
The Potential of Dendritic Cell Subsets in the Development of Personalized Immunotherapy for Cancer Treatment

 , ,
, ,  , , and
, , and 
Abstract
:1. Introduction
2. Biology of Dendritic Cells
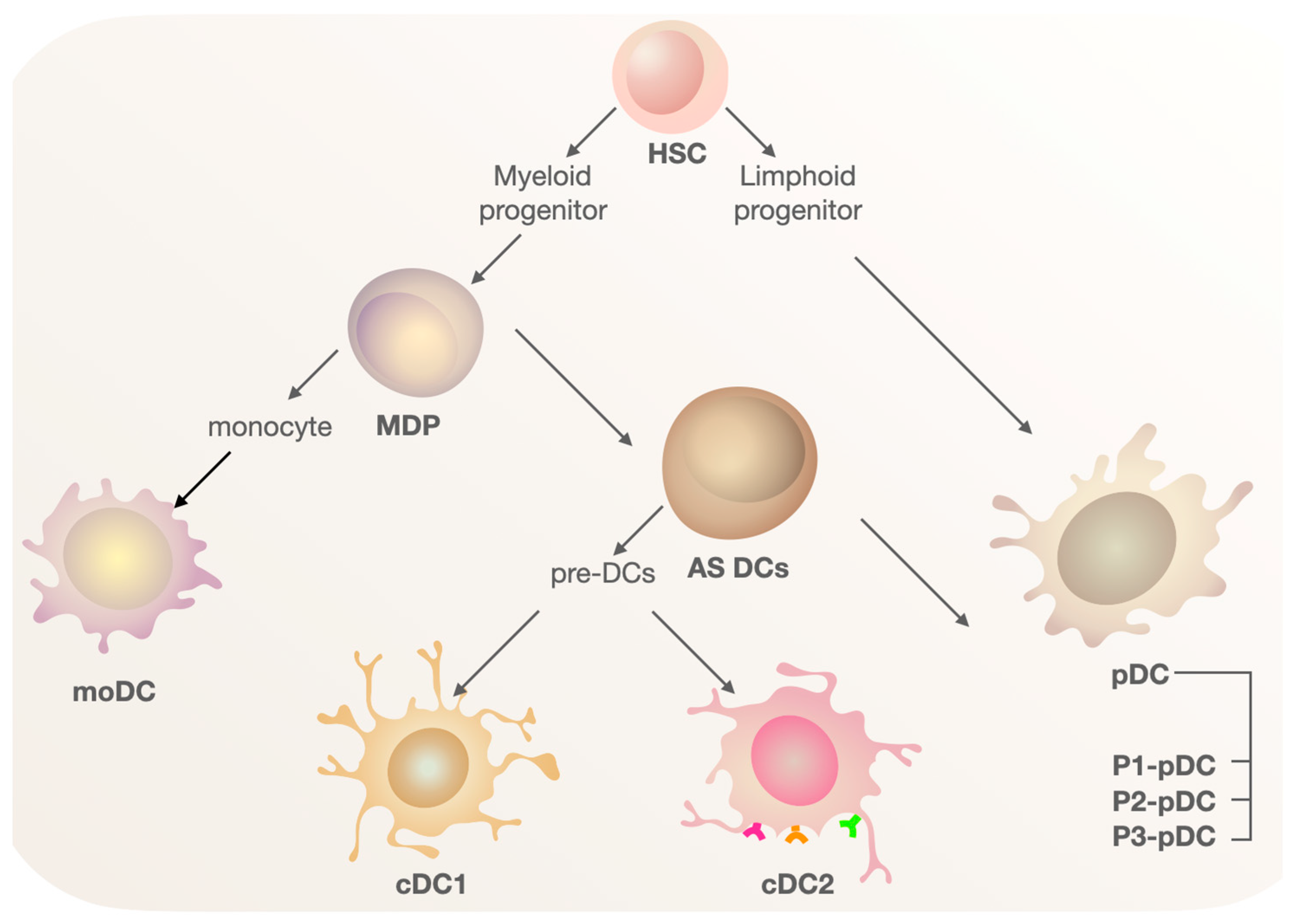
3. Dendritic Cell Populations in the Human Body
3.1. Conventional Dendritic Cells (cDCs)
3.2. Plasmacytoid Dendritic Cells (pDCs)
3.3. Monocyte-Derived Dendritic Cells (moDCs)
4. In Vitro DC Vaccine Design
4.1. DC Differentiation
4.1.1. DCs Derived from Monocytes (moDCs)
4.1.2. DCs Derived from CD34+ Progenitors
4.1.3. Genetic Reprogramming in DCs
4.2. DC Maturation
4.3. Vesicles Derived from Dendritic Cells
4.4. Adaptive Transfer of DC Vaccines
5. Application of DC Vaccines in Cancer Therapy
5.1. Preclinical Studies

{kind=link}
{kind=link}
{kind=link}
| Therapy | Model for Research | Results | References |
|---|---|---|---|
| Comparison of moDC-based therapy and cDC1-based therapy | Irf8+32−/−, Batf3−/− mice C57BL/6, CD45.2+ Irf8+32−/−, mice with subcutaneous methylcholanthrene (MCA)-induced fibrosarcoma injections | Lack of tumor-specific response in the therapy of moDCs in Irf8+32−/− mice | [101] |
| Comparison of CD8α+DC-based therapy and moDC-based therapy | C57BL/6 mice with orthotopic model of ID8 cancer | Reduced volume of ascites in both groups Decreased level of regulatory T-cells (Treg), IL-10, increased expression of CD3, CD4, CD8, and CD11c markers in the CD8α+ DC group | [102] |
| DC-based therapy + inhibitor Arp2/3 CK666 | CD45.2 WT, OT-I and CD45.1 (Ly5.1) mice | The combination of DCs and CK666 inhibitor led to a reduction in phagosomal acidification and an increase in CD8+ T-cell proliferation, compared to the control group | [104] |
| bmDC therapy + DNA vaccine | Human MUC1 transgenic mice | Tumor regression was observed only in mice receiving therapy with bmDCs + DNA vaccine | [105] |
| DC-based vaccine + αPD-1 | C3H/HeJ mice by transplanting murine MBT-2 bladder cancer cells | In the group “DC + αPD-1”, there was higher survival, IFN-γ production, and frequency of CD8+ and CD4+ T-cells in the spleen | [106] |
5.2. Clinical Studies
5.2.1. DC Progenitor-Based Therapy
5.2.2. Therapy Based on Natural DCs
5.2.3. Therapy Based on DC-Derived Vesicles (Dex)
| Therapy | Type of Cancer | Participants | Efficiency, % | References |
|---|---|---|---|---|
| Neoantigen-primed DC vaccine + cyclophosphamide | Non-small cell lung cancer | 12 | 25 OR 75 DCR 100 AE (1–2) | NCT02956551 |
| Adenoviral transduced autologous human epidermal growth factor receptor (AdHER)/neu DC vaccine | Metastatic solid tumors characterized by HER2/Neu expression | 33 | 3 CR 3 PR 15 SD 100 AE | [116] |
| Autologous DCs transduced with AdGMCA9 (DC-AdGMCAIX) | Metastatic renal cell carcinoma | 11 | 45 AE (1–2) 9 SD | NCT01826877 |
| Autologous DCs pulsed with tumor lysate antigen (DCVax®-L) | Newly diagnosed glioblastoma (NDG); recurrent glioblastoma (RG) | 232—NDG 64—RG | 1,5 AE 19.3 mOS (months), NDG 13.2 mOS (months), RG | NCT00045968 |
| IKKβ-matured RNA-transfected DC vaccine + immune checkpoint blockade (ICB) | Metastatic uveal melanoma | 12 | No published results | NCT04335890 |
| Autologous DCs + docetaxel + prednisone | Metastatic castration-resistant prostate cancer | 1182 | 23.9 mOS (months) 24.3 mOS (months, placebo group) | NCT02111577 |
| Alpha-type-1 polarized DC-based vaccination | Newly diagnosed high-grade glioma | 16 | 19 mOS (months) | [114] |
| DCs transduced with MART-1, tyrosinase, and MAGE-A6+ IFN-α | Melanoma | 35 | 5.7 PR 23 SD 40 PD 51.4 AE 36 mOS (months) | NCT01622933 |
| Therapy based on natural autologous cDC2 and pDCs | Melanoma | 15 | 19.4 mDFS (months) 100 AE (1–2) | NCT02574377 |
| Exosomes derived from autologous moDCs | Metastatic melanoma | 15 | 13 PR 13 SD | [58] |
| IFN-γ-exosomes derived from DC (IFN-γ- Dex) | Non-small cell lung cancer | 22 | 15 mOS (months) 19 AE (1–3) 32 SD (for 4 months) | NCT01159288 |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steinman, R.M.; Witmer, M.D. Lymphoid Dendritic Cells Are Potent Stimulators of the Primary Mixed Leukocyte Reaction in Mice. Proc. Natl. Acad. Sci. USA 1978, 75, 5132–5136. [Google Scholar] [CrossRef] [PubMed]
- Stumbles, P.A.; Himbeck, R.; Frelinger, J.A.; Collins, E.J.; Lake, R.A.; Robinson, B.W.S. Cutting Edge: Tumor-Specific CTL Are Constitutively Cross-Armed in Draining Lymph Nodes and Transiently Disseminate to Mediate Tumor Regression Following Systemic CD40 Activation. J. Immunol. 2004, 173, 5923–5928. [Google Scholar] [CrossRef] [PubMed]
- Filin, I.Y.; Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Recent Advances in Experimental Dendritic Cell Vaccines for Cancer. Front. Oncol. 2021, 11, 730824. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Kalinski, P.; Ueda, R.; Hoji, A.; Kohanbash, G.; Donegan, T.E.; Mintz, A.H.; Engh, J.A.; Bartlett, D.L.; Brown, C.K.; et al. Induction of CD8+ T-Cell Responses against Novel Glioma-Associated Antigen Peptides and Clinical Activity by Vaccinations with α-Type 1 Polarized Dendritic Cells and Polyinosinic-Polycytidylic Acid Stabilized by Lysine and Carboxymethylcellulose in Patients with Recurrent Malignant Glioma. J. Clin. Oncol. 2011, 29, 330–336. [Google Scholar] [CrossRef]
- Zhu, P.; Li, S.-Y.; Ding, J.; Fei, Z.; Sun, S.-N.; Zheng, Z.-H.; Wei, D.; Jiang, J.; Miao, J.-L.; Li, S.-Z.; et al. Combination Immunotherapy of Glioblastoma with Dendritic Cell Cancer Vaccines, Anti-PD-1 and Poly I:C. J. Pharm. Anal. 2023, 13, 616–624. [Google Scholar] [CrossRef]
- Vermaelen, K.; Pauwels, R. Accurate and Simple Discrimination of Mouse Pulmonary Dendritic Cell and Macrophage Populations by Flow Cytometry: Methodology and New Insights. Cytom. A 2004, 61, 170–177. [Google Scholar] [CrossRef]
- Bell, D.; Chomarat, P.; Broyles, D.; Netto, G.; Harb, G.M.; Lebecque, S.; Valladeau, J.; Davoust, J.; Palucka, K.A.; Banchereau, J. In Breast Carcinoma Tissue, Immature Dendritic Cells Reside within the Tumor, Whereas Mature Dendritic Cells Are Located in Peritumoral Areas. J. Exp. Med. 1999, 190, 1417–1426. [Google Scholar] [CrossRef]
- Tuna, H.; Avdiushko, R.G.; Sindhava, V.J.; Wedlund, L.; Kaetzel, C.S.; Kaplan, A.M.; Bondada, S.; Cohen, D.A. Regulation of the Mucosal Phenotype in Dendritic Cells by PPARγ: Role of Tissue Microenvironment. J. Leukoc. Biol. 2014, 95, 471–485. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human Dendritic Cell Subsets: An Update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, X.; Jiang, P. Cytokine and Chemokine Signals of T-Cell Exclusion in Tumors. Front. Immunol. 2020, 11, 594609. [Google Scholar] [CrossRef]
- Persson, C.M.; Chambers, B.J. Plasmacytoid Dendritic Cell-Induced Migration and Activation of NK Cells in Vivo. Eur. J. Immunol. 2010, 40, 2155–2164. [Google Scholar] [CrossRef]
- Jacobs, B.; Gebel, V.; Heger, L.; Grèze, V.; Schild, H.; Dudziak, D.; Ullrich, E. Characterization and Manipulation of the Crosstalk Between Dendritic and Natural Killer Cells Within the Tumor Microenvironment. Front. Immunol. 2021, 12, 670540. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, E.S.; Mellman, I. Cell Biology of Antigen Processing in Vitro and in Vivo. Annu. Rev. Immunol. 2005, 23, 975–1028. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Kotsias, F.; Visentin, G.; Bruhns, P.; Savina, A.; Amigorena, S. Autonomous Phagosomal Degradation and Antigen Presentation in Dendritic Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 14556–14561. [Google Scholar] [CrossRef] [PubMed]
- Platt, C.D.; Ma, J.K.; Chalouni, C.; Ebersold, M.; Bou-Reslan, H.; Carano, R.A.D.; Mellman, I.; Delamarre, L. Mature Dendritic Cells Use Endocytic Receptors to Capture and Present Antigens. Proc. Natl. Acad. Sci. USA 2010, 107, 4287–4292. [Google Scholar] [CrossRef]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.-I.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In Vivo Targeting of Antigens to Maturing Dendritic Cells via the DEC-205 Receptor Improves T Cell Vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef]
- Norbury, C.C.; Chambers, B.J.; Prescott, A.R.; Ljunggren, H.G.; Watts, C. Constitutive Macropinocytosis Allows TAP-Dependent Major Histocompatibility Complex Class I Presentation of Exogenous Soluble Antigen by Bone Marrow-Derived Dendritic Cells. Eur. J. Immunol. 1997, 27, 280–288. [Google Scholar] [CrossRef]
- Hilligan, K.L.; Ronchese, F. Antigen Presentation by Dendritic Cells and Their Instruction of CD4+ T Helper Cell Responses. Cell Mol. Immunol. 2020, 17, 587–599. [Google Scholar] [CrossRef]
- MacNabb, B.W.; Tumuluru, S.; Chen, X.; Godfrey, J.; Kasal, D.N.; Yu, J.; Jongsma, M.L.M.; Spaapen, R.M.; Kline, D.E.; Kline, J. Dendritic Cells Can Prime Anti-Tumor CD8+ T Cell Responses through Major Histocompatibility Complex Cross-Dressing. Immunity 2022, 55, 982–997.e8. [Google Scholar] [CrossRef]
- Ukyo, N.; Hori, T.; Yanagita, S.; Ishikawa, T.; Uchiyama, T. Costimulation through OX40 Is Crucial for Induction of an Alloreactive Human T-Cell Response. Immunology 2003, 109, 226–231. [Google Scholar] [CrossRef]
- Heidkamp, G.F.; Sander, J.; Lehmann, C.H.K.; Heger, L.; Eissing, N.; Baranska, A.; Lühr, J.J.; Hoffmann, A.; Reimer, K.C.; Lux, A.; et al. Human Lymphoid Organ Dendritic Cell Identity Is Predominantly Dictated by Ontogeny, Not Tissue Microenvironment. Sci. Immunol. 2016, 1, eaai7677. [Google Scholar] [CrossRef]
- Bowman-Kirigin, J.A.; Desai, R.; Saunders, B.T.; Wang, A.Z.; Schaettler, M.O.; Liu, C.J.; Livingstone, A.J.; Kobayashi, D.K.; Durai, V.; Kretzer, N.M.; et al. The Conventional Dendritic Cell 1 Subset Primes CD8+ T Cells and Traffics Tumor Antigen to Drive Antitumor Immunity in the Brain. Cancer Immunol. Res. 2023, 11, 20–37. [Google Scholar] [CrossRef]
- Villar, J.; Segura, E. Decoding the Heterogeneity of Human Dendritic Cell Subsets. Trends Immunol. 2020, 41, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Granot, T.; Senda, T.; Carpenter, D.J.; Matsuoka, N.; Weiner, J.; Gordon, C.L.; Miron, M.; Kumar, B.V.; Griesemer, A.; Ho, S.-H.; et al. Dendritic Cells Display Subset and Tissue-Specific Maturation Dynamics over Human Life. Immunity 2017, 46, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Schreibelt, G.; Tel, J.; Sliepen, K.H.E.W.J.; Benitez-Ribas, D.; Figdor, C.G.; Adema, G.J.; de Vries, I.J.M. Toll-like Receptor Expression and Function in Human Dendritic Cell Subsets: Implications for Dendritic Cell-Based Anti-Cancer Immunotherapy. Cancer Immunol. Immunother. 2010, 59, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Edelson, B.T.; Kc, W.; Juang, R.; Kohyama, M.; Benoit, L.A.; Klekotka, P.A.; Moon, C.; Albring, J.C.; Ise, W.; Michael, D.G.; et al. Peripheral CD103+ Dendritic Cells Form a Unified Subset Developmentally Related to CD8α+ Conventional Dendritic Cells. J. Exp. Med. 2010, 207, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161.e7. [Google Scholar] [CrossRef]
- Mair, F.; Liechti, T. Comprehensive Phenotyping of Human Dendritic Cells and Monocytes. Cytom. A 2021, 99, 231–242. [Google Scholar] [CrossRef]
- Mishra, G.P.; Jha, A.; Ahad, A.; Sen, K.; Sen, A.; Podder, S.; Prusty, S.; Biswas, V.K.; Gupta, B.; Raghav, S.K. Epigenomics of Conventional Type-I Dendritic Cells Depicted Preferential Control of TLR9 versus TLR3 Response by NCoR1 through Differential IRF3 Activation. Cell Mol. Life Sci. 2022, 79, 429. [Google Scholar] [CrossRef]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 2019, 177, 556–571.e16. [Google Scholar] [CrossRef]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic Cells, Monocytes and Macrophages: A Unified Nomenclature Based on Ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2018, 9, 3176. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef]
- See, P.; Dutertre, C.-A.; Chen, J.; Günther, P.; McGovern, N.; Irac, S.E.; Gunawan, M.; Beyer, M.; Händler, K.; Duan, K.; et al. Mapping the Human DC Lineage through the Integration of High-Dimensional Techniques. Science 2017, 356, eaag3009. [Google Scholar] [CrossRef] [PubMed]
- Hernández, S.S.; Jakobsen, M.R.; Bak, R.O. Plasmacytoid Dendritic Cells as a Novel Cell-Based Cancer Immunotherapy. Int. J. Mol. Sci. 2022, 23, 11397. [Google Scholar] [CrossRef]
- Dzionek, A.; Fuchs, A.; Schmidt, P.; Cremer, S.; Zysk, M.; Miltenyi, S.; Buck, D.W.; Schmitz, J. BDCA-2, BDCA-3, and BDCA-4: Three Markers for Distinct Subsets of Dendritic Cells in Human Peripheral Blood. J. Immunol. 2000, 165, 6037–6046. [Google Scholar] [CrossRef]
- Ju, X.; Zenke, M.; Hart, D.N.J.; Clark, G.J. CD300a/c Regulate Type I Interferon and TNF-α Secretion by Human Plasmacytoid Dendritic Cells Stimulated with TLR7 and TLR9 Ligands. Blood 2008, 112, 1184–1194. [Google Scholar] [CrossRef]
- Bao, M.; Liu, Y.-J. Regulation of TLR7/9 Signaling in Plasmacytoid Dendritic Cells. Protein Cell 2013, 4, 40–52. [Google Scholar] [CrossRef]
- Yun, T.J.; Igarashi, S.; Zhao, H.; Perez, O.A.; Pereira, M.R.; Zorn, E.; Shen, Y.; Goodrum, F.; Rahman, A.; Sims, P.A.; et al. Human Plasmacytoid Dendritic Cells Mount a Distinct Antiviral Response to Virus-Infected Cells. Sci. Immunol. 2021, 6, eabc7302. [Google Scholar] [CrossRef]
- Das, A.; Chauhan, K.S.; Kumar, H.; Tailor, P. Mutation in Irf8 Gene (Irf8R294C) Impairs Type I IFN-Mediated Antiviral Immune Response by Murine pDCs. Front. Immunol. 2021, 12, 758190. [Google Scholar] [CrossRef]
- Deb, P.; Dai, J.; Singh, S.; Kalyoussef, E.; Fitzgerald-Bocarsly, P. Triggering of the cGAS-STING Pathway in Human Plasmacytoid Dendritic Cells Inhibits TLR9-Mediated IFN Production. J. Immunol. 2020, 205, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Sprangers, S.; de Vries, T.J.; Everts, V. Monocyte Heterogeneity: Consequences for Monocyte-Derived Immune Cells. J. Immunol. Res. 2016, 2016, 1475435. [Google Scholar] [CrossRef] [PubMed]
- Goenka, A.; Khan, F.; Verma, B.; Sinha, P.; Dmello, C.C.; Jogalekar, M.P.; Gangadaran, P.; Ahn, B. Tumor Microenvironment Signaling and Therapeutics in Cancer Progression. Cancer Commun. 2023, 43, 525–561. [Google Scholar] [CrossRef] [PubMed]
- Husain, Z.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate and Myeloid-Derived Suppressor Cells. Oncoimmunology 2013, 2, e26383. [Google Scholar] [CrossRef]
- Xia, J.; Miao, Y.; Wang, X.; Huang, X.; Dai, J. Recent Progress of Dendritic Cell-Derived Exosomes (Dex) as an Anti-Cancer Nanovaccine. Biomed. Pharmacother. 2022, 152, 113250. [Google Scholar] [CrossRef]
- Chometon, T.Q.; da Silva Siqueira, M.; Anna, J.C.S.; Almeida, M.R.; Gandini, M.; de Almeida Nogueira, A.C.M.; Antas, P.R.Z. A Protocol for Rapid Monocyte Isolation and Generation of Singular Human Monocyte-Derived Dendritic Cells. PLoS ONE 2020, 15, e0231132. [Google Scholar] [CrossRef]
- Eguíluz-Gracia, I.; Bosco, A.; Dollner, R.; Melum, G.R.; Lexberg, M.H.; Jones, A.C.; Dheyauldeen, S.A.; Holt, P.G.; Bækkevold, E.S.; Jahnsen, F.L. Rapid Recruitment of CD14(+) Monocytes in Experimentally Induced Allergic Rhinitis in Human Subjects. J. Allergy Clin. Immunol. 2016, 137, 1872–1881.e12. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.R.; De Palma, M. Engineering Dendritic Cell Vaccines to Improve Cancer Immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef]
- Kalantari, T.; Kamali-Sarvestani, E.; Zhang, G.-X.; Safavi, F.; Lauretti, E.; Khedmati, M.-E.; Rostami, A. Generation of Large Numbers of Highly Purified Dendritic Cells from Bone Marrow Progenitor Cells after Co-Culture with Syngeneic Murine Splenocytes. Exp. Mol. Pathol. 2013, 94, 336–342. [Google Scholar] [CrossRef]
- Luo, X.; Balan, S.; Arnold-Schrauf, C.; Dalod, M. In Vitro Generation of Human Cross-Presenting Type 1 Conventional Dendritic Cells (cDC1s) and Plasmacytoid Dendritic Cells (pDCs). Methods Mol. Biol. 2023, 2618, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Sprent, J. GM-CSF Culture Revisited: Preparation of Bulk Populations of Highly Pure Dendritic Cells from Mouse Bone Marrow. J. Immunol. 2018, 201, 3129–3139. [Google Scholar] [CrossRef] [PubMed]
- van Eck van der Sluijs, J.; van Ens, D.; Thordardottir, S.; Vodegel, D.; Hermens, I.; van der Waart, A.B.; Falkenburg, J.H.F.; Kester, M.G.D.; de Rink, I.; Heemskerk, M.H.M.; et al. Clinically Applicable CD34+-Derived Blood Dendritic Cell Subsets Exhibit Key Subset-Specific Features and Potently Boost Anti-Tumor T and NK Cell Responses. Cancer Immunol. Immunother. 2021, 70, 3167–3181. [Google Scholar] [CrossRef]
- Nielsen, M.C.; Andersen, M.N.; Møller, H.J. Monocyte Isolation Techniques Significantly Impact the Phenotype of Both Isolated Monocytes and Derived Macrophages in Vitro. Immunology 2020, 159, 63–74. [Google Scholar] [CrossRef]
- Pires, C.F.; Rosa, F.F.; Kurochkin, I.; Pereira, C.-F. Understanding and Modulating Immunity With Cell Reprogramming. Front. Immunol. 2019, 10, 2809. [Google Scholar] [CrossRef]
- Lundberg, K.; Albrekt, A.-S.; Nelissen, I.; Santegoets, S.; de Gruijl, T.D.; Gibbs, S.; Lindstedt, M. Transcriptional Profiling of Human Dendritic Cell Populations and Models—Unique Profiles of in Vitro Dendritic Cells and Implications on Functionality and Applicability. PLoS ONE 2013, 8, e52875. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, S.; Vu Manh, T.-P.; Chelbi, R.; Henri, S.; Malissen, B.; Haniffa, M.; Ginhoux, F.; Dalod, M. Comparative Genomics Analysis of Mononuclear Phagocyte Subsets Confirms Homology between Lymphoid Tissue-Resident and Dermal XCR1(+) DCs in Mouse and Human and Distinguishes Them from Langerhans Cells. J. Immunol. Methods 2016, 432, 35–49. [Google Scholar] [CrossRef]
- Carreno, B.M.; Becker-Hapak, M.; Huang, A.; Chan, M.; Alyasiry, A.; Lie, W.-R.; Aft, R.L.; Cornelius, L.A.; Trinkaus, K.M.; Linette, G.P. IL-12p70-Producing Patient DC Vaccine Elicits Tc1-Polarized Immunity. J. Clin. Investig. 2013, 123, 3383–3394. [Google Scholar] [CrossRef]
- Lopes, A.M.M.; Michelin, M.A.; Murta, E.F.C. Monocyte-Derived Dendritic Cells from Patients with Cervical Intraepithelial Lesions. Oncol. Lett. 2017, 13, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Vopenkova, K.; Mollova, K.; Buresova, I.; Michalek, J. Complex Evaluation of Human Monocyte-Derived Dendritic Cells for Cancer Immunotherapy. J. Cell Mol. Med. 2012, 16, 2827–2837. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef]
- Ding, Z.; Li, Q.; Zhang, R.; Xie, L.; Shu, Y.; Gao, S.; Wang, P.; Su, X.; Qin, Y.; Wang, Y.; et al. Personalized Neoantigen Pulsed Dendritic Cell Vaccine for Advanced Lung Cancer. Signal Transduct. Target. Ther. 2021, 6, 26. [Google Scholar] [CrossRef]
- Koch, E.A.T.; Schaft, N.; Kummer, M.; Berking, C.; Schuler, G.; Hasumi, K.; Dörrie, J.; Schuler-Thurner, B. A One-Armed Phase I Dose Escalation Trial Design: Personalized Vaccination with IKKβ-Matured, RNA-Loaded Dendritic Cells for Metastatic Uveal Melanoma. Front. Immunol. 2022, 13, 785231. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, N.J.; Beer, T.M.; Gerritsen, W.; Oudard, S.; Wiechno, P.; Kukielka-Budny, B.; Samal, V.; Hajek, J.; Feyerabend, S.; Khoo, V.; et al. Efficacy and Safety of Autologous Dendritic Cell-Based Immunotherapy, Docetaxel, and Prednisone vs Placebo in Patients With Metastatic Castration-Resistant Prostate Cancer: The VIABLE Phase 3 Randomized Clinical Trial. JAMA Oncol. 2022, 8, 546–552. [Google Scholar] [CrossRef]
- Vreeland, T.J.; Clifton, G.T.; Hale, D.F.; Chick, R.C.; Hickerson, A.T.; Cindass, J.L.; Adams, A.M.; Bohan, P.M.K.; Andtbacka, R.H.I.; Berger, A.C.; et al. A Phase IIb Randomized Controlled Trial of the TLPLDC Vaccine as Adjuvant Therapy After Surgical Resection of Stage III/IV Melanoma: A Primary Analysis. Ann. Surg. Oncol. 2021, 28, 6126–6137. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. Efficient Presentation of Soluble Antigen by Cultured Human Dendritic Cells Is Maintained by Granulocyte/Macrophage Colony-Stimulating Factor plus Interleukin 4 and Downregulated by Tumor Necrosis Factor Alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef]
- Syme, R.; Bajwa, R.; Robertson, L.; Stewart, D.; Glück, S. Comparison of CD34 and Monocyte-Derived Dendritic Cells from Mobilized Peripheral Blood from Cancer Patients. Stem Cells 2005, 23, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.M. Monocytes Differentiated with GM-CSF and IL-15 Initiate Th17 and Th1 Responses That Are Contact-Dependent and Mediated by IL-15. J. Leukoc. Biol. 2011, 90, 727–734. [Google Scholar] [CrossRef]
- Mohty, M.; Vialle-Castellano, A.; Nunes, J.A.; Isnardon, D.; Olive, D.; Gaugler, B. IFN-α Skews Monocyte Differentiation into Toll-like Receptor 7-Expressing Dendritic Cells with Potent Functional Activities. J. Immunol. 2003, 171, 3385–3393. [Google Scholar] [CrossRef]
- Flörcken, A.; Kopp, J.; van Lessen, A.; Movassaghi, K.; Takvorian, A.; Jöhrens, K.; Möbs, M.; Schönemann, C.; Sawitzki, B.; Egerer, K.; et al. Allogeneic Partially HLA-Matched Dendritic Cells Pulsed with Autologous Tumor Cell Lysate as a Vaccine in Metastatic Renal Cell Cancer: A Clinical Phase I/II Study. Hum. Vaccines Immunother. 2013, 9, 1217–1227. [Google Scholar] [CrossRef]
- Balan, S.; Ollion, V.; Colletti, N.; Chelbi, R.; Montanana-Sanchis, F.; Liu, H.; Vu Manh, T.-P.; Sanchez, C.; Savoret, J.; Perrot, I.; et al. Human XCR1+ Dendritic Cells Derived in Vitro from CD34+ Progenitors Closely Resemble Blood Dendritic Cells, Including Their Adjuvant Responsiveness, Contrary to Monocyte-Derived Dendritic Cells. J. Immunol. 2014, 193, 1622–1635. [Google Scholar] [CrossRef]
- Proietto, A.I.; Mittag, D.; Roberts, A.W.; Sprigg, N.; Wu, L. The Equivalents of Human Blood and Spleen Dendritic Cell Subtypes Can Be Generated in Vitro from Human CD34(+) Stem Cells in the Presence of Fms-like Tyrosine Kinase 3 Ligand and Thrombopoietin. Cell Mol. Immunol. 2012, 9, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Bedke, N.; Swindle, E.J.; Molnar, C.; Holt, P.G.; Strickland, D.H.; Roberts, G.C.; Morris, R.; Holgate, S.T.; Davies, D.E.; Blume, C. A Method for the Generation of Large Numbers of Dendritic Cells from CD34+ Hematopoietic Stem Cells from Cord Blood. J. Immunol. Methods 2020, 477, 112703. [Google Scholar] [CrossRef]
- Kirkling, M.E.; Cytlak, U.; Lau, C.M.; Lewis, K.L.; Resteu, A.; Khodadadi-Jamayran, A.; Siebel, C.W.; Salmon, H.; Merad, M.; Tsirigos, A.; et al. Notch Signaling Facilitates In Vitro Generation of Cross-Presenting Classical Dendritic Cells. Cell Rep. 2018, 23, 3658–3672.e6. [Google Scholar] [CrossRef] [PubMed]
- Balan, S.; Arnold-Schrauf, C.; Abbas, A.; Couespel, N.; Savoret, J.; Imperatore, F.; Villani, A.-C.; Vu Manh, T.-P.; Bhardwaj, N.; Dalod, M. Large-Scale Human Dendritic Cell Differentiation Revealing Notch-Dependent Lineage Bifurcation and Heterogeneity. Cell Rep. 2018, 24, 1902–1915.e6. [Google Scholar] [CrossRef] [PubMed]
- Sundarasetty, B.S.; Chan, L.; Darling, D.; Giunti, G.; Farzaneh, F.; Schenck, F.; Naundorf, S.; Kuehlcke, K.; Ruggiero, E.; Schmidt, M.; et al. Lentivirus-Induced “Smart” Dendritic Cells: Pharmacodynamics and GMP-Compliant Production for Immunotherapy against TRP2-Positive Melanoma. Gene Ther. 2015, 22, 707–720. [Google Scholar] [CrossRef]
- Rosa, F.F.; Pires, C.F.; Kurochkin, I.; Ferreira, A.G.; Gomes, A.M.; Palma, L.G.; Shaiv, K.; Solanas, L.; Azenha, C.; Papatsenko, D.; et al. Direct Reprogramming of Fibroblasts into Antigen-Presenting Dendritic Cells. Sci. Immunol. 2018, 3, eaau4292. [Google Scholar] [CrossRef]
- Rosa, F.F.; Pires, C.F.; Kurochkin, I.; Halitzki, E.; Zahan, T.; Arh, N.; Zimmermannová, O.; Ferreira, A.G.; Li, H.; Karlsson, S.; et al. Single-Cell Transcriptional Profiling Informs Efficient Reprogramming of Human Somatic Cells to Cross-Presenting Dendritic Cells. Sci. Immunol. 2022, 7, eabg5539. [Google Scholar] [CrossRef]
- Hervas-Stubbs, S.; Mancheño, U.; Riezu-Boj, J.-I.; Larraga, A.; Ochoa, M.C.; Alignani, D.; Alfaro, C.; Morales-Kastresana, A.; Gonzalez, I.; Larrea, E.; et al. CD8 T Cell Priming in the Presence of IFN-α Renders CTLs with Improved Responsiveness to Homeostatic Cytokines and Recall Antigens: Important Traits for Adoptive T Cell Therapy. J. Immunol. 2012, 189, 3299–3310. [Google Scholar] [CrossRef]
- Rosalia, R.A.; Quakkelaar, E.D.; Redeker, A.; Khan, S.; Camps, M.; Drijfhout, J.W.; Silva, A.L.; Jiskoot, W.; van Hall, T.; van Veelen, P.A.; et al. Dendritic Cells Process Synthetic Long Peptides Better than Whole Protein, Improving Antigen Presentation and T-Cell Activation. Eur. J. Immunol. 2013, 43, 2554–2565. [Google Scholar] [CrossRef]
- Binder, R.J.; Anderson, K.M.; Basu, S.; Srivastava, P.K. Cutting Edge: Heat Shock Protein Gp96 Induces Maturation and Migration of CD11c+ Cells in Vivo. J. Immunol. 2000, 165, 6029–6035. [Google Scholar] [CrossRef]
- Palucka, A.K.; Ueno, H.; Connolly, J.; Kerneis-Norvell, F.; Blanck, J.-P.; Johnston, D.A.; Fay, J.; Banchereau, J. Dendritic Cells Loaded with Killed Allogeneic Melanoma Cells Can Induce Objective Clinical Responses and MART-1 Specific CD8+ T-Cell Immunity. J. Immunother. 2006, 29, 545–557. [Google Scholar] [CrossRef]
- Geskin, L.J.; Damiano, J.J.; Patrone, C.C.; Butterfield, L.H.; Kirkwood, J.M.; Falo, L.D. Three Antigen-Loading Methods in Dendritic Cell Vaccines for Metastatic Melanoma. Melanoma Res. 2018, 28, 211–221. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and Tumor Antigen mRNA Electroporated Dendritic Cell Vaccination plus Ipilimumab: Link between T-Cell Activation and Clinical Responses in Advanced Melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef] [PubMed]
- Chulpanova, D.S.; Kitaeva, K.V.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Therapeutic Prospects of Extracellular Vesicles in Cancer Treatment. Front. Immunol. 2018, 9, 1534. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Sepúlveda, D.; Tittarelli, A.; Gleisner, M.A.; Ávalos, I.; Pereda, C.; Gallegos, I.; González, F.E.; López, M.N.; Butte, J.M.; Roa, J.C.; et al. Tumor Lysate-Based Vaccines: On the Road to Immunotherapy for Gallbladder Cancer. Cancer Immunol. Immunother. 2018, 67, 1897–1910. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.G.J.V.; de Goeje, P.L.; Cornelissen, R.; Kaijen-Lambers, M.E.H.; Bezemer, K.; van der Leest, C.H.; Mahaweni, N.M.; Kunert, A.; Eskens, F.A.L.M.; Waasdorp, C.; et al. Autologous Dendritic Cells Pulsed with Allogeneic Tumor Cell Lysate in Mesothelioma: From Mouse to Human. Clin. Cancer Res. 2018, 24, 766–776. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Vaccination with an Adenoviral Vector Encoding the Tumor Antigen Directly Linked to Invariant Chain Induces Potent CD4+ T-cell-independent CD8+ T-cell-mediated Tumor Control—Sorensen—2009—European Journal of Immunology—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/eji.200939543 (accessed on 24 September 2023).
- Tacken, P.J.; Figdor, C.G. Targeted Antigen Delivery and Activation of Dendritic Cells in Vivo: Steps towards Cost Effective Vaccines. Semin. Immunol. 2011, 23, 12–20. [Google Scholar] [CrossRef]
- Xie, F.; Zhou, X.; Fang, M.; Li, H.; Su, P.; Tu, Y.; Zhang, L.; Zhou, F. Extracellular Vesicles in Cancer Immune Microenvironment and Cancer Immunotherapy. Adv. Sci. 2019, 6, 1901779. [Google Scholar] [CrossRef]
- Zippoli, M.; Ruocco, A.; Novelli, R.; Rocchio, F.; Miscione, M.S.; Allegretti, M.; Cesta, M.C.; Amendola, P.G. The Role of Extracellular Vesicles and Interleukin-8 in Regulating and Mediating Neutrophil-Dependent Cancer Drug Resistance. Front. Oncol. 2022, 12, 947183. [Google Scholar] [CrossRef]
- Ding, Y.-N.; Ding, H.-Y.; Li, H.; Yang, R.; Huang, J.-Y.; Chen, H.; Wang, L.-H.; Wang, Y.-J.; Hu, C.-M.; An, Y.-L.; et al. Photosensitive Small Extracellular Vesicles Regulate the Immune Microenvironment of Triple Negative Breast Cancer. Acta Biomater. 2023, 167, 534–550. [Google Scholar] [CrossRef]
- Hodge, A.L.; Baxter, A.A.; Poon, I.K.H. Gift Bags from the Sentinel Cells of the Immune System: The Diverse Role of Dendritic Cell-Derived Extracellular Vesicles. J. Leukoc. Biol. 2022, 111, 903–920. [Google Scholar] [CrossRef]
- Pitt, J.M.; André, F.; Amigorena, S.; Soria, J.-C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic Cell–Derived Exosomes for Cancer Therapy. J. Clin. Investig. 2016, 126, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.G.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of Transfer of Functional microRNAs between Mouse Dendritic Cells via Exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Zuo, B.; Zhang, Y.; Zhao, K.; Wu, L.; Qi, H.; Yang, R.; Gao, X.; Geng, M.; Wu, Y.; Jing, R.; et al. Universal Immunotherapeutic Strategy for Hepatocellular Carcinoma with Exosome Vaccines That Engage Adaptive and Innate Immune Responses. J. Hematol. Oncol. 2022, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Escudier, B.; Angevin, E.; Tursz, T.; Zitvogel, L. Exosomes for Cancer Immunotherapy. Ann. Oncol. 2004, 15 (Suppl. S4), 141–144. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; de Vries, I.J.M.; Schreibelt, G.; Lambeck, A.J.A.; Aarntzen, E.H.J.G.; Jacobs, J.F.M.; Scharenborg, N.M.; van de Rakt, M.W.M.M.; de Boer, A.J.; Croockewit, S.; et al. Route of Administration Modulates the Induction of Dendritic Cell Vaccine-Induced Antigen-Specific T Cells in Advanced Melanoma Patients. Clin. Cancer Res. 2011, 17, 5725–5735. [Google Scholar] [CrossRef]
- Edele, F.; Dudda, J.C.; Bachtanian, E.; Jakob, T.; Pircher, H.; Martin, S.F. Efficiency of Dendritic Cell Vaccination against B16 Melanoma Depends on the Immunization Route. PLoS ONE 2014, 9, e105266. [Google Scholar] [CrossRef]
- Filin, I.Y.; Solovyeva, V.V.; Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A. Current Trends in Cancer Immunotherapy. Biomedicines 2020, 8, 621. [Google Scholar] [CrossRef]
- Ferris, S.T.; Ohara, R.A.; Ou, F.; Wu, R.; Huang, X.; Kim, S.; Chen, J.; Liu, T.-T.; Schreiber, R.D.; Murphy, T.L.; et al. cDC1 Vaccines Drive Tumor Rejection by Direct Presentation Independently of Host cDC1. Cancer Immunol. Res. 2022, 10, 920–931. [Google Scholar] [CrossRef]
- Lee, S.-W.; Lee, H.; Lee, K.-W.; Kim, M.-J.; Kang, S.W.; Lee, Y.-J.; Kim, H.; Kim, Y.-M. CD8α+ Dendritic Cells Potentiate Antitumor and Immune Activities against Murine Ovarian Cancers. Sci. Rep. 2023, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Walter, D.L.; Zhang, C. Efficiency of Interferon-γ in Activating Dendritic Cells and Its Potential Synergy with Toll-like Receptor Agonists. Viruses 2023, 15, 1198. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.M.S.; D’Aulerio, R.; Yong, T.; He, M.; Baptista, M.A.P.; Nylén, S.; Westerberg, L.S. Increased Cross-Presentation by Dendritic Cells and Enhanced Anti-Tumour Therapy Using the Arp2/3 Inhibitor CK666. Br. J. Cancer 2023, 128, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Murwanti, R.; Denda-Nagai, K.; Sugiura, D.; Mogushi, K.; Gendler, S.J.; Irimura, T. Prevention of Inflammation-Driven Colon Carcinogenesis in Human MUC1 Transgenic Mice by Vaccination with MUC1 DNA and Dendritic Cells. Cancers 2023, 15, 1920. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Park, J.-H.; Chang, H. Enhanced Anti-Tumor Immunity of Vaccine Combined with Anti-PD-1 Antibody in a Murine Bladder Cancer Model. Investig. Clin. Urol. 2023, 64, 74–81. [Google Scholar] [CrossRef] [PubMed]
- de Mato, F.C.; Barreto, N.; Cordeiro, G.; Munhoz, J.; Bonfanti, A.P.; da Rocha-E-Silva, T.A.A.; Sutti, R.; Cruz, P.B.M.; Sanches, L.R.; Bombeiro, A.L.; et al. Isolated Peptide from Spider Venom Modulates Dendritic Cells In Vitro: A Possible Application in Oncoimmunotherapy for Glioblastoma. Cells 2023, 12, 1023. [Google Scholar] [CrossRef]
- Faiena, I.; Comin-Anduix, B.; Berent-Maoz, B.; Bot, A.; Zomorodian, N.; Sachdeva, A.; Said, J.; Cheung-Lau, G.; Pang, J.; Macabali, M.; et al. A Phase I, Open-Label, Dose-Escalation, and Cohort Expansion Study to Evaluate the Safety and Immune Response to Autologous Dendritic Cells Transduced With AdGMCA9 (DC-AdGMCAIX) in Patients With Metastatic Renal Cell Carcinoma. J. Immunother. 2020, 43, 273–282. [Google Scholar] [CrossRef]
- Maeng, H.M.; Moore, B.N.; Bagheri, H.; Steinberg, S.M.; Inglefield, J.; Dunham, K.; Wei, W.-Z.; Morris, J.C.; Terabe, M.; England, L.C.; et al. Phase I Clinical Trial of an Autologous Dendritic Cell Vaccine Against HER2 Shows Safety and Preliminary Clinical Efficacy. Front. Oncol. 2021, 11, 789078. [Google Scholar] [CrossRef]
- Bloemendal, M.; Bol, K.F.; Boudewijns, S.; Gorris, M.A.J.; de Wilt, J.H.W.; Croockewit, S.A.J.; van Rossum, M.M.; de Goede, A.L.; Petry, K.; Koornstra, R.H.T.; et al. Immunological Responses to Adjuvant Vaccination with Combined CD1c+ Myeloid and Plasmacytoid Dendritic Cells in Stage III Melanoma Patients. Oncoimmunology 2022, 11, 2015113. [Google Scholar] [CrossRef]
- Polyzoidis, S.; Ashkan, K. DCVax®-L—Developed by Northwest Biotherapeutics. Hum. Vaccines Immunother. 2014, 10, 3139–3145. [Google Scholar] [CrossRef]
- Podrazil, M.; Horvath, R.; Becht, E.; Rozkova, D.; Bilkova, P.; Sochorova, K.; Hromadkova, H.; Kayserova, J.; Vavrova, K.; Lastovicka, J.; et al. Phase I/II Clinical Trial of Dendritic-Cell Based Immunotherapy (DCVAC/PCa) Combined with Chemotherapy in Patients with Metastatic, Castration-Resistant Prostate Cancer. Oncotarget 2015, 6, 18192–18205. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, L.H.; Vujanovic, L.; Santos, P.M.; Maurer, D.M.; Gambotto, A.; Lohr, J.; Li, C.; Waldman, J.; Chandran, U.; Lin, Y.; et al. Multiple Antigen-Engineered DC Vaccines with or without IFNα to Promote Antitumor Immunity in Melanoma. J. Immunother. Cancer 2019, 7, 113. [Google Scholar] [CrossRef]
- Mitsuya, K.; Akiyama, Y.; Iizuka, A.; Miyata, H.; Deguchi, S.; Hayashi, N.; Maeda, C.; Kondou, R.; Kanematsu, A.; Watanabe, K.; et al. Alpha-Type-1 Polarized Dendritic Cell-Based Vaccination in Newly Diagnosed High-Grade Glioma: A Phase II Clinical Trial. Anticancer Res. 2020, 40, 6473–6484. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.-P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of Metastatic Melanoma Patients with Autologous Dendritic Cell (DC) Derived-Exosomes: Results of Thefirst Phase I Clinical Trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A Phase I Study of Dexosome Immunotherapy in Patients with Advanced Non-Small Cell Lung Cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic Cell-Derived Exosomes as Maintenance Immunotherapy after First Line Chemotherapy in NSCLC. Oncoimmunology 2016, 5, e1071008. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorodilova, A.V.; Kitaeva, K.V.; Filin, I.Y.; Mayasin, Y.P.; Kharisova, C.B.; Issa, S.S.; Solovyeva, V.V.; Rizvanov, A.A. The Potential of Dendritic Cell Subsets in the Development of Personalized Immunotherapy for Cancer Treatment. Curr. Issues Mol. Biol. 2023, 45, 8053-8070. https://doi.org/10.3390/cimb45100509
Gorodilova AV, Kitaeva KV, Filin IY, Mayasin YP, Kharisova CB, Issa SS, Solovyeva VV, Rizvanov AA. The Potential of Dendritic Cell Subsets in the Development of Personalized Immunotherapy for Cancer Treatment. Current Issues in Molecular Biology. 2023; 45(10):8053-8070. https://doi.org/10.3390/cimb45100509
Chicago/Turabian StyleGorodilova, Anna Valerevna, Kristina Viktorovna Kitaeva, Ivan Yurevich Filin, Yuri Pavlovich Mayasin, Chulpan Bulatovna Kharisova, Shaza S. Issa, Valeriya Vladimirovna Solovyeva, and Albert Anatolyevich Rizvanov. 2023. "The Potential of Dendritic Cell Subsets in the Development of Personalized Immunotherapy for Cancer Treatment" Current Issues in Molecular Biology 45, no. 10: 8053-8070. https://doi.org/10.3390/cimb45100509
APA StyleGorodilova, A. V., Kitaeva, K. V., Filin, I. Y., Mayasin, Y. P., Kharisova, C. B., Issa, S. S., Solovyeva, V. V., & Rizvanov, A. A. (2023). The Potential of Dendritic Cell Subsets in the Development of Personalized Immunotherapy for Cancer Treatment. Current Issues in Molecular Biology, 45(10), 8053-8070. https://doi.org/10.3390/cimb45100509