
Investigating Carvedilol’s Repurposing for the Treatment of Non-Small Cell Lung Cancer via Aldehyde Dehydrogenase Activity Modulation in the Presence of β-Adrenergic Agonists

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
2.1. Measuring the Basal Level of ALDH in A549 and H1299 Cell Lines
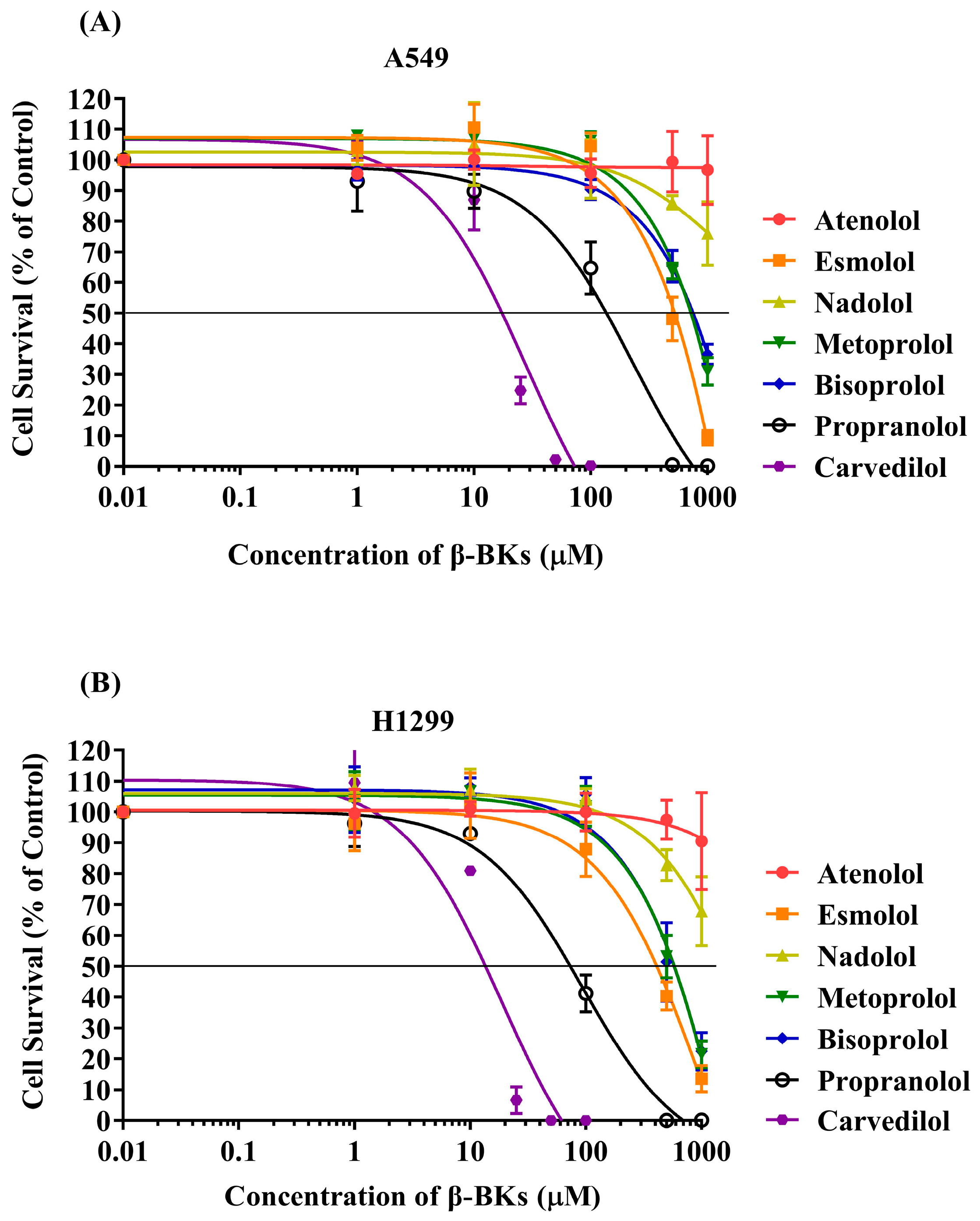
2.2. Measuring Cytotoxicity of Drugs in A549 and H1299 Cell Lines
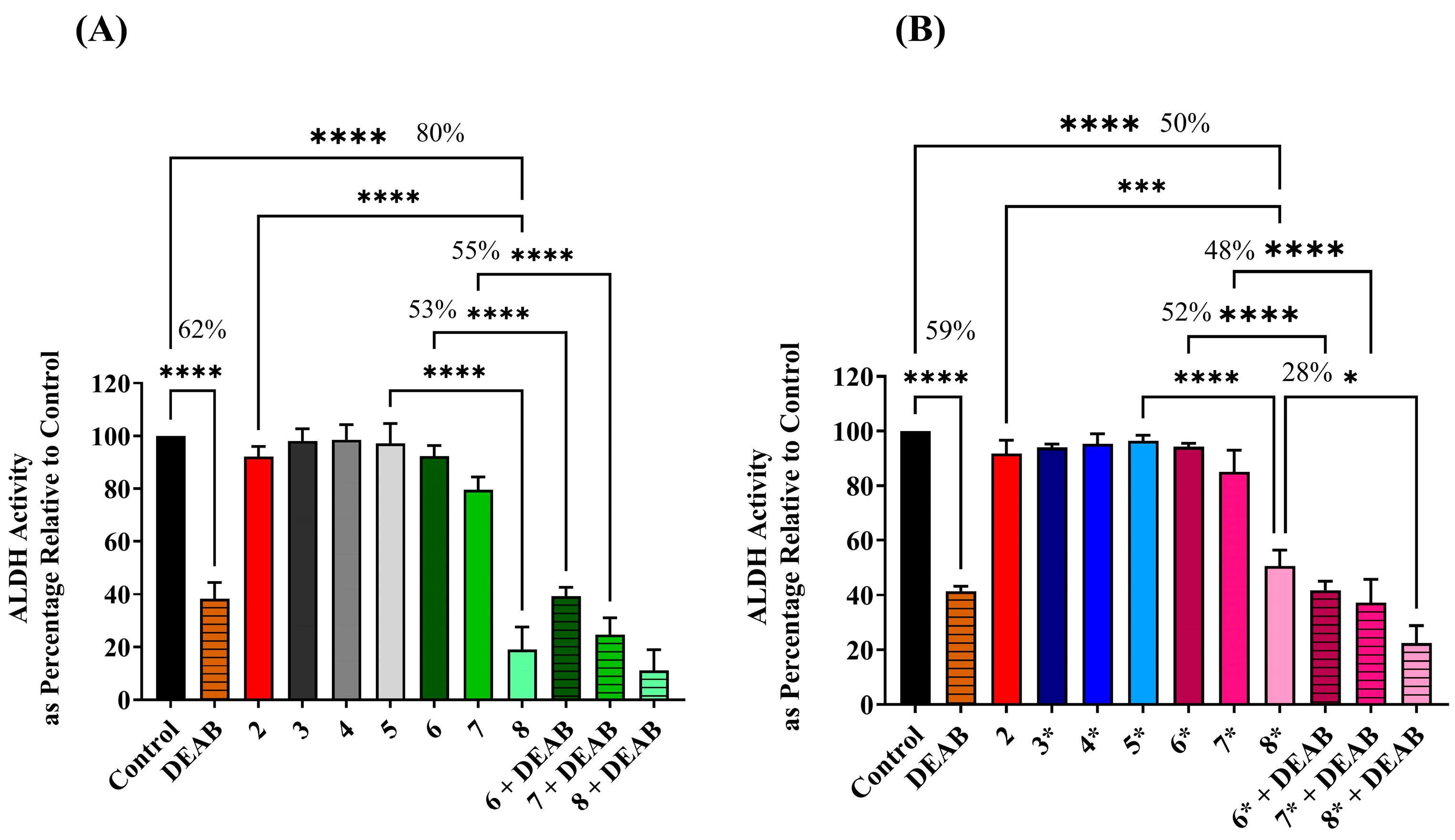
2.3. Assessing ALDH Activity Level following Exposure to Different Treatment Conditions
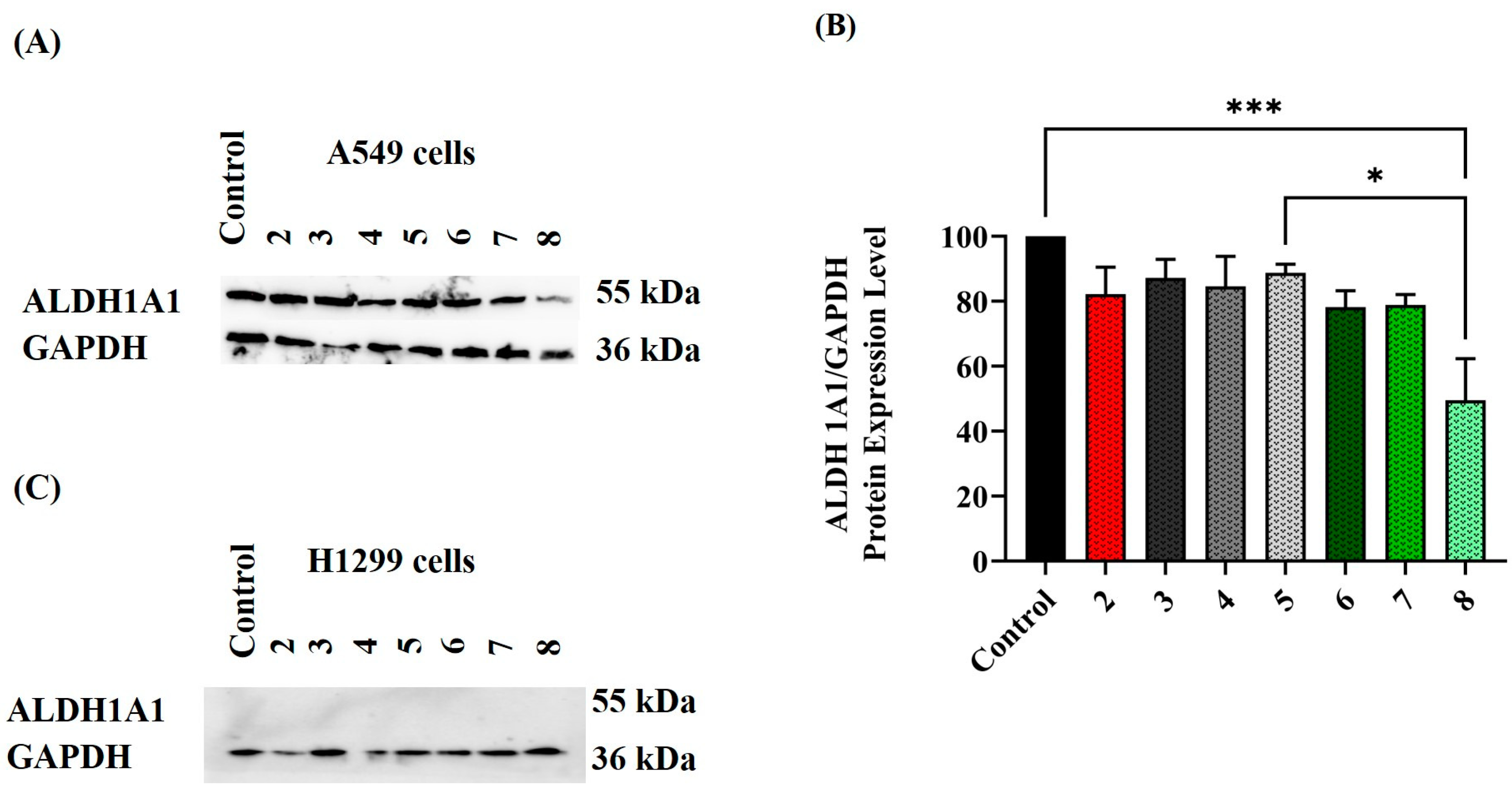
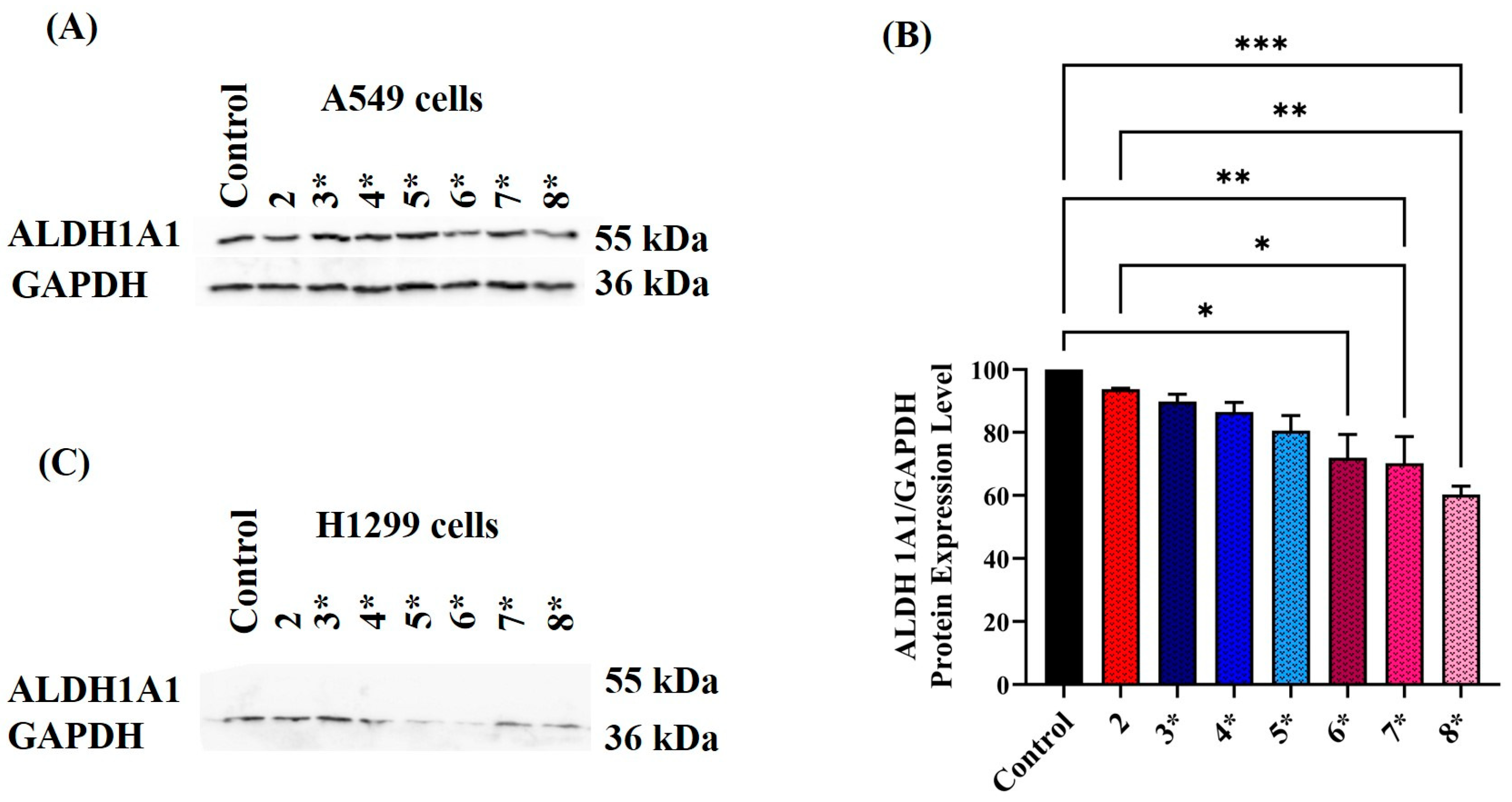
2.4. Assessing ALDH Protein Expression Level following Exposure to Various Treatment Conditions
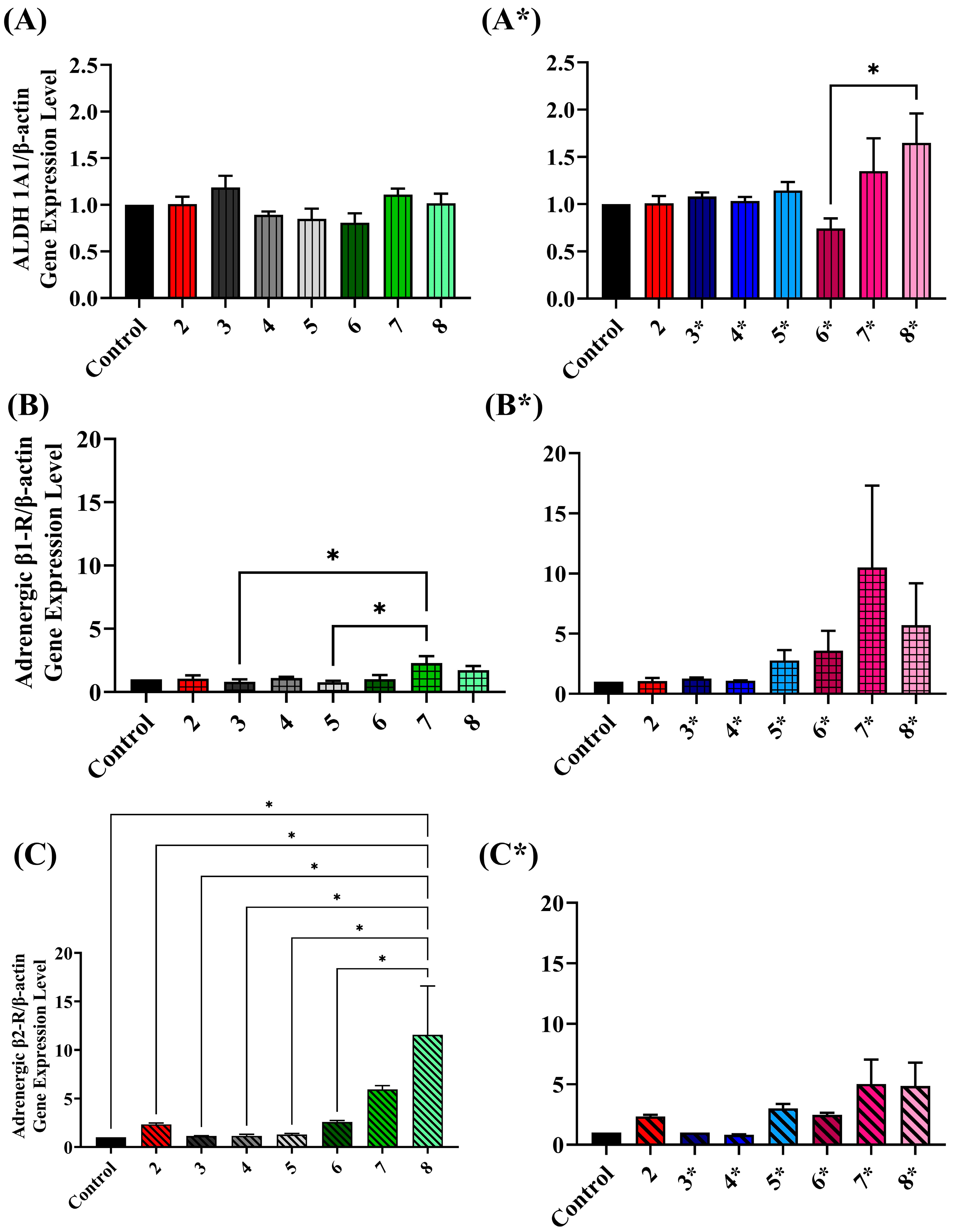
2.5. Assessing ALDH Gene Expression Level upon Exposure to Various Treatment Conditions
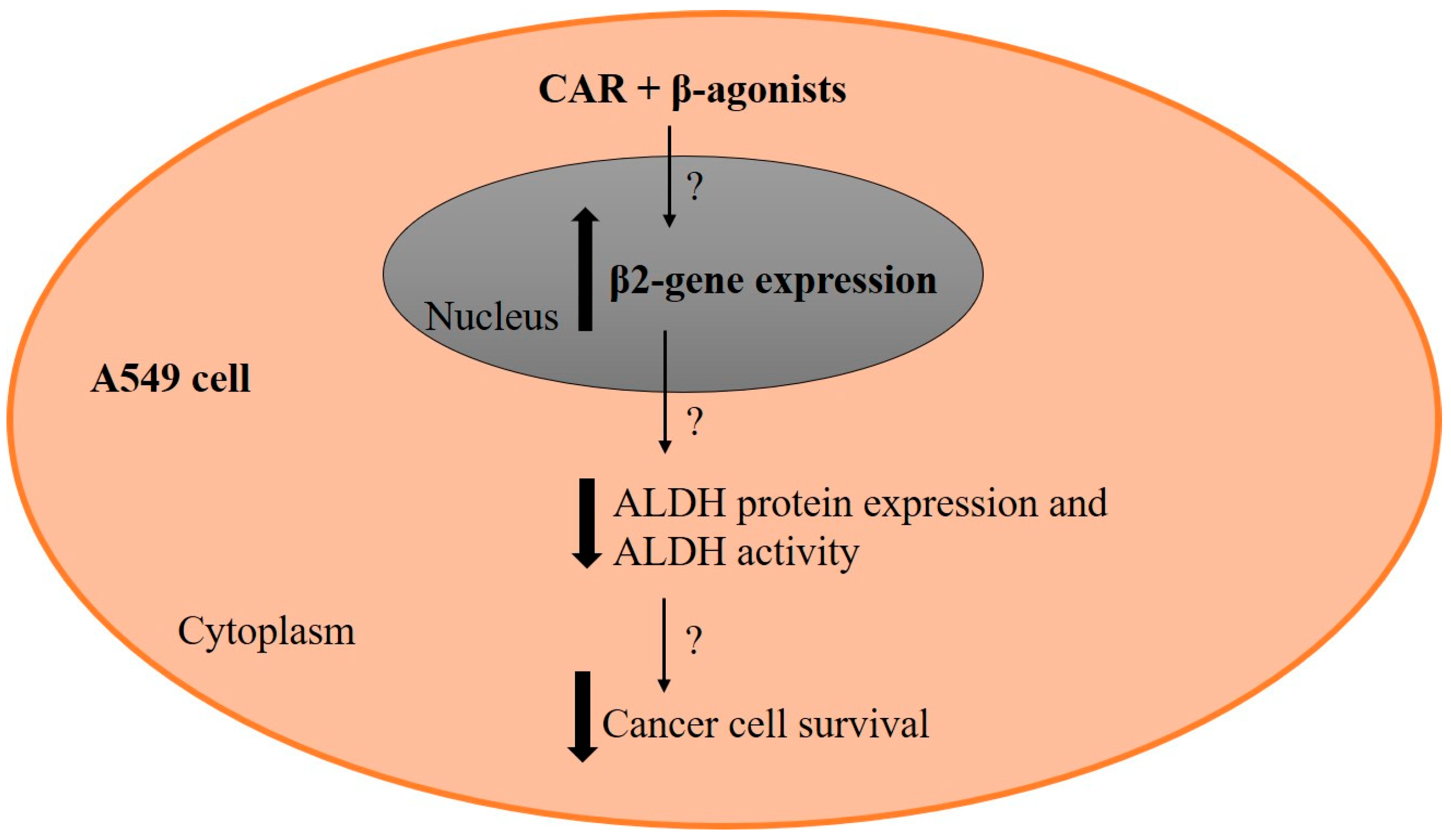
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Cell Culture
4.2.2. Sample Preparation for Measuring ALDH Levels
4.2.3. Quantification of Total Protein Concentration in Cell Lysate
4.2.4. NADH Fluorescence Spectrophotometric Activity Assay
4.2.5. Western Blot
4.2.6. MTT Cytotoxicity Assay
4.2.7. Gene Expression Analysis
4.2.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Lung Cancer. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/lung-cancer#:~:text=Lung%20cancer%20is%20the%20leading,when%20treatment%20options%20are%20limited (accessed on 29 August 2023).
- Zheng, M. Classification and Pathology of Lung Cancer. Surg. Oncol. Clin. N. Am. 2016, 25, 447–468. [Google Scholar] [CrossRef] [PubMed]
- Sosa Iglesias, V.; Giuranno, L.; Dubois, L.J.; Theys, J.; Vooijs, M. Drug Resistance in Non-Small Cell Lung Cancer: A Potential for NOTCH Targeting? Front. Oncol. 2018, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Doumat, G.; Daher, D.; Zerdan, M.B.; Nasra, N.; Bahmad, H.F.; Recine, M.; Poppiti, R. Drug Repurposing in Non-Small Cell Lung Carcinoma: Old Solutions for New Problems. Curr. Oncol. 2023, 30, 704–719. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Kim, S.Y.; Cheng, H. Update 2020: Management of Non-Small Cell Lung Cancer. Lung 2020, 198, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.R.; Liao, P.F.; Wu, T.W. Efficacy and safety of anaplastic lymphoma kinase inhibitors for non-small cell lung cancer: A systematic review and network meta-analysis. Thorac. Cancer 2023, 14, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Mohsenzadegan, M.; Peng, R.-W.; Roudi, R. Dendritic cell/cytokine-induced killer cell-based immunotherapy in lung cancer: What we know and future landscape. J. Cell. Physiol. 2020, 235, 74–86. [Google Scholar] [CrossRef]
- Lu, C.; Li, X.; Ren, Y.; Zhang, X. Disulfiram: A novel repurposed drug for cancer therapy. Cancer Chemother. Pharmacol. 2021, 87, 159–172. [Google Scholar] [CrossRef]
- Cole, S.W.; Nagaraja, A.S.; Lutgendorf, S.K.; Green, P.A.; Sood, A.K. Sympathetic nervous system regulation of the tumour microenvironment. Nat. Rev. Cancer 2015, 15, 563–572. [Google Scholar] [CrossRef]
- Kamiya, A.; Hayama, Y.; Kato, S.; Shimomura, A.; Shimomura, T.; Irie, K.; Kaneko, R.; Yanagawa, Y.; Kobayashi, K.; Ochiya, T. Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat. Neurosci. 2019, 22, 1289–1305. [Google Scholar] [CrossRef]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell 2018, 33, 75–90.e7. [Google Scholar] [CrossRef]
- Chida, Y.; Hamer, M.; Wardle, J.; Steptoe, A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat. Clin. Pract. Oncol. 2008, 5, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Liu, D.; Duan, H.; Qian, L.; Wang, L.; Niu, L.; Zhang, H.; Yong, Z.; Gong, Z.; Song, L.; et al. The β2-adrenergic receptor and Her2 comprise a positive feedback loop in human breast cancer cells. Breast Cancer Res. Treat. 2011, 125, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Hein, L.; Kobilka, B.K. Adrenergic receptor signal transduction and regulation. Neuropharmacology 1995, 34, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.W.; Sood, A.K. Molecular pathways: Beta-adrenergic signaling in cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Armaiz-Pena, G.N.; Allen, J.K.; Cruz, A.; Stone, R.L.; Nick, A.M.; Lin, Y.G.; Han, L.Y.; Mangala, L.S.; Villares, G.J.; Vivas-Mejia, P.; et al. Src activation by β-adrenoreceptors is a key switch for tumour metastasis. Nat. Commun. 2013, 4, 1403. [Google Scholar] [CrossRef]
- Yang, E.V.; Eubank, T.D. The impact of adrenergic signaling in skin cancer progression: Possible repurposing of β-blockers for treatment of skin cancer. Cancer Biomark. 2013, 13, 155–160. [Google Scholar] [CrossRef]
- Chen, M.; Liang, S.; Shahid, A.; Andresen, B.T.; Huang, Y. Prevention of Skin Carcinogenesis by the β-Blocker Carvedilol. Cancer Prev. Res. 2015, 8, 27–36. [Google Scholar] [CrossRef]
- Shahid, A.; Chen, M.; Lin, C.; Andresen, B.T.; Parsa, C.; Orlando, R.; Huang, Y. The β-Blocker Carvedilol Prevents Benzo(a)pyrene-Induced Lung Toxicity, Inflammation and Carcinogenesis. Cancers 2023, 15, 583. [Google Scholar] [CrossRef]
- Wang, H.M.; Liao, Z.X.; Komaki, R.; Welsh, J.W.; O’Reilly, M.S.; Chang, J.Y.; Zhuang, Y.; Levy, L.B.; Lu, C.; Gomez, D.R. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann. Oncol. 2013, 24, 1312–1319. [Google Scholar] [CrossRef]
- Yamamoto, H.; Hamasaki, T.; Onda, K.; Nojiri, T.; Aragaki, M.; Horie, N.; Sato, N.; Hida, Y. Landiolol, an ultra-short acting beta-1 blocker, for preventing postoperative lung cancer recurrence: Study protocol for a phase III, multicenter randomized trial with two parallel groups of patients. Trials 2019, 20, 715. [Google Scholar] [CrossRef]
- Sidorova, M.; Petrikaitė, V. The Effect of Beta Adrenoreceptor Blockers on Viability and Cell Colony Formation of Non-Small Cell Lung Cancer Cell Lines A549 and H1299. Molecules 2022, 27, 1938. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, A.S.; Sadaoui, N.C.; Lutgendorf, S.K.; Ramondetta, L.M.; Sood, A.K. β-blockers: A new role in cancer chemotherapy? Expert Opin. Investig. Drugs 2013, 22, 1359–1363. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Le, X.; Heymach, J.V. β-Adrenergic Signaling in Lung Cancer: A Potential Role for Beta-Blockers. J. Neuroimmune Pharmacol. 2020, 15, 27–36. [Google Scholar] [PubMed]
- Goldfarb, Y.; Sorski, L.; Benish, M.; Levi, B.; Melamed, R.; Ben-Eliyahu, S. Improving postoperative immune status and resistance to cancer metastasis: A combined perioperative approach of immunostimulation and prevention of excessive surgical stress responses. Ann. Surg. 2011, 253, 798–810. [Google Scholar] [CrossRef]
- Yang, E.V.; Kim, S.J.; Donovan, E.L.; Chen, M.; Gross, A.C.; Webster Marketon, J.I.; Barsky, S.H.; Glaser, R. Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: Implications for stress-related enhancement of tumor progression. Brain Behav. Immun. 2009, 23, 267–275. [Google Scholar] [CrossRef]
- Yue, T.L.; Cheng, H.Y.; Lysko, P.G.; McKenna, P.J.; Feuerstein, R.; Gu, J.L.; Lysko, K.A.; Davis, L.L.; Feuerstein, G. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J. Pharmacol. Exp. Ther. 1992, 263, 92–98. [Google Scholar]
- Dandona, P.; Ghanim, H.; Brooks, D.P. Antioxidant activity of carvedilol in cardiovascular disease. J. Hypertens. 2007, 25, 731–741. [Google Scholar] [CrossRef]
- Lysko, P.G.; Webb, C.L.; Gu, J.L.; Ohlstein, E.H.; Ruffolo, R.R., Jr.; Yue, T.L. A comparison of carvedilol and metoprolol antioxidant activities in vitro. J. Cardiovasc. Pharmacol. 2000, 36, 277–281. [Google Scholar] [CrossRef]
- Yue, T.L.; McKenna, P.J.; Lysko, P.G.; Ruffolo, R.R., Jr.; Feuerstein, G.Z. Carvedilol, a new antihypertensive, prevents oxidation of human low density lipoprotein by macrophages and copper. Atherosclerosis 1992, 97, 209–216. [Google Scholar] [CrossRef]
- Bhat, K.P.; Pezzuto, J.M. Cancer chemopreventive activity of resveratrol. Ann. N. Y. Acad. Sci. 2002, 957, 210–229. [Google Scholar] [CrossRef]
- Schevzov, G.; Kee, A.J.; Wang, B.; Sequeira, V.B.; Hook, J.; Coombes, J.D.; Lucas, C.A.; Stehn, J.R.; Musgrove, E.A.; Cretu, A.; et al. Regulation of cell proliferation by ERK and signal-dependent nuclear translocation of ERK is dependent on Tm5NM1-containing actin filaments. Mol. Biol. Cell 2015, 26, 2475–2490. [Google Scholar] [CrossRef]
- Bryant, J.A.; Finn, R.S.; Slamon, D.J.; Cloughesy, T.F.; Charles, A.C. EGF activates intracellular and intercellular calcium signaling by distinct pathways in tumor cells. Cancer Biol. Ther. 2004, 3, 1243–1249. [Google Scholar] [CrossRef]
- Cleveland, K.H.; Liang, S.; Chang, A.; Huang, K.M.; Chen, S.; Guo, L.; Huang, Y.; Andresen, B.T. Carvedilol inhibits EGF-mediated JB6 P+ colony formation through a mechanism independent of adrenoceptors. PLoS ONE 2019, 14, e0217038. [Google Scholar] [CrossRef]
- Nakamura, K.; Kusano, K.; Nakamura, Y.; Kakishita, M.; Ohta, K.; Nagase, S.; Yamamoto, M.; Miyaji, K.; Saito, H.; Morita, H.; et al. Carvedilol Decreases Elevated Oxidative Stress in Human Failing Myocardium. Circulation 2002, 105, 2867–2871. [Google Scholar] [CrossRef] [PubMed]
- Nagase, I.; Yoshida, T.; Saito, M. Up-regulation of uncoupling proteins by beta-adrenergic stimulation in L6 myotubes. FEBS Lett. 2001, 494, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Guivernau, M.; Quintanilla, M.E.; Tampier, L. Investigations on the ethanol-induced flushing reaction: Effects of propranolol and dipyridamole on acetaldehyde and prostacyclin metabolism. Toxicology 1994, 90, 1–9. [Google Scholar] [CrossRef]
- Moreb, J.S.; Baker, H.V.; Chang, L.J.; Amaya, M.; Lopez, M.C.; Ostmark, B.; Chou, W. ALDH isozymes downregulation affects cell growth, cell motility and gene expression in lung cancer cells. Mol. Cancer 2008, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.A.; Parajuli, B.; Buchman, C.D.; Dria, K.; Hurley, T.D. N,N-diethylaminobenzaldehyde (DEAB) as a substrate and mechanism-based inhibitor for human ALDH isoenzymes. Chem. Biol. Interact. 2015, 234, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.; Brocker, C.; Thompson, D.C.; Black, W.; Vasiliou, K.; Nebert, D.W.; Vasiliou, V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genom. 2011, 5, 283–303. [Google Scholar] [CrossRef]
- Xia, J.; Li, S.; Liu, S.; Zhang, L. Aldehyde dehydrogenase in solid tumors and other diseases: Potential biomarkers and therapeutic targets. MedComm 2023, 4, e195. [Google Scholar] [CrossRef]
- Ibrahim, A.I.M.; Ikhmais, B.; Batlle, E.; AbuHarb, W.K.; Jha, V.; Jaradat, K.T.; Jiménez, R.; Pequerul, R.; Parés, X.; Farrés, J.; et al. Design, Synthesis, Biological Evaluation and In Silico Study of Benzyloxybenzaldehyde Derivatives as Selective ALDH1A3 Inhibitors. Molecules 2021, 26, 5770. [Google Scholar] [CrossRef] [PubMed]
- Marchitti, S.A.; Brocker, C.; Stagos, D.; Vasiliou, V. Non-P450 aldehyde oxidizing enzymes: The aldehyde dehydrogenase superfamily. Expert Opin. Drug Metab. Toxicol. 2008, 4, 697–720. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, J.; Dumas, F.; Lacroix, A.; Bhat, P.V. A novel isoenzyme of aldehyde dehydrogenase specifically involved in the biosynthesis of 9-cis and all-trans retinoic acid. Biochem. J. 1995, 305 Pt 2, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef]
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A.; et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef]
- Visus, C.; Ito, D.; Amoscato, A.; Maciejewska-Franczak, M.; Abdelsalem, A.; Dhir, R.; Shin, D.M.; Donnenberg, V.S.; Whiteside, T.L.; DeLeo, A.B. Identification of human aldehyde dehydrogenase 1 family member A1 as a novel CD8+ T-cell-defined tumor antigen in squamous cell carcinoma of the head and neck. Cancer Res. 2007, 67, 10538–10545. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chen, Y.W.; Hsu, H.S.; Tseng, L.M.; Huang, P.I.; Lu, K.H.; Chen, D.T.; Tai, L.K.; Yung, M.C.; Chang, S.C. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem. Biophys. Res. Commun. 2009, 385, 307–313. [Google Scholar] [CrossRef]
- Li, T.; Su, Y.; Mei, Y.; Leng, Q.; Leng, B.; Liu, Z.; Stass, S.A.; Jiang, F. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Investig. 2010, 90, 234–244. [Google Scholar] [CrossRef]
- Ioannou, M.; Serafimidis, I.; Arnes, L.; Sussel, L.; Singh, S.; Vasiliou, V.; Gavalas, A. ALDH1B1 is a potential stem/progenitor marker for multiple pancreas progenitor pools. Dev. Biol. 2013, 374, 153–163. [Google Scholar] [CrossRef]
- Su, Y.; Qiu, Q.; Zhang, X.; Jiang, Z.; Leng, Q.; Liu, Z.; Stass, S.A.; Jiang, F. Aldehyde dehydrogenase 1 A1-positive cell population is enriched in tumor-initiating cells and associated with progression of bladder cancer. Cancer Epidemiol. Biomark. Prev. 2010, 19, 327–337. [Google Scholar] [CrossRef]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Feng, Y.; Cheng, L. Cellular Reprogramming as a Therapeutic Target in Cancer. Trends Cell Biol. 2019, 29, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Spinola, M.; Dodge, M.; Raso, M.G.; Behrens, C.; Gao, B.; Schuster, K.; Shao, C.; Larsen, J.E.; Sullivan, L.A.; et al. Aldehyde Dehydrogenase Activity Selects for Lung Adenocarcinoma Stem Cells Dependent on Notch Signaling. Cancer Res. 2010, 70, 9937–9948. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, S.H.; Hong, D.; Lee, J.S.; Ahn, H.S.; Ahn, J.H.; Seong, T.W.; Lee, C.H.; Jang, H.; Hong, K.M.; et al. Aldehyde dehydrogenase is used by cancer cells for energy metabolism. Exp. Mol. Med. 2016, 48, e272. [Google Scholar] [CrossRef]
- Moreb, J.S.; Zucali, J.R.; Ostmark, B.; Benson, N.A. Heterogeneity of aldehyde dehydrogenase expression in lung cancer cell lines is revealed by Aldefluor flow cytometry-based assay. Cytom. B Clin. Cytom. 2007, 72, 281–289. [Google Scholar] [CrossRef]
- Ismail, W.H.; Abusara, O.H.; Ikhmais, B.; Abul-Futouh, H.; Sunoqrot, S.; Ibrahim, A.I.M. Design, Synthesis, and Biological Activity of Coniferyl Aldehyde Derivatives as Potential Anticancer and Antioxidant Agents. Jordan J. Pharm. Sci. 2023, 16, 368–380. [Google Scholar] [CrossRef]
- Moreb, J.S.; Ucar, D.; Han, S.; Amory, J.K.; Goldstein, A.S.; Ostmark, B.; Chang, L.J. The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by Aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance. Chem. Biol. Interact. 2012, 195, 52–60. [Google Scholar] [CrossRef]
- Lawal, B.; Kuo, Y.C.; Wu, A.T.; Huang, H.S. Therapeutic potential of EGFR/mTOR/Nf-kb targeting small molecule for the treatment of non-small cell lung cancer. Am. J. Cancer Res. 2023, 13, 2598–2616. [Google Scholar] [PubMed]
- Parajuli, B.; Fishel, M.L.; Hurley, T.D. Selective ALDH3A1 inhibition by benzimidazole analogues increase mafosfamide sensitivity in cancer cells. J. Med. Chem. 2014, 57, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Frishman, W.H. Beta-Adrenergic Blockers. Circulation 2003, 107, e117–e119. [Google Scholar] [CrossRef] [PubMed]
- Okudela, K.; Woo, T.; Mitsui, H.; Suzuki, T.; Tajiri, M.; Sakuma, Y.; Miyagi, Y.; Tateishi, Y.; Umeda, S.; Masuda, M.; et al. Downregulation of ALDH1A1 expression in non-small cell lung carcinomas–its clinicopathologic and biological significance. Int. J. Clin. Exp. Pathol. 2013, 6, 1. [Google Scholar]
- Coelho, M.; Imperatori, A.; Chiaravalli, A.M.; Franzi, F.; Castiglioni, M.; Rasini, E.; Luini, A.; Legnaro, M.; Marino, F.; Ribeiro, L.; et al. Beta1- and Beta2-Adrenoceptors Expression Patterns in Human Non-small Cell Lung Cancer: Relationship with Cancer Histology. J. Neuroimmune Pharmacol. 2019, 14, 697–708. [Google Scholar] [CrossRef]
- Kim, D.; Pauer, S.H.; Yong, H.M.; An, S.S.; Liggett, S.B. β2-Adrenergic Receptors Chaperone Trapped Bitter Taste Receptor 14 to the Cell Surface as a Heterodimer and Exert Unidirectional Desensitization of Taste Receptor Function. J. Biol. Chem. 2016, 291, 17616–17628. [Google Scholar] [CrossRef]
- Smith, C.; Teitler, M. Beta-blocker selectivity at cloned human beta 1- and beta 2-adrenergic receptors. Cardiovasc. Drugs Ther. 1999, 13, 123–126. [Google Scholar] [CrossRef]
- Molenaar, P.; Christ, T.; Ravens, U.; Kaumann, A. Carvedilol blocks β2- more than β1-adrenoceptors in human heart. Cardiovasc. Res. 2006, 69, 128–139. [Google Scholar] [CrossRef]
- Ma, Z.; Liu, X.; Zhang, Q.; Yu, Z.; Gao, D. Carvedilol suppresses malignant proliferation of mammary epithelial cells through inhibition of the ROS-mediated PI3K/AKT signaling pathway. Oncol. Rep. 2019, 41, 811–818. [Google Scholar] [CrossRef]
- Peixoto, R.; Pereira, M.D.L.; Oliveira, M. Beta-Blockers and Cancer: Where Are We? Pharmaceuticals 2020, 13, 105. [Google Scholar] [CrossRef]
- Ji, Y.; Chen, S.; Xiao, X.; Zheng, S.; Li, K. β-blockers: A novel class of antitumor agents. OncoTargets Ther. 2012, 5, 391–401. [Google Scholar] [CrossRef]
- Rodríguez-Zavala, J.S.; Calleja, L.F.; Moreno-Sánchez, R.; Yoval-Sánchez, B. Role of Aldehyde Dehydrogenases in Physiopathological Processes. Chem. Res. Toxicol. 2019, 32, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.; Kwon, M.; Yu, J.S.; Jang, M.; Kim, G.-H.; Kim, K.H.; Ko, S.-K.; Ahn, J.S. Ent-Peniciherqueinone Suppresses Acetaldehyde-Induced Cytotoxicity and Oxidative Stress by Inducing ALDH and Suppressing MAPK Signaling. Pharmaceutics 2020, 12, 1229. [Google Scholar] [CrossRef]
- Yen, H.C.; Xu, Q.; Chou, D.M.; Zhao, Z.; Elledge, S.J. Global protein stability profiling in mammalian cells. Science 2008, 322, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Liu, Y.Q.; Sun, Z.H.; Zhangsun, D.T.; Luo, S.L. Identification of nicotinic acetylcholine receptor subunits in different lung cancer cell lines and the inhibitory effect of alpha-conotoxin TxID on lung cancer cell growth. Eur. J. Pharmacol. 2019, 865, 172674. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-S.; Lin, W.-S.; Lin, C.-L.; Kao, C.-H. Carvedilol use is associated with reduced cancer risk: A nationwide population-based cohort study. Int. J. Cardiol. 2015, 184, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.M.; Liang, S.; Yeung, S.; Oiyemhonlan, E.; Cleveland, K.H.; Parsa, C.; Orlando, R.; Meyskens, F.L.; Andresen, B.T., Jr.; Huang, Y. Topically Applied Carvedilol Attenuates Solar Ultraviolet Radiation Induced Skin Carcinogenesis. Cancer Prev. Res. 2017, 10, 598–606. [Google Scholar] [CrossRef]
- Montoya, A.; Varela-Ramirez, A.; Dickerson, E.; Pasquier, E.; Torabi, A.; Aguilera, R.; Nahleh, Z.; Bryan, B. The beta adrenergic receptor antagonist propranolol alters mitogenic and apoptotic signaling in late stage breast cancer. Biomed. J. 2019, 42, 155–165. [Google Scholar] [CrossRef]
- Albiñana, V.; Villar Gómez de Las Heras, K.; Serrano-Heras, G.; Segura, T.; Perona-Moratalla, A.B.; Mota-Pérez, M.; de Campos, J.M.; Botella, L.M. Propranolol reduces viability and induces apoptosis in hemangioblastoma cells from von Hippel-Lindau patients. Orphanet J. Rare Dis. 2015, 10, 118. [Google Scholar] [CrossRef]
- Ji, Y.; Li, K.; Xiao, X.; Zheng, S.; Xu, T.; Chen, S. Effects of propranolol on the proliferation and apoptosis of hemangioma-derived endothelial cells. J. Pediatr. Surg. 2012, 47, 2216–2223. [Google Scholar] [CrossRef]
- Wolter, J.K.; Wolter, N.E.; Blanch, A.; Partridge, T.; Cheng, L.; Morgenstern, D.A.; Podkowa, M.; Kaplan, D.R.; Irwin, M.S. Anti-tumor activity of the beta-adrenergic receptor antagonist propranolol in neuroblastoma. Oncotarget 2014, 5, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Wolter, N.E.; Wolter, J.K.; Enepekides, D.J.; Irwin, M.S. Propranolol as a novel adjunctive treatment for head and neck squamous cell carcinoma. J. Otolaryngol. Head Neck Surg. 2012, 41, 334–344. [Google Scholar] [PubMed]
- Zhang, D.; Ma, Q.; Shen, S.; Hu, H. Inhibition of Pancreatic Cancer Cell Proliferation by Propranolol Occurs Through Apoptosis Induction: The Study of β-Adrenoceptor Antagonist’s Anticancer Effect in Pancreatic Cancer Cell. Pancreas 2009, 38, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.A.; Hurley, T.D. Characterization of two distinct structural classes of selective aldehyde dehydrogenase 1A1 inhibitors. J. Med. Chem. 2015, 58, 1964–1975. [Google Scholar] [CrossRef]
- Ibrahim, A.I.M.; Abul-Futouh, H.; Bourghli, L.M.S.; Abu-Sini, M.; Sunoqrot, S.; Ikhmais, B.; Jha, V.; Sarayrah, Q.; Abulebdah, D.H.; Ismail, W.H. Design and Synthesis of Thionated Levofloxacin: Insights into a New Generation of Quinolones with Potential Therapeutic and Analytical Applications. Curr. Issues Mol. Biol. 2022, 44, 4626–4638. [Google Scholar] [CrossRef]
- Markossian, S.; Grossman, A.; Brimacombe, K.; Arkin, M.; Auld, D.; Austin, C.P.; Baell, J.; Chung, T.D.; Coussens, N.P.; Dahlin, J.L.; et al. (Eds.) Assay Guidance Manual [Internet]; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. Available online: https://www.ncbi.nlm.nih.gov/books/NBK53196/ (accessed on 29 August 2023).
- Hussein, B.; Ikhmais, B.; Kadirvel, M.; Magwaza, R.N.; Halbert, G.; Bryce, R.A.; Stratford, I.J.; Freeman, S. Discovery of potent 4-aminoquinoline hydrazone inhibitors of NRH: Quinoneoxidoreductase-2 (NQO2). Eur. J. Med. Chem. 2019, 182, 111649. [Google Scholar] [CrossRef]
- Ladage, D.; Schwinger, R.H.; Brixius, K. Cardio-selective beta-blocker: Pharmacological evidence and their influence on exercise capacity. Cardiovasc. Ther. 2013, 31, 76–83. [Google Scholar] [CrossRef]
- Khalil, R.; Al-Awaida, W.J.; Al-Ameer, H.J.; Jarrar, Y.; Imraish, A.; Al Bawareed, O.; Qawadri, R.; Al Madhoon, F.; Obeidat, L. Investigation of ACE rs4646994, MTHFR rs1801133 and VDR rs2228570 Genotypes in Jordanian Patients with Fibromyalgia Syndrome. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 1920–1928. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Compounds | Toxicity (IC50 (µM) ± SE) in NSCLC Cell Lines at 96 h | |
|---|---|---|
| A549 | H1299 | |
| DEAB | >100 | >100 |
| Isoproterenol | 88.3 ± 6 | 38 ± 4 |
| Epinephrine | 78.7 ± 2.3 | 23.5 ± 3.3 |
| Atenolol | >1000 | >1000 |
| Esmolol | 513.3 ± 51.7 | 400 ± 30.6 |
| Nadolol | >1000 | >1000 |
| Metoprolol | 706.7 ± 31.8 | 570 ± 60.3 |
| Bisoprolol | 730.0 ± 79.4 | 560 ± 112.4 |
| Propranolol | 146.3 ± 29.3 | 76 ± 13.3 |
| Carvedilol | 18 ± 2.1 | 13.7 ± 0.3 |
| Genes | Sequence of Nucleotides (5′-3′) | |
|---|---|---|
| ALDH1A1 | Forward | CAA GAT CCA GGG CCG TAC AA |
| Reverse | CAG TGC AGG CCC TAT CTT CC | |
| ADRB2 | Forward | CAA GAA TAA GGC CCG GGT GA |
| Reverse | CCG GTA CCA GTG CAT CTG AA | |
| B1AR | Forward | CCG GGA ACA GGA ACA CAC |
| Reverse | GAA AGC AAA AGG AAA TAT GTC | |
| ACTNB | Forward | TTC CTT CCT GGG CAT GGA GT |
| Reverse | GCA ATG ATC TTG ATC TTC ATT | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikhmais, B.A.; Hammad, A.M.; Abusara, O.H.; Hamadneh, L.; Abumansour, H.; Abdallah, Q.M.; Ibrahim, A.I.M.; Elsalem, L.; Awad, M.; Alshehada, R. Investigating Carvedilol’s Repurposing for the Treatment of Non-Small Cell Lung Cancer via Aldehyde Dehydrogenase Activity Modulation in the Presence of β-Adrenergic Agonists. Curr. Issues Mol. Biol. 2023, 45, 7996-8012. https://doi.org/10.3390/cimb45100505
Ikhmais BA, Hammad AM, Abusara OH, Hamadneh L, Abumansour H, Abdallah QM, Ibrahim AIM, Elsalem L, Awad M, Alshehada R. Investigating Carvedilol’s Repurposing for the Treatment of Non-Small Cell Lung Cancer via Aldehyde Dehydrogenase Activity Modulation in the Presence of β-Adrenergic Agonists. Current Issues in Molecular Biology. 2023; 45(10):7996-8012. https://doi.org/10.3390/cimb45100505
Chicago/Turabian StyleIkhmais, Balqis A., Alaa M. Hammad, Osama H. Abusara, Lama Hamadneh, Hamza Abumansour, Qasem M. Abdallah, Ali I. M. Ibrahim, Lina Elsalem, Mariam Awad, and Rahaf Alshehada. 2023. "Investigating Carvedilol’s Repurposing for the Treatment of Non-Small Cell Lung Cancer via Aldehyde Dehydrogenase Activity Modulation in the Presence of β-Adrenergic Agonists" Current Issues in Molecular Biology 45, no. 10: 7996-8012. https://doi.org/10.3390/cimb45100505
APA StyleIkhmais, B. A., Hammad, A. M., Abusara, O. H., Hamadneh, L., Abumansour, H., Abdallah, Q. M., Ibrahim, A. I. M., Elsalem, L., Awad, M., & Alshehada, R. (2023). Investigating Carvedilol’s Repurposing for the Treatment of Non-Small Cell Lung Cancer via Aldehyde Dehydrogenase Activity Modulation in the Presence of β-Adrenergic Agonists. Current Issues in Molecular Biology, 45(10), 7996-8012. https://doi.org/10.3390/cimb45100505