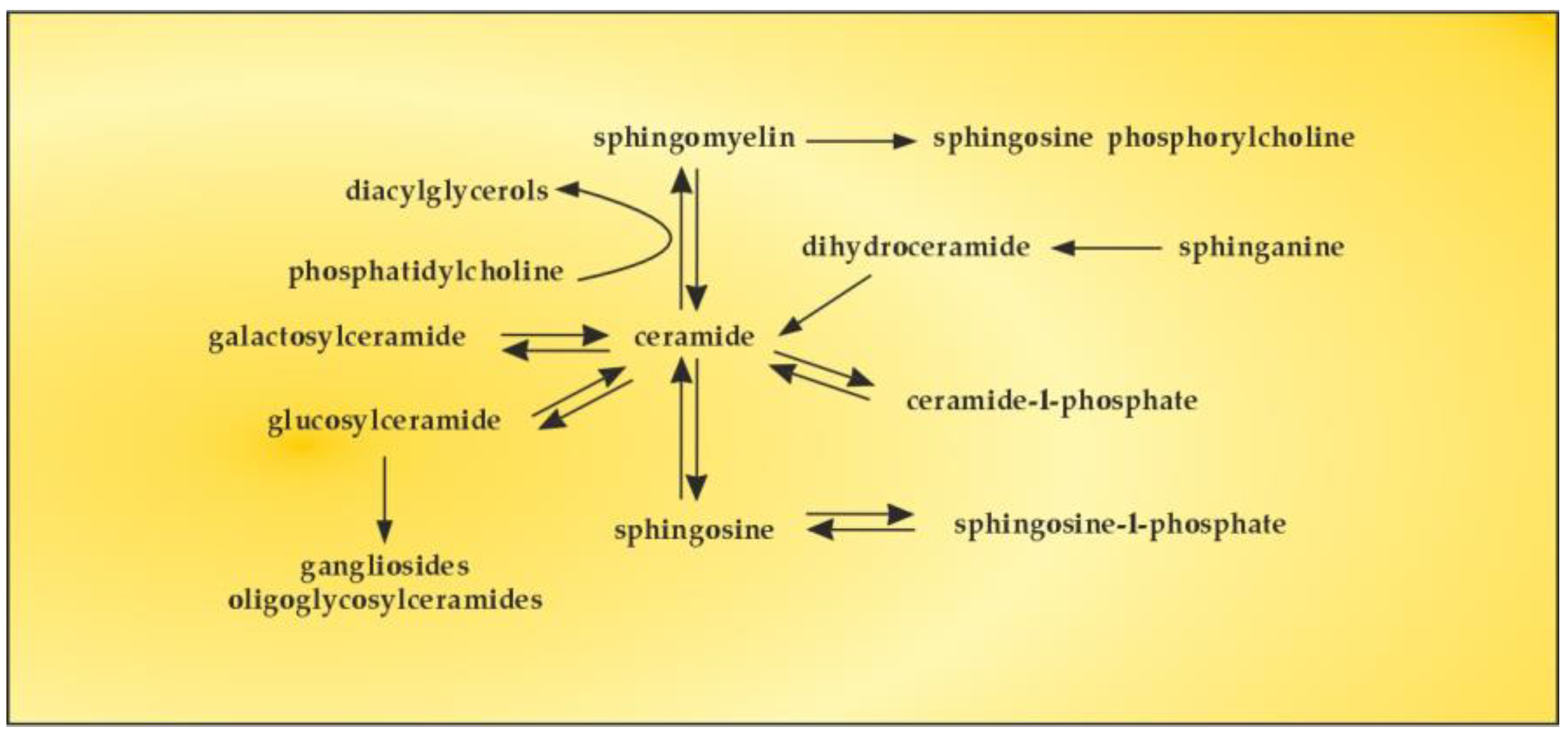
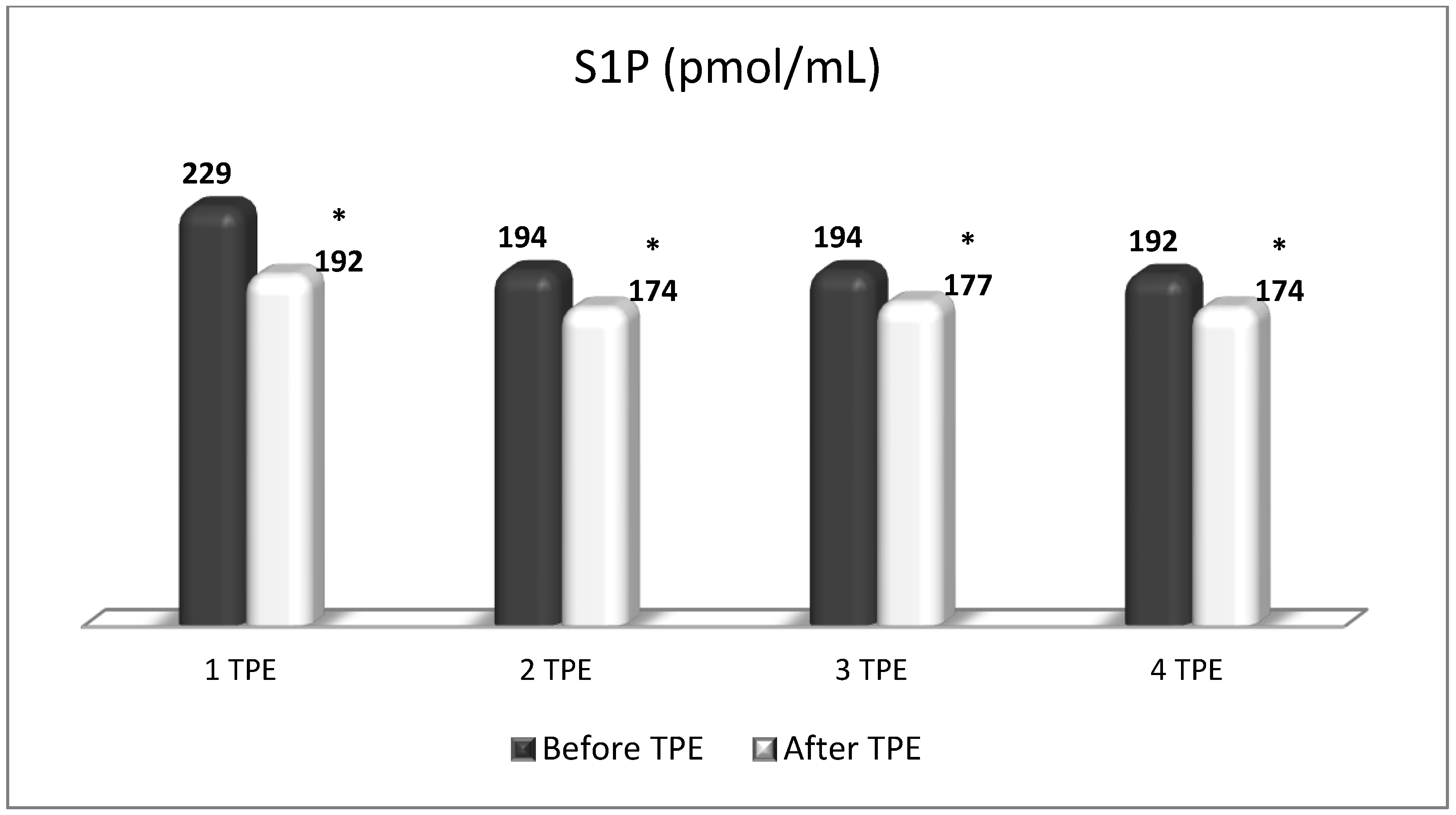
6. The Role of S1P
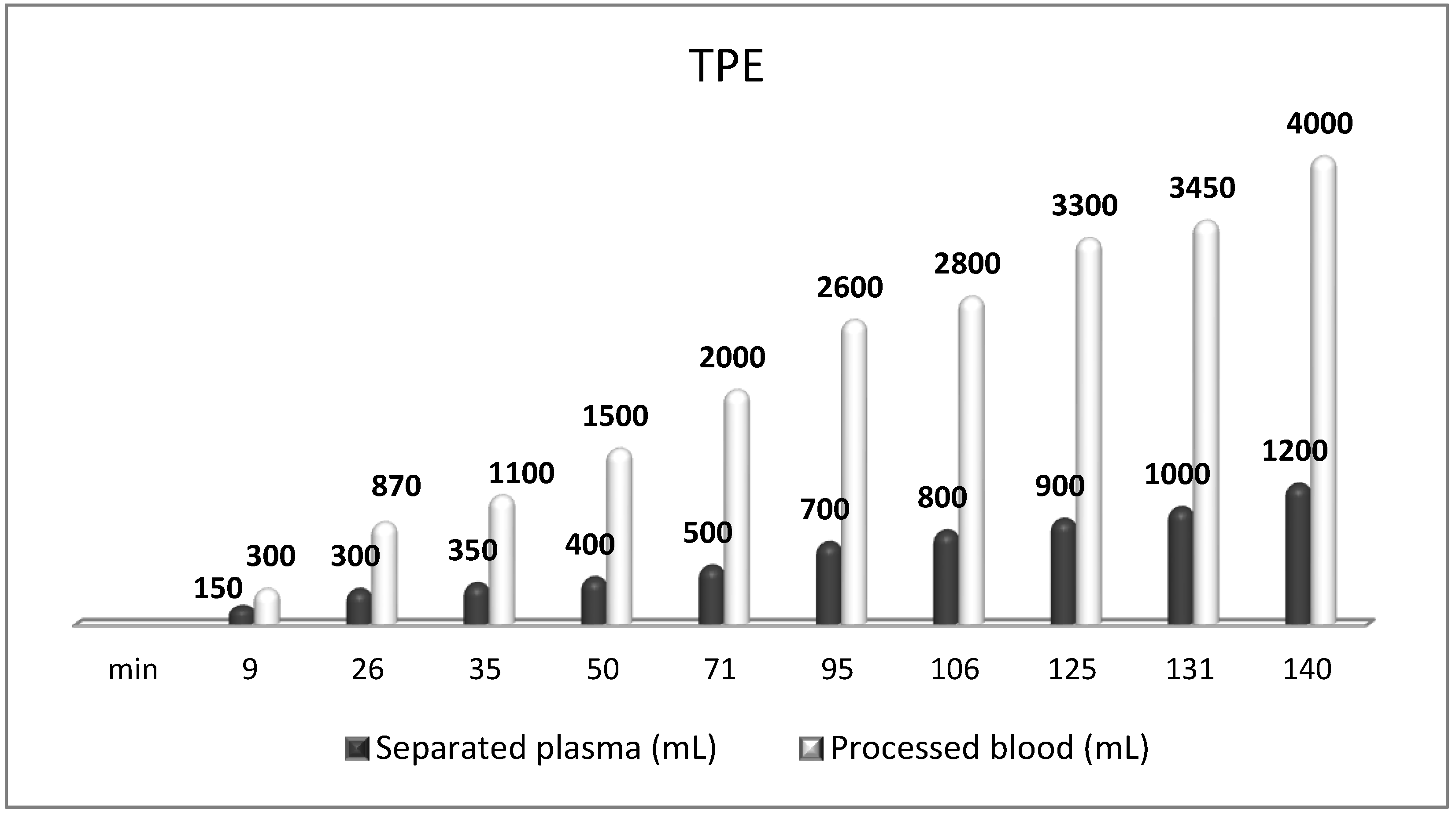
7. The Role of TPE
8. Summary of Achieving Neuroprotection in MS
9. Limitations and Future Directions
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zahoor, I.; Rui, B.; Khan, J.; Datta, I.; Giri, S. An emerging potential of metabolomics in multiple sclerosis: A comprehensive overview. Cell. Mol. Life Sci. 2021, 78, 3181–3203. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front. Immunol. 2018, 9, 3116. [Google Scholar] [CrossRef]
- Sandi, D.; Kokas, Z.; Biernacki, T.; Bencsik, K.; Klivényi, P.; Vécsei, L. Proteomics in Multiple Sclerosis: The Perspective of the Clinician. Int. J. Mol. Sci. 2022, 23, 5162. [Google Scholar] [CrossRef]
- Vyshkina, T.; Kalman, B. Autoantibodies and neurodegeneration in multiple sclerosis. Lab. Investig. 2008, 88, 796–807. [Google Scholar] [CrossRef]
- Li, Y.; Noto, D.; Hoshino, Y.; Mizuno, M.; Yoshikawa, S.; Miyake, S. Immunoglobulin directly enhances differentiation of oli-godendrocyte-precursor cells and remyelination. Sci. Rep. 2023, 13, 9394. [Google Scholar] [CrossRef]
- Muñoz, U.; Sebal, C.; Escudero, E.; Esiri, M.; Tzartos, J.; Sloan, C.; Sadaba, M.C. Main Role of Antibodies in Demyelination and Axonal Damage in Multiple Sclerosis. Cell. Mol. Neurobiol. 2022, 42, 1809–1827. [Google Scholar] [CrossRef]
- Höftberger, R.; Lassmann, H.; Berger, T.; Reindl, M. Pathogenic autoantibodies in multiple sclerosis—From a simple idea to a complex concept. Nat. Rev. Neurol. 2022, 18, 681–688. [Google Scholar] [CrossRef]
- Cunniffe, N.; Coles, A. Promoting remyelination in multiple sclerosis. J. Neurol. 2021, 268, 30–44. [Google Scholar] [CrossRef]
- Davies, A.J.; Fehmi, J.; Senel, M.; Tumani, H.; Dorst, J.; Rinaldi, S. Immunoadsorption and Plasma Exchange in Seropositive and Seronegative Immune-Mediated Neuropathies. J. Clin. Med. 2020, 9, 2025. [Google Scholar] [CrossRef]
- Farrokhi, M. Plasmapheresis for Multiple Sclerosis in the Twenty-First Century: Take It or Leave It? J. Rev. Med. Sci. 2021, 1, e1. [Google Scholar]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Brück, W.; Lucchinetti, C.F. The Immunopathology of Multiple Sclerosis: An Overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Danikowski, K.M.; Jayaraman, S.; Prabhakar, B.S. Regulatory T cells in multiple sclerosis and myasthenia gravis. J. Neuroinflammation 2017, 14, 117. [Google Scholar] [CrossRef]
- Haase, S.; Linker, R.A. Inflammation in multiple sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211007687. [Google Scholar] [CrossRef]
- Bordet, R.; Camu, W.; De Seze, J.; Laplaud, D.-A.; Ouallet, J.-C.; Thouvenot, E. Mechanism of action of s1p receptor modulators in multiple sclerosis: The double requirement. Rev. Neurol. 2020, 176, 100–112. [Google Scholar] [CrossRef]
- Stadelmann, C.; Wegner, C.; Bruck, W. Inflammation, demyelination, and degeneration—Recent insights from MS pathology. Biochim. Biophys. Acta 2011, 1812, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Perdaens, O.; van Pesch, V. Molecular Mechanisms of Immunosenescene and Inflammaging: Relevance to the Immunopath-ogenesis and Treatment of Multiple Sclerosis. Front. Neurol. 2022, 12, 811518. [Google Scholar] [CrossRef]
- Martino, G.; Adorini, L.; Rieckmann, P.; Hillert, J.; Kallmann, B.; Comi, G.; Filippi, M. Inflammation in multiple sclerosis: The good, the bad, and the complex. Lancet Neurol. 2002, 1, 499–509. [Google Scholar] [CrossRef]
- Billiau, A.; Heremans, H.; Vandekerckhove, F.; Dijkmans, R.; Sobis, H.; Meulepas, E.; Carton, H. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J. Immunol. 1988, 140, 1506–1510. [Google Scholar] [CrossRef]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P. TNF alpha promotes proliferation of oligoden-drocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Acosta, M.; Cortes, C.; MacPhee, H.; Namaka, M.P. Exploring the role of nerve growth factor in multiple sclerosis: Implications in myelin repair. CNS Neurol. Disord.—Drug Targets 2013, 12, 1242–1256. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Marino, M.W.; Wong, G.; Grail, D.; Dunn, A.; Bettadapura, J.; Slavin, A.J.; Old, L.; Bernard, C.C. TNF is a potent an-ti-inflammatory cytokine in autoimmune-mediated demyelination. Nat. Med. 1998, 4, 78–83. [Google Scholar] [CrossRef]
- Wang, K.; Song, F.; Fernandez-Escobar, A.; Luo, G.; Wang, J.-H.; Sun, Y. The Properties of Cytokines in Multiple Sclerosis: Pros and Cons. Am. J. Med. Sci. 2018, 356, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Hulshof, S.; Montagne, L.; De Groot, C.J.; Van Der Valk, P. Cellular localization and expression patterns of interleukin-10, interleukin-4, and their receptors in multiple sclerosis lesions. Glia 2002, 38, 24–35. [Google Scholar] [CrossRef]
- Kerschensteiner, M.; Gallmeier, E.; Behrens, L.; Leal, V.V.; Misgeld, T.; Klinkert, W.E.; Kolbeck, R.; Hoppe, E.; Oropeza-Wekerle, R.L.; Bartke, I.; et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: A neuroprotective role of inflammation? J. Exp. Med. 1999, 189, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Hartung, H.-P.; Freedman, M.S.; Boyko, A.; Radü, E.W.; Mikol, D.D.; Lamarine, M.; Hyvert, Y.; Freudensprung, U.; Plitz, T.; et al. Atacicept in multiple sclerosis (ATAMS): A randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014, 13, 353–363. [Google Scholar] [CrossRef]
- Nociti, V.; Romozzi, M. The Role of BDNF in Multiple Sclerosis Neuroinflammation. Int. J. Mol. Sci. 2023, 24, 8447. [Google Scholar] [CrossRef]
- Rodríguez, A.M.; Rodríguez, J.; Giambartolomei, G.H. Microglia at the Crossroads of Pathogen-Induced Neuroinflammation. ASN Neuro 2022, 14, 175909142211045. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ma, J.; Ding, G.; Gong, Q.; Wang, Y.; Yu, H.; Cheng, X. Microglia Polarization from M1 toward M2 Phenotype Is Promoted by Astragalus Polysaccharides Mediated through Inhibition of MiR-155 in Experimental Autoimmune Encephalo-myelitis. Oxidative Med. Cell. Longev. 2021, 2021, 5753452. [Google Scholar] [CrossRef] [PubMed]
- Qie, S.; Ran, Y.; Lu, X.; Su, W.; Li, W.; Xi, J.; Gong, W.; Liu, Z. Candesartan Modulates Microglia Activation and Polarization via NF-KB Signaling Pathway. Int. J. Immunopathol. Pharmacol. 2020, 34, 205873842097490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wei, Y.-Z.; Wang, G.-Q.; Li, D.-D.; Shi, J.-S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Mi-croglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell Neurosci. 2019, 12, 531. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Kang, J.; Liu, S.; Xu, Y.; Shao, B. The Protective Effects of Peroxisome Proliferator-Activated Receptor Gamma in Cerebral Ischemia-Reperfusion Injury. Front. Neurol. 2020, 11, 588516. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11, 374. [Google Scholar] [CrossRef]
- Gandhi, R.; Laroni, A.; Weiner, H.L. Role of the innate immune system in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2010, 221, 7–14. [Google Scholar] [CrossRef]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef]
- Palasz, E.; Wilkaniec, A.; Stanaszek, L.; Andrzejewska, A.; Adamczyk, A. Glia-Neurotrophic Factor Relationships: Possible Role in Pathobiology of Neuroinflammation-Related Brain Disorders. Int. J. Mol. Sci. 2023, 24, 6321. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Correale, J.; Farez, M.F. The Role of Astrocytes in Multiple Sclerosis Progression. Front. Neurol. 2015, 6, 180. [Google Scholar] [CrossRef] [PubMed]
- Voss, E.V.; Škuljec, J.; Gudi, V.; Skripuletz, T.; Pul, R.; Trebst, C.; Stangel, M. Characterisation of microglia during de- and remyelination: Can they create a repair promoting environment? Neurobiol. Dis. 2012, 45, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Liu, K.; Zhou, L.; Chen, P. Precious but convenient means of prevention and treatment: Physiological molecular mechanisms of interaction between exercise and motor factors and Alzheimer’s disease. Front. Physiol. 2023, 14, 1193031. [Google Scholar] [CrossRef]
- Archelos, J.J.; Storch, M.K.; Hartung, H.P. The role of B cells and autoantibodies in multiple sclerosis. Ann. Neurol. 2000, 47, 694–706. [Google Scholar] [CrossRef]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic b-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef]
- Castillo-Trivino, T.; Braithwaite, D.; Bacchetti, P.; Waubant, E. Rituximab in relapsing and progressive forms of multiple sclerosis: A systematic review. PLoS ONE 2013, 8, e66308. [Google Scholar] [CrossRef]
- Nourbakhsh, B.; Mowry, E.M. Multiple Sclerosis Risk Factors and Pathogenesis. Contin. Lifelong Learn. Neurol. 2019, 25, 596–610. [Google Scholar] [CrossRef]
- Lassmann, H. Multiple sclerosis pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a028936. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.; Lindner, M.; Arthur, A.; Brennan, K.; Jarius, S.; Hussey, J.; Chan, A.; Stroet, A.; Olsson, T.; Willison, H.; et al. Functional identification of pathogenic autoantibody responses in patients with multiple sclerosis. Brain 2012, 135, 1819–1833. [Google Scholar] [CrossRef]
- Pröbstel, A.-K.; Sanderson, N.S.R.; Derfuss, T. B Cells and autoantibodies in multiple sclerosis. Int. J. Mol. Sci. 2015, 16, 16576–16592. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Given, K.S.; Harlow, D.E.; Matschulat, A.M.; Macklin, W.B.; Bennett, J.L.; Owens, G.P. Myelin-specific multiple sclerosis antibodies cause complement-dependent oligodendrocyte loss and demyelination. Acta Neuropathol. Commun. 2017, 5, 25. [Google Scholar] [CrossRef]
- Miyachi, Y.; Fujii, T.; Yamasaki, R.; Tsuchimoto, D.; Iinuma, K.; Sakoda, A.; Fukumoto, S.; Matsushita, T.; Masaki, K.; Isobe, N.; et al. Serum Anti-oligodendrocyte Autoantibodies in Patients With Multiple Sclerosis Detected by a Tissue-Based Immunofluores-cence Assay. Front. Neurol. 2021, 12, 681980. [Google Scholar] [CrossRef] [PubMed]
- van der Poel, M.; Hoepel, W.; Hamann, J.; Huitinga, I.; Dunnen, J.D. IgG Immune Complexes Break Immune Tolerance of Human Microglia. J. Immunol. 2020, 205, 2511–2518. [Google Scholar] [CrossRef]
- Zhou, W.; Graner, M.; Paucek, P.; Beseler, C.; Boisen, M.; Bubak, A.; Asturias, F.; George, W.; Graner, A.; Ormond, D.; et al. Multiple sclerosis plasma IgG aggregates induce complement-dependent neuronal apoptosis. Cell Death Dis. 2023, 14, 254. [Google Scholar] [CrossRef] [PubMed]
- Tonev, D.G.; Momchilova, A.B. Therapeutic Plasma Exchange in Certain Immune-Mediated Neurological Disorders: Focus on a Novel Nanomembrane-Based Technology. Biomedicines 2023, 11, 328. [Google Scholar] [CrossRef] [PubMed]
- Zaprianova, E.; Deleva, D.; Ilinov, P.; Sultanov, E.; Filchev, A.; Christova, L.; Sultanov, B. Serum ganglioside patterns in multiple sclerosis. Neurochem. Res. 2001, 26, 95–100. [Google Scholar] [CrossRef]
- Kanda, T.; Iwasaki, T.; Yamawaki, M.; Tai, T.; Mizusawa, H. Anti-GM1 antibody facilitates leakage in an in vitro blood-nerve barrier model. Neurology 2000, 55, 585–587. [Google Scholar] [CrossRef]
- Lehmann, H.C.; Lopez, P.H.H.; Zhang, G.; Ngyuen, T.; Zhang, J.; Kieseier, B.C.; Mori, S.; Sheikh, K.A. Passive immunization with anti-ganglioside antibodies directly inhibits axon regeneration in an animal model. J. Neurosci. 2007, 27, 27–34. [Google Scholar] [CrossRef]
- Ravindranath, M.H.; Muthugounder, S.; Saravanan, T.S.; Presser, N.; Morton, D.L. Human antiganglioside autoantibodies: Validation of ELISA. Ann. N. Y. Acad. Sci. 2005, 1050, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R.; Skaper, S.D.; Dal Toso, R.; Petrelli, L.; Leon, A. Nerve growth factor: From neurotrophin to neurokine. Trends Neurosci. 1996, 19, 514–520. [Google Scholar] [CrossRef]
- Bibel, M.; Barde, Y.-A. Neurotrophins: Key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000, 14, 2919–2937. [Google Scholar] [CrossRef] [PubMed]
- Barbacid, M. Neurotrophic factors and their receptors. Curr. Opin. Cell Biol. 1995, 7, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Patapoutian, A.; Reichardt, L.F. Trk receptors: Mediators of neurotrophin action. Curr. Opin. Neurobiol. 2001, 11, 272–280. [Google Scholar] [CrossRef]
- Chao, M.V. The p75 neurotrophin receptor. J. Neurobiol. 1994, 25, 1373–1385. [Google Scholar] [CrossRef]
- Barker, P.A. p75NTR is positively promiscuous: Novel partners and new insights. Neuron 2004, 42, 529–533. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Chao, M.V.; Hempstead, B.L. p75 and Trk: A two-receptor system. Trends Neurosci. 1995, 18, 321–326. [Google Scholar] [CrossRef]
- Alcover-Sanchez, B.; Garcia-Martin, G.; Wandosell, F.; Cubelos, B. R-Ras GTPases Signaling Role in Myelin Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5911. [Google Scholar] [CrossRef]
- Gaesser, J.M.; Fyffe-Maricich, S.L. Intracellular signaling pathway regulation of myelination and remyelination in the CNS. Exp. Neurol. 2016, 283, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Gonsalvez, D.; Ferner, A.H.; Peckham, H.; Murray, S.S.; Xiao, J. The roles of extracellular related-kinases 1 and 2 signaling in CNS myelination. Neuropharmacology 2016, 110, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Fyffe-Maricich, S.L.; Furusho, M.; Miller, R.H.; Bansal, R. ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J. Neurosci. 2012, 32, 8855–8864. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Furusho, M.; Dupree, J.L.; Bansal, R. Role of ERK1/2 MAPK signaling in the maintenance of myelin and axonal in-tegrity in the adult CNS. J. Neurosci. 2014, 34, 16031–16045. [Google Scholar] [CrossRef]
- Duncan, G.J.; Plemel, J.R.; Assinck, P.; Manesh, S.B.; Muir, F.G.W.; Hirata, R.; Berson, M.; Liu, J.; Wegner, M.; Emery, B.; et al. Myelin regulatory factor drives remyelination in multiple sclerosis. Acta Neuropathol. 2017, 134, 403–422. [Google Scholar] [CrossRef]
- Niewiadomska, G.; Mietelska-Porowska, A.; Mazurkiewicz, M. The Cholinergic System, Nerve Growth Factor and the Cyto-skeleton. Behav. Brain Res. 2011, 221, 515–526. [Google Scholar] [CrossRef]
- Du, Y.; Dreyfus, C.F. Oligodendrocytes as Providers of Growth Factors. J. Neurosci. Res. 2002, 68, 647–654. [Google Scholar] [CrossRef]
- Bracci-Laudiero, L.; Bonini, S.; Manni, L.; Aloe, L. The expanding role of nerve growth factor: From neurotrophic activity to immunologic diseases. Allergy 1997, 52, 883–894. [Google Scholar] [CrossRef]
- Kalinowska-Lyszczarz, A.; Losy, J. The role of neurotrophins in multiple sclerosis-pathological and clinical implications. Int. J. Mol. Sci. 2012, 13, 13713–13725. [Google Scholar] [CrossRef]
- Linker, R.; Gold, R.; Luhder, F. Function of neurotrophic factors beyond the nervous system: Infammation and autoimmune demyelination. Crit. Rev. Immunol. 2009, 29, 43–68. [Google Scholar] [CrossRef] [PubMed]
- Hohlfeld, R. Neurotrophic cross-talk between the nervous and immune systems: Relevance for repair strategies in multiple sclerosis? J. Neurol. Sci. 2008, 265, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Sawada, J.; Itakura, A.; Tanaka, A.; Furusaka, T.; Matsuda, H. Nerve growth factor functions as a chemoattractant for mast cells through both mitogen-activated protein kinase and phosphatidylinositol 3-kinase signaling pathways. Blood 2000, 95, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Samah, B.; Porcheray, F.; Gras, G. Neurotrophins modulate monocyte chemotaxis without affecting macrophage function. Clin. Exp. Immunol. 2008, 151, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Susaki, Y.; Shimizu, S.; Katakura, K.; Watanabe, N.; Kawamoto, K.; Matsumoto, M.; Tsudzuki, M.; Furusaka, T.; Kitamura, Y.; Matsuda, H. Functional properties of murine macrophages promoted by nerve growth factor. Blood 1996, 88, 4630–4637. [Google Scholar] [CrossRef]
- Beigelman, A.; Levy, J.; Hadad, N.; Pinsk, V.; Haim, A.; Fruchtman, Y.; Levy, R. Abnormal neutrophil chemotactic activity in children with congenital insensitivity to pain with anhidrosis (CIPA): The role of nerve growth factor. Clin. Immunol. 2009, 130, 365–372. [Google Scholar] [CrossRef]
- Kannan, Y.; Ushio, H.; Koyama, H.; Okada, M.; Oikawa, M.; Yoshihara, T.; Kaneko, M.; Matsuda, H. Nerve growth factor enhances survival, phagocytosis, and superoxide production of murine neutrophils. Blood 1991, 77, 1320–1325. [Google Scholar] [CrossRef]
- Hamada, A.; Watanabe, N.; Ohtomo, H.; Matsuda, H. Nerve growth factor enhances survival and cytotoxic activity of human eosinophils. Br. J. Haematol. 1996, 93, 299–302. [Google Scholar] [CrossRef]
- Mazurek, N.; Weskamp, G.; Erne, P.; Otten, U. Nerve growth factor induces mast cell degranulation without changing intra-cellular calcium levels. FEBS Lett. 1986, 198, 315–320. [Google Scholar] [CrossRef]
- Kawamoto, K.; Okada, T.; Kannan, Y.; Ushio, H.; Matsumoto, M.; Matsuda, H. Nerve growth factor prevents apoptosis of rat peritoneal mast cells through the Trk proto-oncogene receptor. Blood 1995, 86, 4638–4644. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; U, H.S.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 2005, 11, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Hadaczek, P.; Eberling, J.L.; Pivirotto, P.; Bringas, J.; Forsayeth, J.; Bankiewicz, K.S. Eight years of clinical improvement in mptp-lesioned primates after gene therapy with AAV2-hAADC. Mol. Ther. 2010, 18, 1458–1461. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Yang, J.H.; Barba, D.; Hoi-Sang, U.; Bakay, R.A.E.; Pay, M.M.; Masliah, E.; Conner, J.M.; Kobalka, P.; Roy, S.; et al. Nerve growth factor gene therapy activation of neuronal responses in Alzheimer disease. JAMA Neurol. 2015, 72, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Tuszynski, M.H. Intraparenchymal NGF infusions rescue degenerating cholinergic neurons. Cell Transplant. 2000, 9, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Jönhagen, M.E.; Nordberg, A.; Amberla, K.; Bäckman, L.; Ebendal, T.; Meyerson, B.; Olson, L.; Seiger, Å.; Shigeta, M.; Theodorsson, E.; et al. Intracerebroventricular infusion of nerve growth factor in three patients with alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 1998, 9, 246–257. [Google Scholar] [CrossRef]
- Delivanoglou, N.; Boziki, M.; Theotokis, P.; Kesidou, E.; Touloumi, O.; Dafi, N.; Nousiopoulou, E.; Lagoudaki, R.; Grigoriadis, N.; Charalampopoulos, I.; et al. Spatio-temporal expression profile of NGF and the two-receptor system, TrkA and p75NTR, in experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2020, 17, 41. [Google Scholar] [CrossRef]
- Minnone, G.; De Benedetti, F.; Bracci-Laudiero, L. NGF and its receptors in the regulation of inflammatory response. Int. J. Mol. Sci. 2017, 18, 1028. [Google Scholar] [CrossRef]
- Cohen, I.R.; Schwartz, M. Autoimmune maintenance and neuroprotection of the central nervous system. J. Neuroimmunol. 1999, 100, 111–114. [Google Scholar] [CrossRef]
- Schwartz, M.; Raposo, C. Protective autoimmunity: A unifying model for the immune network involved in CNS repair. Neuroscientist 2014, 20, 343–358. [Google Scholar] [CrossRef]
- Lee, X.; Yang, Z.; Shao, Z.; Rosenberg, S.S.; Levesque, M.; Pepinsky, R.B.; Qiu, M.; Miller, R.H.; Chan, J.R.; Mi, S. NGF regulates the expression of axonal LINGO-1 to inhibit oligodendrocyte differentiation and myelination. J. Neurosci. 2007, 27, 220–225. [Google Scholar] [CrossRef]
- Plemel, J.R.; Liu, W.Q.; Yong, V.W. Remyelination therapies: A new direction and challenge in multiple sclerosis. Nat. Rev. Drug Discov. 2017, 16, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Geoffroy, C.G.; Chan, A.F.; Tolentino, K.E.; Crawford, M.J.; Leal, M.A.; Kang, B.; Zheng, B. Assessing spinal axon regen-eration and sprouting in Nogo-, MAG-, and OMgp-deficient mice. Neuron 2010, 66, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Alatrash, R.; Golubenko, M.; Martynova, E.; Garanina, E.; Mukhamedshina, Y.; Khaiboullina, S.; Rizvanov, A.; Salafutdinov, I.; Arkhipova, S. Genetically Engineered Artificial Microvesicles Carrying Nerve Growth Factor Restrains the Progression of Au-toimmune Encephalomyelitis in an Experimental Mouse Model. Int. J. Mol. Sci. 2023, 24, 8332. [Google Scholar] [CrossRef] [PubMed]
- Baldassarro, V.A.; Cescatti, M.; Rocco, M.L.; Aloe, L.; Lorenzini, L.; Giardino, L.; Calzà, L. Nerve growth factor promotes differ-entiation and protects the oligodendrocyte precursor cells from in vitro hypoxia/ischemia. Front Neurosci. 2023, 17, 1111170. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, L.; Baldassarro, V.A.; Stanzani, A.; Giardino, L. Nerve Growth Factor: The First Molecule of the Neurotrophin Family. Adv. Exp. Med. Biol. 2021, 1331, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Loy, R.; Taglialatela, G.; Angelucci, L.; Heyer, D.; Perez-Polo, R. Regional CNS uptake of blood-borne nerve growth factor. J. Neurosci. Res. 1994, 39, 339–346. [Google Scholar] [CrossRef]
- Tiberi, A.; Carucci, N.M.; Testa, G.; Rizzi, C.; Pacifico, P.; Borgonovo, G.; Arisi, I.; D’onofrio, M.; Brandi, R.; Gan, W.-B.; et al. Reduced levels of NGF shift astrocytes toward a neurotoxic phenotype. Front. Cell Dev. Biol. 2023, 11, 1165125. [Google Scholar] [CrossRef]
- Rizzi, C.; Tiberi, A.; Giustizieri, M.; Marrone, M.C.; Gobbo, F.; Carucci, N.M.; Meli, G.; Arisi, I.; D’Onofrio, M.; Marinelli, S.; et al. NGF steers microglia toward a neuroprotective phenotype. Glia 2018, 66, 1395–1416. [Google Scholar] [CrossRef]
- Villoslada, P.; Genain, C.P. Role of nerve growth factor and other trophic factors in brain inflammation. Prog. Brain Res. 2004, 146, 403–414. [Google Scholar] [CrossRef]
- Guarnieri, G.; Sarchielli, E.; Comeglio, P.; Herrera-Puerta, E.; Piaceri, I.; Nacmias, B.; Benelli, M.; Kelsey, G.; Maggi, M.; Gallina, P.; et al. Tumor necrosis factor α influences phenotypic plasticity and promotes epigenetic changes in human basal forebrain cholinergic neuroblasts. Int. J. Mol. Sci. 2020, 21, 6128. [Google Scholar] [CrossRef]
- Micera, A.; Properzi, F.; Triaca, V.; Aloe, L. Nerve growth factor antibody exacerbates neuropathological signs of experimental allergic encephalomyelitis in adult Lewis rats. J. Neuroimmunol. 2000, 104, 116–123. [Google Scholar] [CrossRef]
- Villoslada, P.; Hauser, S.L.; Bartke, I.; Unger, J.; Heald, N.; Rosenberg, D.; Cheung, S.W.; Mobley, W.C.; Fisher, S.; Genain, C.P. Human nerve growth factor protects common marmosets against autoimmune encephalomyelitis by switching the balance of t helper cell type 1 and 2 cytokines within the central nervous system. J. Exp. Med. 2000, 191, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Kenarov, P.; Petrov, N.; Voinov, V.; Daskalov, M.; Anaya, F.; Russo, G.; Momchilova, A. A new approach using nanomem-brane—Based therapeutic plasmapheresis for treatment of patients with multiple sclerosis. A case report. J. Pharmacol. Clin. Toxicol. 2014, 2, 1031. [Google Scholar]
- Hannun, Y.A. The sphingomyelin cycle and the second messenger function of ceramide. J. Biol. Chem. 1994, 269, 3125–3128. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y. Functional roles of sphingosine and sphingosine-1-phosphate in regard to membrane sphingolipid signaling pathways. J. Biochem. 1997, 122, 1080–1087. [Google Scholar] [CrossRef]
- Lucaciu, A.; Brunkhorst, R.; Pfeilschifter, J.M.; Pfeilschifter, W.; Subburayalu, J. The S1P-S1PR Axis in Neurological Disor-ders-Insights into Current and Future Therapeutic Perspectives. Cells 2020, 9, 1515. [Google Scholar] [CrossRef]
- Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G.F.; Spiegel, S.; Proia, R.L. Essential Role for Sphingosine Kinases in Neural and Vascular Development. Mol. Cell. Biol. 2005, 25, 11113–11121. [Google Scholar] [CrossRef]
- Riganti, L.; Antonucci, F.; Gabrielli, M.; Prada, I.; Giussani, P.; Viani, P.; Valtorta, F.; Menna, E.; Matteoli, M.; Verderio, C. Sphingosine-1-Phosphate (S1P) Impacts Presynaptic Functions by Regulating Synapsin I Localization in the Presynaptic Compartment. J. Neurosci. 2016, 36, 4624–4634. [Google Scholar] [CrossRef]
- Moore, A.N.; Kampfl, A.W.; Zhao, X.; Hayes, R.L.; Dash, P.K. Sphingosine-1-phosphate induces apoptosis of cultured hippo-campal neurons that requires protein phosphatases and activator protein-1 complexes. Neuroscience 1999, 94, 405–415. [Google Scholar] [CrossRef]
- Giussani, P.C.; Ferraretto, A.; Gravaghi, C.; Bassi, R.; Tettamanti, G.; Riboni, L.; Viani, P. Sphingosine-1-Phosphate and Cal-cium Signaling in Cerebellar Astrocytes and Differentiated Granule Cells. Neurochem. Res. 2006, 32, 27–37. [Google Scholar] [CrossRef]
- Mitroi, D.N.; Deutschmann, A.U.; Raucamp, M.; Karunakaran, I.; Glebov, K.; Hans, M.; Walter, J.; Saba, J.; Gräler, M.; Ehninger, D.; et al. Sphingosine 1-phosphate lyase ablation disrupts presynaptic architecture and function via an ubiquitin- proteasome mediated mechanism. Sci. Rep. 2016, 6, 37064. [Google Scholar] [CrossRef]
- Obinata, H.; Hla, T. Sphingosine 1-phosphate and inflammation. Int. Immunol. 2019, 31, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Jana, A.; Pahan, K. Sphingolipids in multiple sclerosis. Neuromolecular Med. 2010, 12, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Kułakowska, A.; Żendzian-Piotrowska, M.; Baranowski, M.; Konończuk, T.; Drozdowski, W.; Górski, J.; Bucki, R. Intrathecal increase of sphingosine 1-phosphate at early stage multiple sclerosis. Neurosci. Lett. 2010, 477, 149–152. [Google Scholar] [CrossRef]
- Hopkin, S.J.; Lewis, J.W.; Krautter, F.; Chimen, M.; McGettrick, H.M. Triggering the Resolution of Immune Mediated Inflammatory Diseases: Can Targeting Leukocyte Migration Be the Answer? Front. Pharmacol. 2019, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-Phosphate Receptor 1 Signaling in Mammalian Cells. Molecules 2017, 22, 344. [Google Scholar] [CrossRef] [PubMed]
- Momchilova, A.; Pankov, R.; Alexandrov, A.; Markovska, T.; Pankov, S.; Krastev, P.; Staneva, G.; Vassileva, E.; Krastev, N.; Pinkas, A. Sphingolipid Catabolism and Glycerophospholipid Levels Are Altered in Erythrocytes and Plasma from Multiple Sclerosis Patients. Int. J. Mol. Sci. 2022, 23, 7592. [Google Scholar] [CrossRef]
- Bravo, G.; Cedeño, R.R.; Casadevall, M.P.; Ramió-Torrentà, L. Sphingosine-1-Phosphate (S1P) and S1P Signaling Pathway Modulators, from Current Insights to Future Perspectives. Cells 2022, 11, 2058. [Google Scholar] [CrossRef]
- Garris, C.S.; Wu, L.; Acharya, S.; Arac, A.; Blaho, V.A.; Huang, Y.; Moon, B.S.; Axtell, R.C.; Ho, P.P.; Steinberg, G.K.; et al. Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroin-flammation. Nat. Immunol. 2013, 14, 1166–1172. [Google Scholar] [CrossRef]
- Assi, E.; Cazzato, D.; De Palma, C.; Perrotta, C.; Clementi, E.; Cervia, D. Sphingolipids and Brain Resident Macrophages in Neuroinflammation: An Emerging Aspect of Nervous System Pathology. J. Immunol. Res. 2013, 2013, 309302. [Google Scholar] [CrossRef]
- Noda, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J. Neuroimmunol. 2013, 256, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Cohan, S.; Lucassen, E.; Smoot, K.; Brink, J.; Chen, C. Sphingosine-1-Phosphate: Its Pharmacological Regulation and the Treatment of Multiple Sclerosis: A Review Article. Biomedicines 2020, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Proia, R.L.; Hla, T. Emerging biology of sphingosine-1-phosphate: Its role in pathogenesis and therapy. J. Clin. Invest. 2015, 125, 1379–1387. [Google Scholar] [CrossRef]
- Grassi, S.; Mauri, L.; Prioni, S.; Cabitta, L.; Sonnino, S.; Prinetti, A.; Giussani, P. Sphingosine 1-Phosphate Receptors and Met-abolic Enzymes as Druggable Targets for Brain Diseases. Front. Pharmacol. 2019, 10, 807. [Google Scholar] [CrossRef]
- Fischer, I.; Alliod, C.; Martinier, N.; Newcombe, J.; Brana, C.; Pouly, S. Sphingosine Kinase 1 and sphingosine 1-Phosphate receptor 3 are functionally upregulated on astrocytes under pro-inflammatory conditions. PLoS ONE 2011, 6, e23905. [Google Scholar] [CrossRef]
- Dusaban, S.S.; Chun, J.; Rosen, H.; Purcell, N.H.; Brown, J.H. Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J. Neuroinflamm. 2017, 14, 111. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, K.; Hla, T. Vascular and Immunobiology of the Circulatory Sphingosine 1-Phosphate Gradient. Annu. Rev. Physiol. 2017, 79, 67–91. [Google Scholar] [CrossRef]
- Marfia, G.; Navone, S.; Guarnaccia, L.; Campanella, R.; Mondoni, M.; Locatelli, M.; Barassi, A.; Fontana, L.; Palumbo, F.; Garzia, E.; et al. Decreased serum level of sphingosine-1-phosphate: A novel predictor of clinical severity in COVID-19. EMBO Mol. Med. 2021, 13, e13424. [Google Scholar] [CrossRef]
- Connelly-Smith, L.; Alquist, C.R.; Aqui, N.A.; Hofmann, J.C.; Klingel, R.; Onwuemene, O.A.; Patriquin, C.J.; Pham, H.P.; Sanchez, A.P.; Schneiderman, J.; et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice—Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Ninth Special Issue. J. Clin. Apher. 2023, 38, 77–278. [Google Scholar] [CrossRef]
- Redant, S.; De Bels, D.; Ismaili, K.; Honoré, P.M. Membrane-based therapeutic plasma exchange in intensive care. Blood Purif. 2021, 50, 290–297. [Google Scholar] [CrossRef]
- Reeves, H.M.; Winters, J.L. The mechanisms of action of plasma exchange. Br. J. Haematol. 2014, 164, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Oji, S.; Miyamoto, K.; Narita, T.; Kameyama, M.; Matsuo, H. Real-world application of plasmapheresis for neurological disease: Results from the Japan-Plasmapheresis Outcome and Practice Patterns Study. Ther. Apher. Dial. 2023, 27, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Yamakova, Y.; Ilieva, V.A.; Petkov, R.; Yankov, G. Nanomembrane-Based Therapeutic Plasmapheresis after Non-Invasive Ventilation Failure for Treatment of a Patient with Acute Respiratory Distress Syndrome and Myasthenia Gravis: A Case Re-port. Blood Purif. 2019, 48, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, A.; Vassileva, P.; Momchilova, A.; Tsonchev, Z.; Kirilova, Y.; Ivanova, R.; Sapundzhiev, P.; Petkova, D.; Tzoneva, R.; Daskalov, M.; et al. A new approach using nanomembrane-based therapeutic plasmapheresis for treatment of patients with multiple sclerosis and neuromyelitis optica. Comptes Rendus L’academie Bulg. Sci. 2016, 69, 373–384. [Google Scholar]
- Momchilova, A.; Tsonchev, Z.; Hadzhilazova, M.; Tzoneva, R.; Alexandrov, A.; Nikolakov, D.; Ilieva, V.; Pankov, R. Sphin-golipid Metabolism Is Dysregulated in Erythrocytes from Multiple Sclerosis Patient. Comptes Rendus L’academie Bulg. Des Sci. 2020, 73, 426–432. [Google Scholar]
- Sapundzhiev, P.; Momchilova, A.; Vassileva, P.; Kirilova, Y.; Ivanova, R.; Bozhilova, M.; Orozova, M.; Staneva, G.; Krastev, P.; Pankov, R.; et al. Plasmapheresis Affects Ophthalmological Parameters and Oxidative Stress in Patients with Multiple Sclerosis and Neuromyelitis Optica. Arch. Biomed. Eng. Biotechnol. 2021, 5, 000617. [Google Scholar]
- Tenchov, B.; Koynova, R.; Antonova, B.; Zaharinova, S.; Abarova, S.; Tsonchev, Z.; Komsa-Penkova, R.; Momchilova, A. Blood plasma thermal behavior and protein oxidation as indicators of multiple sclerosis clinical status and plasma exchange therapy progression. Thermochim. Acta 2019, 671, 193–199. [Google Scholar] [CrossRef]
- Tsonchev, Z.; Alexandrov, A.; Momchilova, A.; Pankov, R.; Orozova, M.; Georgieva, R.; Georgiev, S.; Alexandrov, S.; Voinov, V.; Anaya, F.; et al. Therapeutic Apheresis with Nanotechnology Membrane for Human Diseases; Bulgarian Academy of Science Prof. Marin Drinov Publishing House: Sofia, Bulgaria, 2020; Volume 2, pp. 1–163. [Google Scholar]
- Tonev, D.; Georgieva, R.; Vavrek, E. Our Clinical Experience in the Treatment of Myasthenia Gravis Acute Exacerbations with a Novel Nanomembrane-Based Therapeutic Plasma Exchange Technology. J. Clin. Med. 2022, 11, 4021. [Google Scholar] [CrossRef]
- Escolar, G.; Páez, A.; Cid, J. Conventional Therapeutic Plasma Exchange Versus Low Volume Plasma Exchange in Chronic Pathologies: Potential Benefit in Alzheimer’s Disease. Plasmatology 2022, 16, 26348535221131685. [Google Scholar] [CrossRef]
- Klingele, M.; Allmendinger, C.; Thieme, S.; Baerens, L.; Fliser, D.; Jan, B. Therapeutic apheresis within immune-mediated neurological disorders: Dosing and its effectiveness. Sci. Rep. 2020, 10, 7925. [Google Scholar] [CrossRef]
- Dorst, J.; Fillies, F.; Dreyhaupt, J.; Senel, M.; Tumani, H. Safety and Tolerability of Plasma Exchange and Immunoadsorption in Neuroinflammatory Diseases. J. Clin. Med. 2020, 9, 2874. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Connelly-Smith, L.; Aqui, N.; Balogun, R.A.; Klingel, R.; Meyer, E.; Pham, H.P.; Schneiderman, J.; Witt, V.; Wu, Y.; et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice—Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Eighth Special Issue. J. Clin. Apher. 2019, 34, 171–354. [Google Scholar] [CrossRef]
- Strasser, E. Principles of Therapeutic Apheresis in Neurological Disease. Transfus. Med. Hemotherapy 2023, 50, 88–97. [Google Scholar] [CrossRef]
- Chang, X.; Gorbet, M. The effect of shear on in vitro platelet and leukocyte material-induced activation. J. Biomater. Appl. 2013, 28, 407–415. [Google Scholar] [CrossRef]
- Yeh, J.H.; Chien, P.J.; Hsueh, Y.M.; Shih, C.M.; Chiu, H.C. Changes in the lymphocyte subset after double-filtration plasmapheresis. Am. J. Clin. Pathol. 2007, 128, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Adamson, R.H.; Clark, J.F.; Radeva, M.; Kheirolomoom, A.; Ferrara, K.W.; Curry, F.E. Albumin modulates S1P delivery from red blood cells in perfused microvessels: Mechanism of the protein effect. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1011–H1017. [Google Scholar] [CrossRef] [PubMed]
- Hardersen, R.; Enebakk, T.; Christiansen, D.; Ludviksen, J.K.; Mollnes, T.E.; Lappegård, K.T.; Hovland, A. Comparison of cytokine changes in three different lipoprotein apheresis systems in an ex vivo whole blood model. J. Clin. Apher. 2020, 35, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, A.; Momchilova, A.; Orozova, M.; Alexandrov, S.; Krastev, P.; Stanev, G.; Nikolova, B.; Tsonchev, Z. Therapeutic Apheresis; Bulgarian Academy of Science Prof. Marin Drinov Publishing House: Sofia, Bulgaria, 2022; pp. 1–120. [Google Scholar]
- Dixit, D.; Okuniewska, M.; Schwab, S.R. Secrets and lyase: Control of sphingosine 1-phosphate distribution. Immunol. Rev. 2019, 289, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Pyne, S.; Rakhit, S.; Conway, A.M.; McKie, A.; Darroch, P.; Tate, R.; Pyne, N. Extracellular actions of sphingosine I-phosphate through endothelial differentiation gene products in mammalian cells: Role in regulating proliferation and apoptosis. Biochem. Soc. Trans. 1999, 27, 404–409. [Google Scholar] [CrossRef]
- Ghasemi, R.; Dargahi, L.; Ahmadiani, A. Integrated sphingosine-1 phosphate signaling in the central nervous system: From physiological equilibrium to pathological damage. Pharmacol. Res. 2016, 104, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Petrou, P.; Ben-Hur, T.; Vaknin-Dembinsky, A.; Abramsky, O.; Karussis, D. Clinical Efficacy of Plasma-Exchange in Patients with Progressive forms of Multiple Sclerosis and NMO-Spectrum Disease. J. Mult. Scler. 2016, 3, 1–6. [Google Scholar] [CrossRef]
- Jacob, S.; Mazibrada, G.; Irani, S.R.; Jacob, A.; Yudina, A. The Role of Plasma Exchange in the Treatment of Refractory Au-toimmune Neurological Diseases: A Narrative Review. J. Neuroimmune Pharmacol. 2021, 16, 806–817. [Google Scholar] [CrossRef] [PubMed]
- Cortese, I.; Chaudhry, V.; So, Y.T.; Cantor, F.; Cornblath, D.R.; Rae-Grant, A. Evidence-based guideline update: Plasmapheresis in neurologic disorders: Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2011, 76, 294–300. [Google Scholar] [CrossRef]
- Lipphardt, M.; Mühlhausen, J.; Kitze, B.; Heigl, F.; Mauch, E.; Helms, J.; Müller, G.A.; Koziolek, M.J. Immunoadsorption or plasma exchange in steroid-refractory multiple sclerosis and neuromyelitis optica. J. Clin. Apher. 2019, 34, 381–391. [Google Scholar] [CrossRef]
- Blechinger, S.; Ehler, J.; Bsteh, G.; Winkelmann, A.; Leutmezer, F.; Meister, S.; Santer, A.; Hecker, M.; Berger, T.; Rommer, P.; et al. Therapeutic plasma exchange in steroid-refractory multiple sclerosis relapses. A retrospective two-center study. Ther. Adv. Neurol. Disord. 2021, 14, 1756286420975642. [Google Scholar] [CrossRef]
- Meca-Lallana, J.E.; Hernández-Clares, R.; León-Hernández, A.; Genovés Aleixandre, A.; Cacho Pérez, M.; Martín-Fernández, J.J. Plasma exchange for steroidrefractory relapses in multiple sclerosis: An observational, MRI pilot study. Clin. Ther. 2013, 35, 474–485. [Google Scholar] [CrossRef]
- Bunganic, R.; Blahutova, S.; Revendova, K.; Zapletalova, O.; Hradilek, P.; Hrdlickova, R.; Ganesh, A.; Cermakova, Z.; Bar, M.; Volny, O. Therapeutic plasma exchange in multiple sclerosis patients with an aggressive relapse: An observational analysis in a high-volume center. Sci. Rep. 2022, 12, 18374. [Google Scholar] [CrossRef]
- Kenarov, P.; Momchilova, A.; Anaya, F.; Voynov, V.; Alexandrov, A.; Tsonchev, Z.; Daskalov, M. Therapeutic Apheresis with Nanotechnology Membrane for Human Diseases; St. Kliment Ohridski University Press: Sofia, Bulgaria, 2014; Volume 1, pp. 1–155. [Google Scholar]
- Kolev, O.I.; Kenarov, P. Vestibular and ocular motor function prior to and after therapeutic apheresis with small plasmafilter in multiple sclerosis. J. Clin. Apher. 2016, 31, 470–472. [Google Scholar] [CrossRef]
- Das, J.; Chauhan, V.D.; Mills, D.; Johal, N.J.; Tan, M.; Matthews, R.; Keh, R.; Lilleker, J.B.; Gosal, D.; Sharaf, N. Therapeutic plasma exchange in neurological dis-orders: Experience from a tertiary neuroscience centre. Transfus. Apher. Sci. 2019, 58, 102654. [Google Scholar] [CrossRef]
- Podbielska, M.; Ariga, T.; Pokryszko-Dragan, A. Sphingolipid Players in Multiple Sclerosis: Their Influence on the Initiation and Course of the Disease. Int. J. Mol. Sci. 2022, 23, 5330. [Google Scholar] [CrossRef] [PubMed]
- Collongues, N.; Becker, G.; Jolivel, V.; Ayme-Dietrich, E.; de Seze, J.; Binamé, F.; Patte-Mensah, C.; Monassier, L.; Mensah-Nyagan, A.G. A Narrative Review on Axonal Neuroprotection in Multiple Sclerosis. Neurol. Ther. 2022, 11, 981–1042. [Google Scholar] [CrossRef]
- Krämer, J.; Wiendl, H. What Have Failed, Interrupted, and Withdrawn Antibody Therapies in Multiple Sclerosis Taught Us? Neurotherapeutics 2022, 19, 785–807. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.A.; Denkova, M.; Ramanathan, S.; Dale, R.C.; Brilot, F. Pathogenesis of autoimmune demyelination: From multiple scle-rosis to neuromyelitis optica spectrum disorders and myelin oligodendrocyte glycoprotein antibody-associated disease. Clin. Transl. Immunol. 2021, 10, e1316. [Google Scholar] [CrossRef] [PubMed]
- Barizzone, N.; Leone, M.; Pizzino, A.; Kockum, I.; MultipleMS Consortium; Martinelli-Boneschi, F.; D’alfonso, S. A Scoping Review on Body Fluid Biomarkers for Prognosis and Disease Activity in Patients with Multiple Sclerosis. J. Pers. Med. 2022, 12, 1430. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fournier, M.; Lacruz, L.; Nozal, P.; Chico, J.L.; Barranco, A.T.; Otero-Ortega, L.; Corral, I.; Carrasco, A. The study of neural antibodies in neurology: A practical summary. Front. Immunol. 2022, 13, 1043723. [Google Scholar] [CrossRef]
- David, S.; Russell, L.; Castro, P.; van de Louw, A.; Zafrani, L.; Pirani, T.; Nielsen, N.D.; Mariotte, E.; Ferreyro, B.L.; Kielstein, J.T.; et al. Research priorities for therapeutic plasma exchange in critically ill patients. Intensiv. Care Med. Exp. 2023, 11, 26. [Google Scholar] [CrossRef]


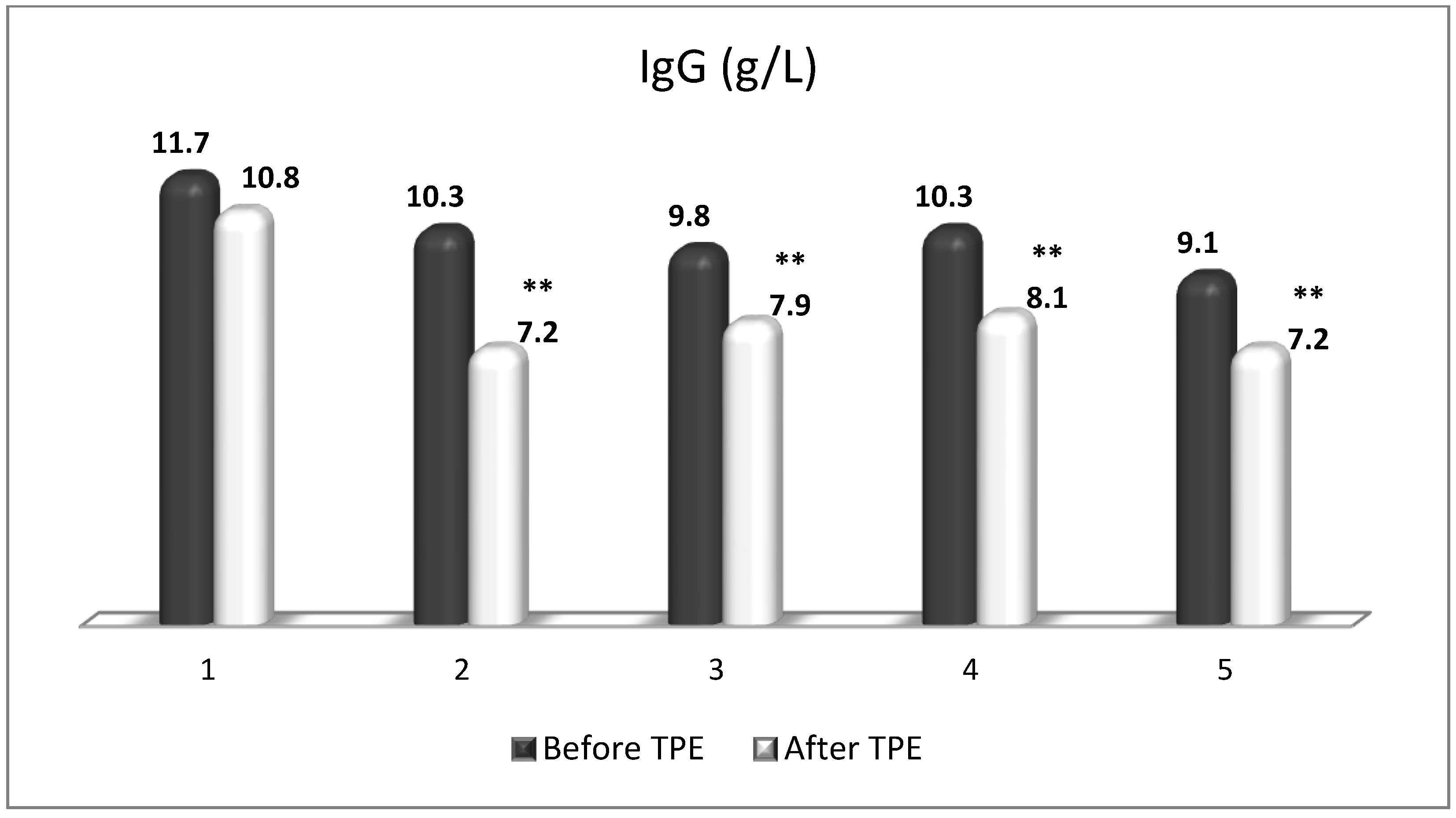
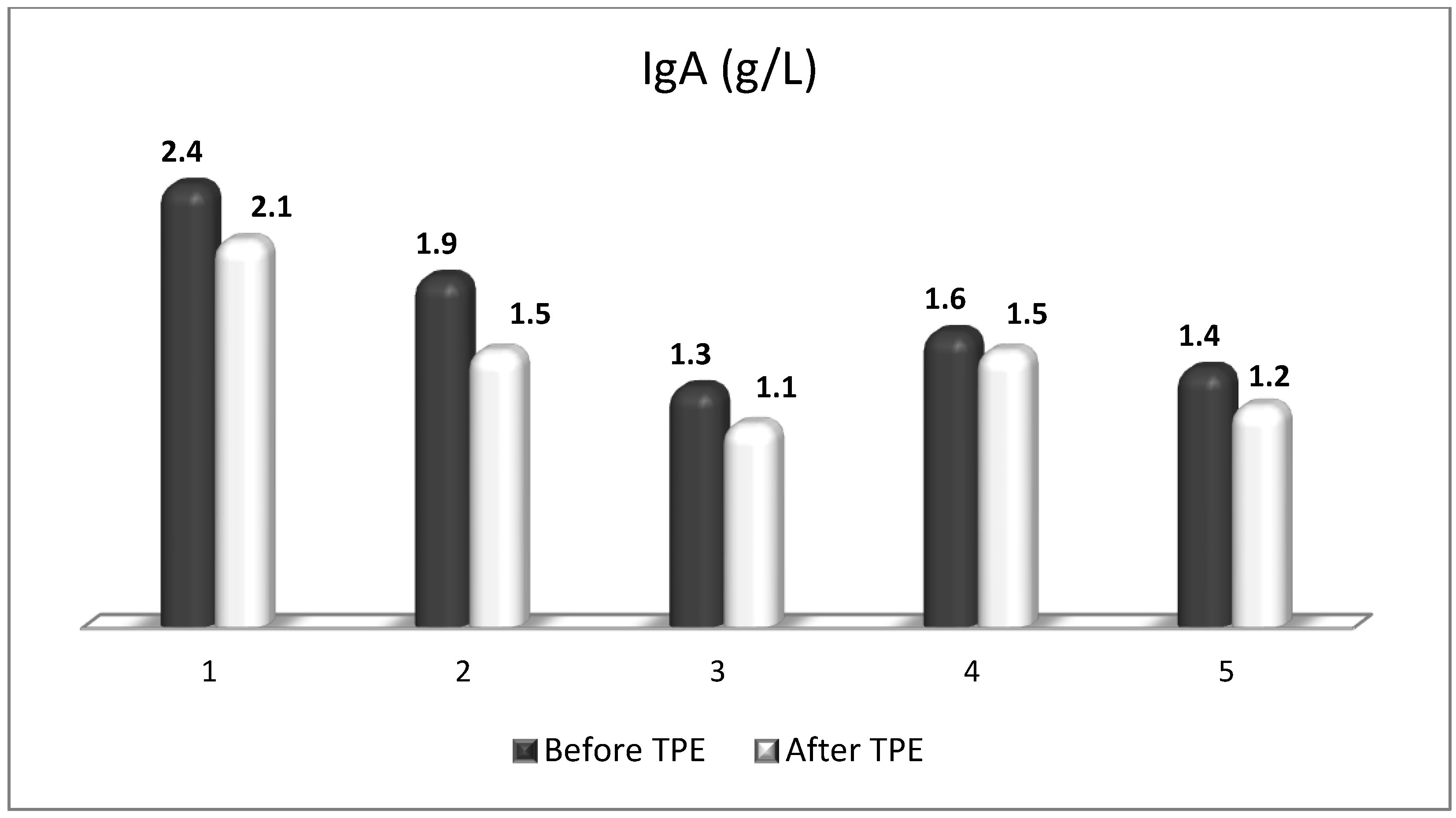
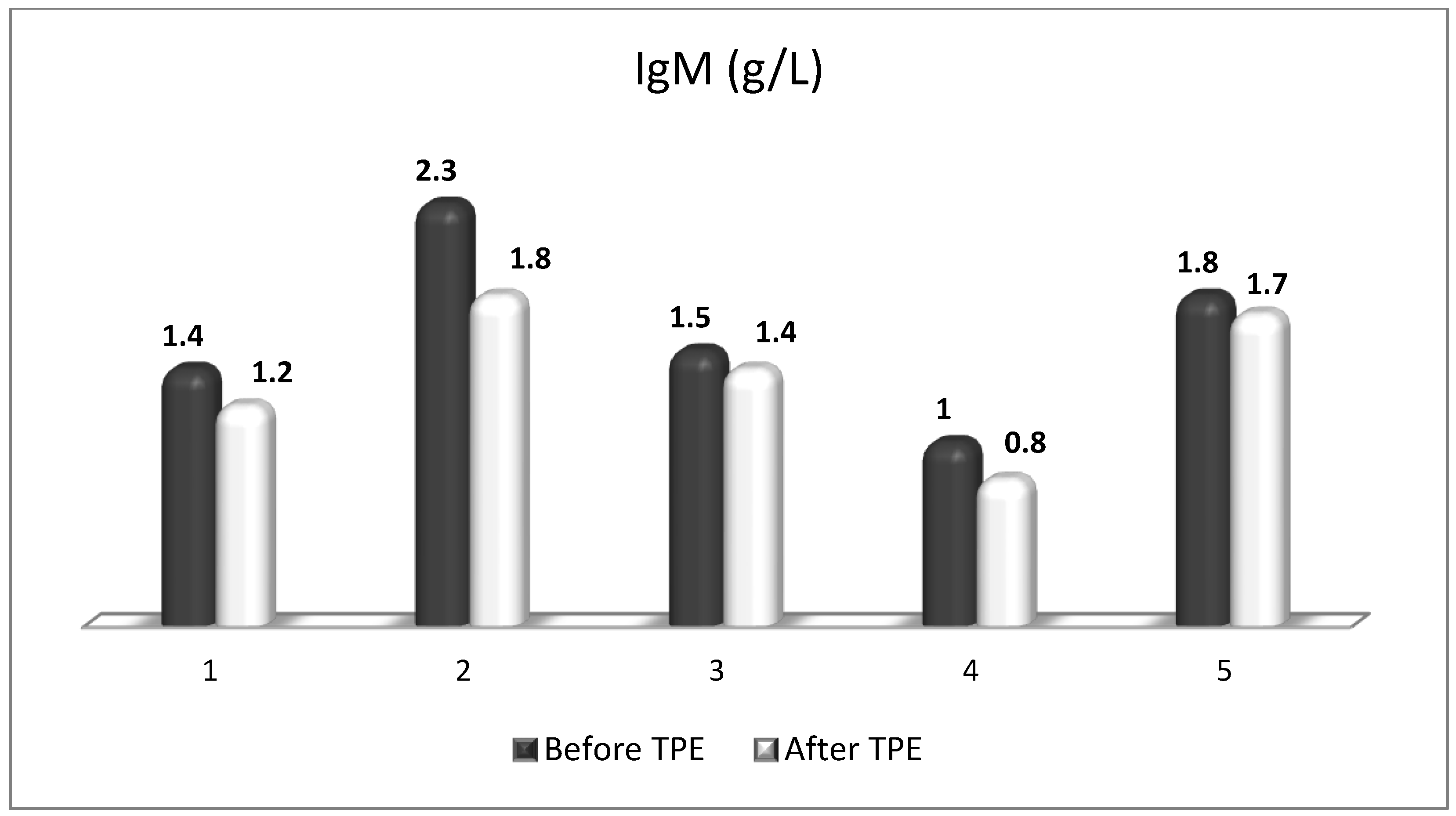

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular Source | Target Receptor | Target Cell | |
|---|---|---|---|
| β-NGF (mature) | mast cells, monocytes, macrophages, eosinophils, granulocytes, basophiles, | TrkA | T cells, macrophages |
| T cells, B cells | p75NTR and TrkA | B cells, mast cells |
| Target | Effect |
|---|---|
| Immune system | Modulation of immune system via enhanced sympathetic innervation of lymph nodes with indirect effect decreasing CD4+ and CD8+ proliferation |
| BBB | Maintenance of BBB integrity |
| Lymphocytes | Switch to the anti-inflammatory phenotype by avoiding cytotoxicity and inducing immunosuppressive cytokines (IL-10, TGF-β) |
| Macrophages/ microglia | Decrease in antigen presentation by macrophage/microglia by reducing the expression of major histocompatibility complex (MHC) molecules; Shift from pro-inflammatory M1 to anti-inflammatory M2 phenotype |
| Astrocytes | Inactivation of toxic astrocytes mediators; Attenuation of astrogliosis, shift from pro-inflammatory A1 to anti-inflammatory A2 phenotype |
| OLs | Promotion of proliferation, migration, maturation, and survival of OPCs |
| Neurons | Promotion of axonal survival during inflammation; Upregulation of axonal LINGO1 with inhibition of axonal receptivity to oligodendrocyte myelination |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonev, D.; Momchilova, A. Therapeutic Plasma Exchange and Multiple Sclerosis Dysregulations: Focus on the Removal of Pathogenic Circulatory Factors and Altering Nerve Growth Factor and Sphingosine-1-Phosphate Plasma Levels. Curr. Issues Mol. Biol. 2023, 45, 7749-7774. https://doi.org/10.3390/cimb45100489
Tonev D, Momchilova A. Therapeutic Plasma Exchange and Multiple Sclerosis Dysregulations: Focus on the Removal of Pathogenic Circulatory Factors and Altering Nerve Growth Factor and Sphingosine-1-Phosphate Plasma Levels. Current Issues in Molecular Biology. 2023; 45(10):7749-7774. https://doi.org/10.3390/cimb45100489
Chicago/Turabian StyleTonev, Dimitar, and Albena Momchilova. 2023. "Therapeutic Plasma Exchange and Multiple Sclerosis Dysregulations: Focus on the Removal of Pathogenic Circulatory Factors and Altering Nerve Growth Factor and Sphingosine-1-Phosphate Plasma Levels" Current Issues in Molecular Biology 45, no. 10: 7749-7774. https://doi.org/10.3390/cimb45100489
APA StyleTonev, D., & Momchilova, A. (2023). Therapeutic Plasma Exchange and Multiple Sclerosis Dysregulations: Focus on the Removal of Pathogenic Circulatory Factors and Altering Nerve Growth Factor and Sphingosine-1-Phosphate Plasma Levels. Current Issues in Molecular Biology, 45(10), 7749-7774. https://doi.org/10.3390/cimb45100489