
Autophagy Modulation and Its Implications on Glioblastoma Treatment
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Exploring the Mechanism and Application of Autophagy Modulation through the JAK/STAT Pathway
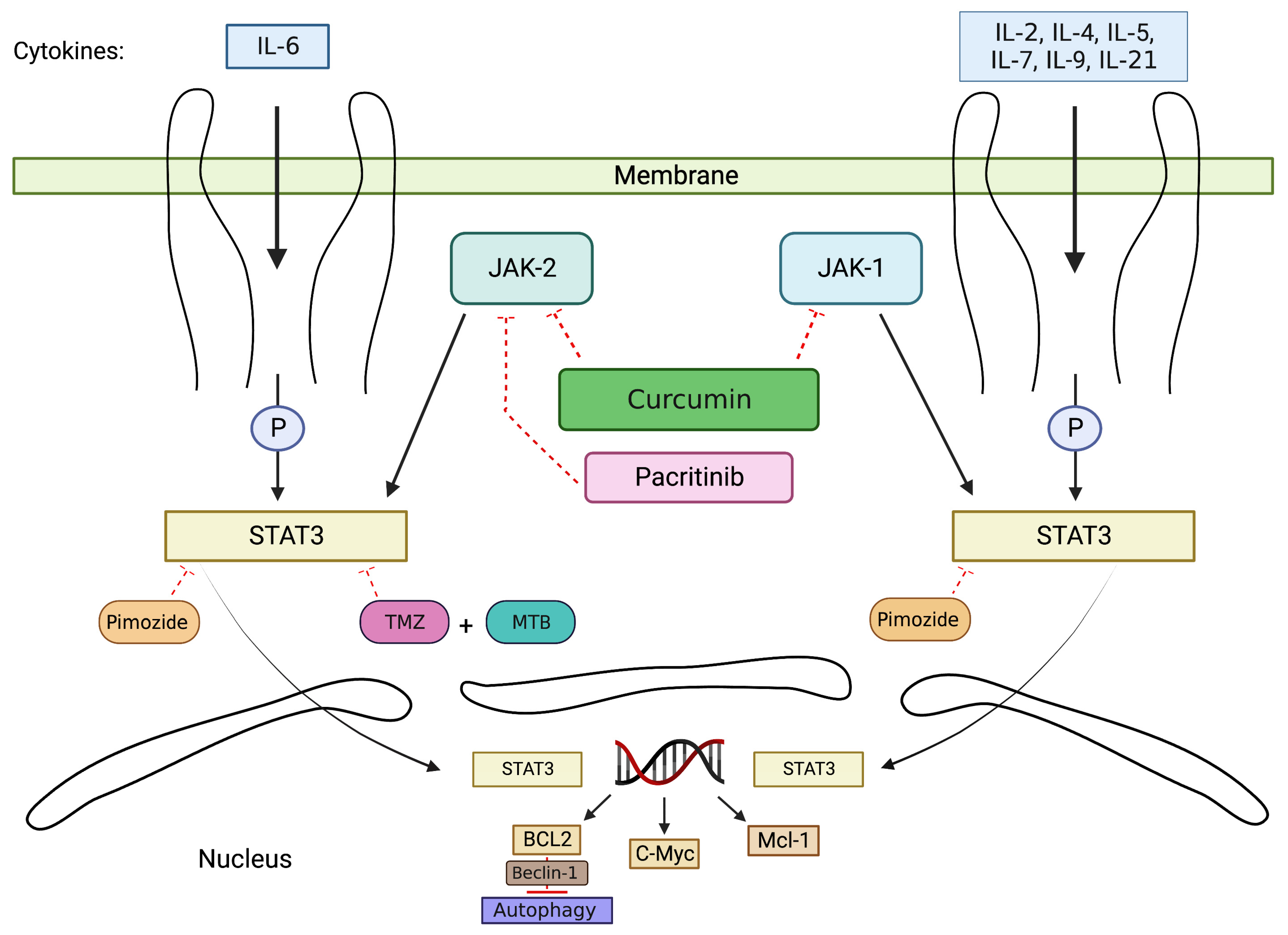
2.1. JAK/STAT Signaling Pathway
2.2. Drugs Induce Cell Apoptosis via Inhibition of JAK/STAT Pathway
2.3. Drugs Enhance the Efficacy of TMZ via JAK/STAT Pathway
3. Unveiling Autophagy Modulation Mechanism and Application via PI3K/AKT/mTOR Pathway
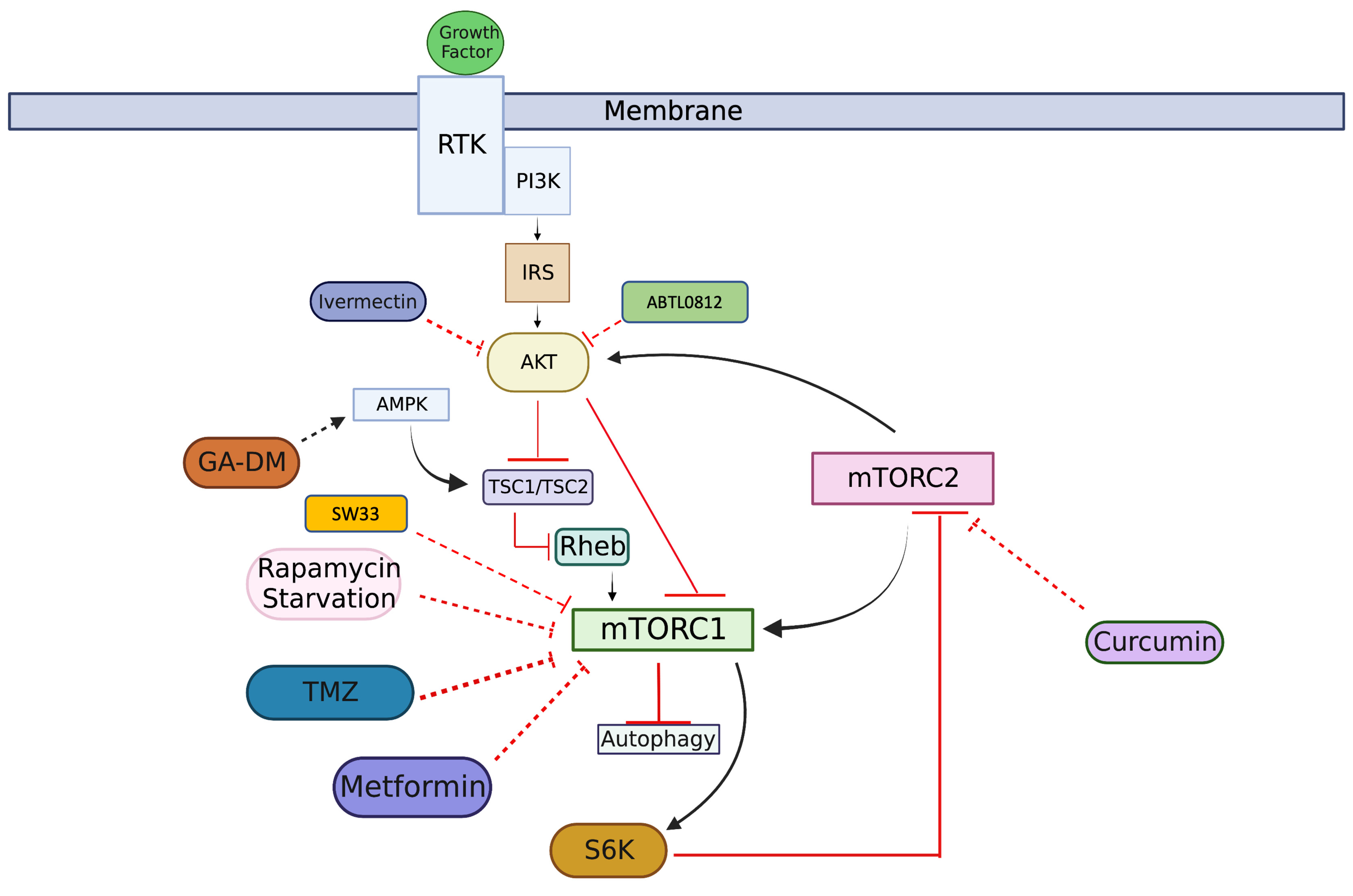
3.1. PI3K/AKT/mTOR Signaling Pathway
3.2. Drugs Induce Autophagy-Mediated Cell Death by Inhibiting AKT/mTOR Pathway
3.3. Drugs Decrease Cell Proliferation by Targeting AKT/mTOR Pathway
4. Future Perspectives for Autophagy Studies
Author Contributions
Funding
Conflicts of Interest
References
- Lefranc, F.; Kiss, R. Autophagy, the Trojan horse to combat glioblastomas. Neurosurg. Focus 2006, 20, E7. [Google Scholar] [CrossRef]
- Cahill, D.; Turcan, S. Origin of Gliomas. Semin. Neurol. 2018, 38, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Epidemiology and Overview of Gliomas. Semin. Oncol. Nurs. 2018, 34, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Batara, D.C.R.; Choi, M.; Shin, H.; Kim, H.; Kim, S. Friend or Foe: Paradoxical Roles of Autophagy in Gliomagenesis. Cells 2021, 10, 1411. [Google Scholar] [CrossRef] [PubMed]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef] [PubMed]
- Wirsching, H.; Galanis, E.; Weller, M. Glioblastoma. Handb. Clin. Neurol. 2016, 134, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Mischel, P.S.; Cloughesy, T.F. Targeted molecular therapy of GBM. Brain Pathol. 2003, 13, 52–61. [Google Scholar] [CrossRef]
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef]
- Kohsaka, S.; Wang, L.; Yachi, K.; Mahabir, R.; Narita, T.; Itoh, T.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol. Cancer Ther. 2012, 11, 1289–1299. [Google Scholar] [CrossRef]
- Bryukhovetskiy, I.; Ponomarenko, A.; Lyakhova, I.; Zaitsev, S.; Zayats, Y.; Korneyko, M.; Eliseikina, M.; Mischenko, P.; Shevchenko, V.; Shanker Sharma, H.; et al. Personalized regulation of glioblastoma cancer stem cells based on biomedical technologies: From theory to experiment (Review). Int. J. Mol. Med. 2018, 42, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Campos, B.; Olsen, L.R.; Urup, T.; Poulsen, H.S. A comprehensive profile of recurrent glioblastoma. Oncogene 2016, 35, 5819–5825. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. CNS cancer: Glioblastoma subtypes revisited. Nat. Rev. Clin. Oncol. 2017, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.; Colapietro, A.; Cristiano, L.; Rossetti, A.; Mattei, V.; Gravina, G.L.; Perez-Montoyo, H.; Yeste-Velasco, M.; Alfon, J.; Domenech, C.; et al. Anticancer effects of ABTL0812, a clinical stage drug inducer of autophagy-mediated cancer cell death, in glioblastoma models. Front. Oncol. 2022, 12, 943064. [Google Scholar] [CrossRef]
- Jiang, H.; White, E.J.; Conrad, C.; Gomez-Manzano, C.; Fueyo, J. Chapter 13 Autophagy Pathways in Glioblastoma. Meth. Enzymol. 2009, 453, 273–286. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, H.; Zhao, Z.; Zhang, Y.; Wang, Z.; Jiang, T.; Bao, Z. Prognostic Correlation of Autophagy-Related Gene Expression-Based Risk Signature in Patients with Glioblastoma. Onco. Targets Ther. 2020, 13, 95–107. [Google Scholar] [CrossRef]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017, 32, 840–855.e8. [Google Scholar] [CrossRef]
- Danial, N.; Hockenbery, D. Hematology: Basic Principles and Practice; Cell Death; Hoffman, R., Ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2018; Volume 18, pp. 186–196. [Google Scholar]
- Wang, L.; Ye, X.; Zhao, T. The physiological roles of autophagy in the mammalian life cycle. Biol. Rev. 2019, 94, 503–516. [Google Scholar] [CrossRef]
- Wang, L.; Klionsky, D.J.; Shen, H. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 2023, 24, 186–203. [Google Scholar] [CrossRef]
- Griffey, C.J.; Yamamoto, A. Macroautophagy in CNS health and disease. Nat. Rev. Neurosci. 2022, 23, 411–427. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Cuttler, K.; Hassan, M.; Carr, J.; Cloete, R.; Bardien, S. Emerging evidence implicating a role for neurexins in neurodegenerative and neuropsychiatric disorders. Open Biol. 2021, 11, 210091. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Broz, D.K.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 Transcriptional Programs Dictate Acute DNA-Damage Responses and Tumor Suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Bourdenx, M.; Gavathiotis, E.; Cuervo, A.M. Chaperone-mediated autophagy: A gatekeeper of neuronal proteostasis. Autophagy 2021, 17, 2040–2042. [Google Scholar] [CrossRef]
- Andrade-Tomaz, M.; de Souza, I.; Rocha, C.R.R.; Gomes, L.R. The Role of Chaperone-Mediated Autophagy in Cell Cycle Control and Its Implications in Cancer. Cells 2020, 9, 2140. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Park, J.; Kim, D.; Yoon, S. Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 2016, 49, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; Di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef] [PubMed]
- Bourdenx, M.; Martín-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 2021, 184, 2696–2714.e25. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Yang, Y.; Klionsky, D.J. Moments in autophagy and disease: Past and present. Mol. Aspects Med. 2021, 82, 100966. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.; Seok, S.; Kim, Y.; Zhang, Y.; Yau, P.; Iwamori, N.; Xu, H.E.; Ma, J.; Kemper, B.; Kemper, J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020, 11, 807. [Google Scholar] [CrossRef] [PubMed]
- Liśkiewicz, D.; Liśkiewicz, A.; Grabowski, M.; Nowacka-Chmielewska, M.M.; Jabłońska, K.; Wojakowska, A.; Marczak, Ł.; Barski, J.J.; Małecki, A. Upregulation of hepatic autophagy under nutritional ketosis. J. Nutr. Biochem. 2021, 93, 108620. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, M.; Fan, J.; Yan, W.; Zha, X.; Song, H.; Wan, R.; Yin, Y.; Wang, W. Ischemia-induced upregulation of autophagy preludes dysfunctional lysosomal storage and associated synaptic impairments in neurons. Autophagy 2021, 17, 1519–1542. [Google Scholar] [CrossRef]
- He, L.; Lu, J.; Yue, Z. Autophagy in ageing and ageing-associated diseases. Acta. Pharmacol. Sin. 2013, 34, 605–611. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Hikita, H.; Tatsumi, T.; Sakamori, R.; Nozaki, Y.; Sakane, S.; Shiode, Y.; Nakabori, T.; Saito, Y.; Hiramatsu, N.; et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 2016, 64, 1994–2014. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Chao, X.; Williams, J.; Fulte, S.; Li, T.; Yang, L.; Ding, W. Autophagy in liver diseases: A review. Mol. Aspects Med. 2021, 82, 100973. [Google Scholar] [CrossRef] [PubMed]
- Bishop, E.; Bradshaw, T.D. Autophagy modulation: A prudent approach in cancer treatment? Cancer Chemother. Pharmacol. 2018, 82, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.R.; Thorburn, A. Autophagy and organelle homeostasis in cancer. Dev. Cell. 2021, 56, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, E.; Fares, J.; Cordero, A.; Rashidi, A.; Arrieta, V.A.; Kanojia, D.; Lesniak, M.S. Repurposing autophagy regulators in brain tumors. Int. J. Cancer 2022, 151, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Zhu, C. The Role of Autophagy in Childhood Central Nervous System Tumors. Curr. Treat. Options Oncol. 2022, 23, 1535–1547. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhang, H. Core autophagy genes and human diseases. Curr. Opin. Cell. Biol. 2019, 61, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, M.; Yue, Z. Autophagy and Parkinson’s Disease. In Autophagy: Biology and Diseases: Clinical Science; Le, W., Ed.; Springer: Singapore, 2020; pp. 21–51. [Google Scholar]
- Hippert, M.M.; O’Toole, P.S.; Thorburn, A. Autophagy in Cancer: Good, Bad, or Both? Cancer Res. 2006, 66, 9349–9351. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, S.K.; Mukhopadhyay, S.; Sinha, N.; Das, D.N.; Panda, P.K.; Patra, S.K.; Maiti, T.K.; Mandal, M.; Dent, P.; Wang, X.; et al. Autophagy: Cancer’s friend or foe? Adv. Cancer Res. 2013, 118, 61–95. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Fan, S.; Qin, T.; Yang, J.; Sun, Y.; Lu, Y.; Mao, J.; Li, L. Role of autophagy in breast cancer and breast cancer stem cells (Review). Int. J. Oncol. 2018, 52, 1057–1070. [Google Scholar] [CrossRef] [PubMed]
- Koustas, E.; Trifylli, E.; Sarantis, P.; Kontolatis, N.I.; Damaskos, C.; Garmpis, N.; Vallilas, C.; Garmpi, A.; Papavassiliou, A.G.; Karamouzis, M.V. The Implication of Autophagy in Gastric Cancer Progression. Life 2021, 11, 1304. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Mohamad Hanif, E.A.; Chin, S. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi-Dehlaghi, F.; Mohammadi, P.; Valipour, E.; Pournaghi, P.; Kiani, S.; Mansouri, K. Autophagy: A challengeable paradox in cancer treatment. Cancer Med. 2023, 12, 11542–11569. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Perez-Montoyo, H. Therapeutic Potential of Autophagy Modulation in Cholangiocarcinoma. Cells 2020, 9, 614. [Google Scholar] [CrossRef]
- Cj, P.; Hv, E.; Vijayakurup, V.; R Menon, G.; Nair, S.; Gopala, S. High LC3/Beclin Expression Correlates with Poor Survival in Glioma: A Definitive Role for Autophagy as Evidenced by In Vitro Autophagic Flux. Pathol. Oncol. Res. 2019, 25, 137–148. [Google Scholar] [CrossRef]
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016, 12, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhang, Y.; Jiang, Y.; Wang, K.; Wang, X.; Zhou, D.; Wang, Y.; Yu, R.; Zhou, X. YAP promotes autophagy and progression of gliomas via upregulating HMGB1. J. Exp. Clin. Cancer Res. 2021, 40, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Perego, C.; Vanoni, C.; Massari, S.; Raimondi, A.; Pola, S.; Cattaneo, M.G.; Francolini, M.; Vicentini, L.M.; Pietrini, G. Invasive behaviour of glioblastoma cell lines is associated with altered organisation of the cadherin-catenin adhesion system. J. Cell Sci. 2002, 115, 3331–3340. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef]
- Camand, E.; Peglion, F.; Osmani, N.; Sanson, M.; Etienne-Manneville, S. N-cadherin expression level modulates integrin-mediated polarity and strongly impacts on the speed and directionality of glial cell migration. J. Cell Sci. 2012, 125, 844–857. [Google Scholar] [CrossRef]
- Kma, L.; Baruah, T.J. The interplay of ROS and the PI3K/Akt pathway in autophagy regulation. Biotechnol. Appl. Biochem. 2022, 69, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kong, Y.; Sun, Y.; Qian, Z.; Gao, C.; Shi, X.; Li, S.; Piao, Y.; Piao, F. Bone marrow mesenchymal stem cells conditioned medium protects VSC4.1 cells against 2,5-hexanedione-induced autophagy via NGF-PI3K/Akt/mTOR signaling pathway. Brain Res. 2018, 1696, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, J.; Mao, Q.; Wu, J.; Wang, Y. The Interaction Between Autophagy and JAK/STAT3 Signaling Pathway in Tumors. Front. Genet. 2022, 13, 880359. [Google Scholar] [CrossRef]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Ou, A.; Ott, M.; Fang, D.; Heimberger, A.B. The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma. Cancers 2021, 13, 437. [Google Scholar] [CrossRef]
- J J O’Shea Targeting the Jak/STAT pathway for immunosuppression. Ann. Rheum. Dis. 2004, 63, ii67–ii71. [CrossRef]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [PubMed]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E.T.3.; Silvennoinen, O.; O’Shea, J.J. The Janus kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Kaushik, I.; Srivastava, S.K. Pimozide Suppresses the Growth of Brain Tumors by Targeting STAT3-Mediated Autophagy. Cells. 2020, 9, 2141. [Google Scholar] [CrossRef] [PubMed]
- Clevenger, C.V. Roles and regulation of stat family transcription factors in human breast cancer. Am. J. Pathol. 2004, 165, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.; Arya, A.; Naema, A.F.; Wong, W.F.; Sethi, G.; Looi, C.Y. Signal Transducer and Activator of Transcription (STATs) Proteins in Cancer and Inflammation: Functions and Therapeutic Implication. Front. Oncol. 2019, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef]
- Abroun, S.; Saki, N.; Ahmadvand, M.; Asghari, F.; Salari, F.; Rahim, F. STATs: An Old Story, Yet Mesmerizing. Cell J. 2015, 17, 395–411. [Google Scholar]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Mukthavaram, R.; Ouyang, X.; Saklecha, R.; Jiang, P.; Nomura, N.; Pingle, S.C.; Guo, F.; Makale, M.; Kesari, S. Effect of the JAK2/STAT3 inhibitor SAR317461 on human glioblastoma tumorspheres. J. Transl. Med. 2015, 13, 269. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, C.; Mori, N. The antipsychotic drug pimozide is effective against human T-cell leukemia virus type 1-infected T cells. Eur. J. Pharmacol. 2021, 908, 174373. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Henkel, L.; Linder, B.; Zielke, S.; Tascher, G.; Trautmann, S.; Geisslinger, G.; Münch, C.; Fulda, S.; Tegeder, I.; et al. Autophagy activation, lipotoxicity and lysosomal membrane permeabilization synergize to promote pimozide- and loperamide-induced glioma cell death. Autophagy 2021, 17, 3424–3443. [Google Scholar] [CrossRef] [PubMed]
- Kaza, N.; Kohli, L.; Roth, K.A. Autophagy in brain tumors: A new target for therapeutic intervention. Brain Pathol. 2012, 22, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Solís, C.; Serrano-Garcia, N.; Escamilla-Ramírez, Á.; Castillo-Rodríguez, R.A.; Jimenez-Farfan, D.; Palencia, G.; Calvillo, M.; Alvarez-Lemus, M.A.; Flores-Nájera, A.; Cruz-Salgado, A.; et al. Autophagic and Apoptotic Pathways as Targets for Chemotherapy in Glioblastoma. Int. J. Mol. Sci. 2018, 19, 3773. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold. Spring Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef]
- Feng, Y.; Ke, C.; Tang, Q.; Dong, H.; Zheng, X.; Lin, W.; Ke, J.; Huang, J.; Yeung, S.J.; Zhang, H. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis. 2014, 5, e1088. [Google Scholar] [CrossRef]
- Tai, W.; Shiau, C.; Chen, H.; Liu, C.; Lin, C.; Cheng, A.; Chen, P.; Chen, K. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef] [PubMed]
- Marquez, R.T.; Xu, L. Bcl-2:Beclin 1 complex: Multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am. J. Cancer Res. 2012, 2, 214–221. [Google Scholar] [PubMed]
- Mohamadian, M.; Ahmadi, S.S.; Bahrami, A.; Ferns, G.A. Review on the Therapeutic Potential of Curcumin and its Derivatives on Glioma Biology. Neurochem. Res. 2022, 47, 2936–2953. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Palanivelu, K. The effect of curcumin (turmeric) on Alzheimer’s disease: An overview. Ann. Indian Acad. Neurol. 2008, 11, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Askarizadeh, A.; Barreto, G.E.; Henney, N.C.; Majeed, M.; Sahebkar, A. Neuroprotection by curcumin: A review on brain delivery strategies. Int. J. Pharm. 2020, 585, 119476. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Biagioni, F.; Busceti, C.L.; Lazzeri, G.; Frati, A.; Fornai, F. The Multi-Faceted Effect of Curcumin in Glioblastoma from Rescuing Cell Clearance to Autophagy-Independent Effects. Molecules 2020, 25, 4839. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.C.; Kamarudin, M.N.A.; Naidu, R. Anticancer Mechanism of Curcumin on Human Glioblastoma. Nutrients 2021, 13, 950. [Google Scholar] [CrossRef]
- Porro, C.; Cianciulli, A.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. Curcumin Regulates Anti-Inflammatory Responses by JAK/STAT/SOCS Signaling Pathway in BV-2 Microglial Cells. Biology 2019, 8, 51. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Rafiei, H.; Mohammadinejad, R.; Afshar, E.G.; Farkhondeh, T.; Samarghandian, S. Potential therapeutic effects of curcumin mediated by JAK/STAT signaling pathway: A review. Phytother. Res. 2020, 34, 1745–1760. [Google Scholar] [CrossRef]
- Klinger, N.V.; Mittal, S. Therapeutic Potential of Curcumin for the Treatment of Brain Tumors. Oxid. Med. Cell Longev. 2016, 2016, 9324085. [Google Scholar] [CrossRef]
- Weissenberger, J.; Priester, M.; Bernreuther, C.; Rakel, S.; Glatzel, M.; Seifert, V.; Kögel, D. Dietary curcumin attenuates glioma growth in a syngeneic mouse model by inhibition of the JAK1,2/STAT3 signaling pathway. Clin. Cancer Res. 2010, 16, 5781–5795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J. Cellular and Molecular Mechanisms in Idiopathic Pulmonary Fibrosis. Adv. Respir. Med. 2023, 91, 5. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.J.; Ray, S.K. Regulation of autophagy as a therapeutic option in glioblastoma. Apoptosis 2021, 26, 574–599. [Google Scholar] [CrossRef] [PubMed]
- Karachi, A.; Dastmalchi, F.; Mitchell, D.A.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro. Oncol. 2018, 20, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug. Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, A.; Xu, Y.; Xin, Y. Momelotinib sensitizes glioblastoma cells to temozolomide by enhancement of autophagy via JAK2/STAT3 inhibition. Oncol. Rep. 2019, 41, 1883–1892. [Google Scholar] [CrossRef]
- Sanati, M.; Binabaj, M.M.; Ahmadi, S.S.; Aminyavari, S.; Javid, H.; Mollazadeh, H.; Bibak, B.; Mohtashami, E.; Jamialahmadi, T.; Afshari, A.R.; et al. Recent advances in glioblastoma multiforme therapy: A focus on autophagy regulation. Biomed. Pharmacother. 2022, 155, 113740. [Google Scholar] [CrossRef]
- Escamilla-Ramírez, A.; Castillo-Rodríguez, R.A.; Zavala-Vega, S.; Jimenez-Farfan, D.; Anaya-Rubio, I.; Briseño, E.; Palencia, G.; Guevara, P.; Cruz-Salgado, A.; Sotelo, J.; et al. Autophagy as a Potential Therapy for Malignant Glioma. Pharmaceuticals 2020, 13, 156. [Google Scholar] [CrossRef]
- Jensen, K.V.; Cseh, O.; Aman, A.; Weiss, S.; Luchman, H.A. The JAK2/STAT3 inhibitor pacritinib effectively inhibits patient-derived GBM brain tumor initiating cells in vitro and when used in combination with temozolomide increases survival in an orthotopic xenograft model. PLoS ONE 2017, 12, e0189670. [Google Scholar] [CrossRef]
- Verstovsek, S.; Odenike, O.; Singer, J.W.; Granston, T.; Al-Fayoumi, S.; Deeg, H.J. Phase 1/2 study of pacritinib, a next generation JAK2/FLT3 inhibitor, in myelofibrosis or other myeloid malignancies. J. Hematol. Oncol. 2016, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ma, H.; Wang, Z.L.; Li, W.H.; Liu, H.; Zhao, Y.X. The PI3K/AKT/mTOR pathway regulates autophagy to induce apoptosis of alveolar epithelial cells in chronic obstructive pulmonary disease caused by PM2.5 particulate matter. J. Int. Med. Res. 2020, 48, 300060520927919. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Qiu, H.; Lin, L.; Zhang, S.; Li, D.; Jin, D. Inhibition of PI3K/AKT/mTOR Signalling Pathway Activates Autophagy and Suppresses Peritoneal Fibrosis in the Process of Peritoneal Dialysis. Front. Physiol. 2022, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, S.; Zhang, R.; Lin, J.; Tian, Y.; Yang, Y.; Liu, J.; Ma, Y. Neuroprotective effect of astragalin via activating PI3K/Akt-mTOR-mediated autophagy on APP/PS1 mice. Cell Death Discov. 2023, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.; Mier, J.W.; Atkins, M.B. PI3K/Akt/mTOR Pathway: A Growth and Proliferation Pathway. In Renal Cell Carcinoma: Molecular Targets and Clinical Applications; Bukowski, R.M., Figlin, R.A., Motzer, R.J., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 267–285. [Google Scholar]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Tsuji-Tamura, K.; Sato, M.; Fujita, M.; Tamura, M. The role of PI3K/Akt/mTOR signaling in dose-dependent biphasic effects of glycine on vascular development. Biochem. Biophys. Res. Commun. 2020, 529, 596–602. Available online: https://www.sciencedirect.com/science/article/pii/S0006291X20313036 (accessed on 25 September 2023). [CrossRef]
- Ryskalin, L.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Gambardella, S.; Frati, A.; Fornai, F. mTOR-Dependent Cell Proliferation in the Brain. Biomed. Res. Int. 2017, 2017, 7082696. [Google Scholar] [CrossRef]
- Xu, W.; Hua, H.; Chiu, Y.; Li, G.; Zhi, H.; Yu, Z.; Ren, F.; Luo, Y.; Cui, W. CD146 Regulates Growth Factor-Induced mTORC2 Activity Independent of the PI3K and mTORC1 Pathways. Cell Rep. 2019, 29, 1311–1322.e5. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. [Google Scholar] [CrossRef] [PubMed]
- Marinković, M.; Šprung, M.; Buljubašić, M.; Novak, I. Autophagy Modulation in Cancer: Current Knowledge on Action and Therapy. Oxid. Med. Cell Longev. 2018, 2018, 8023821. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Bastidas, R.J.; Shertz, C.A.; Lee, S.C.; Heitman, J.; Cardenas, M.E. Rapamycin exerts antifungal activity in vitro and in vivo against Mucor circinelloides via FKBP12-dependent inhibition of Tor. Eukaryot. Cell 2012, 11, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005, 65, 3336–3346. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Kim, S.Y.; Kim, S.W. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy 2013, 9, 1631–1632. [Google Scholar] [CrossRef] [PubMed]
- Göktuna, S.İ. IKBKE inhibits TSC1 to activate the mTOR/S6K pathway for oncogenic transformation. Turk. J. Biol. 2018, 42, 268–278. [Google Scholar] [CrossRef]
- Tavares, M.R.; Pavan, I.C.B.; Amaral, C.L.; Meneguello, L.; Luchessi, A.D.; Simabuco, F.M. The S6K protein family in health and disease. Life Sci. 2015, 131, 1–10. [Google Scholar] [CrossRef]
- Xia, Q.; Xu, M.; Zhang, P.; Liu, L.; Meng, X.; Dong, L. Therapeutic Potential of Autophagy in Glioblastoma Treatment With Phosphoinositide 3-Kinase/Protein Kinase B/Mammalian Target of Rapamycin Signaling Pathway Inhibitors. Front. Oncol. 2020, 10, 572904. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Zhang, P.; Zhang, C.; Wang, W.; Wu, M.; Xu, W.; Tao, L.; Li, Z.; Zhang, Y. Cytotoxicity and Autophagy Induced by Ivermectin via AMPK/mTOR Signaling Pathway in RAW264.7 Cells. Molecules 2023, 28, 2201. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, T.; Li, M.; Lin, Y.; Liu, Y.; Tang, S.; Dai, C. Ivermectin-Induced Apoptotic Cell Death in Human SH-SY5Y Cells Involves the Activation of Oxidative Stress and Mitochondrial Pathway and Akt/mTOR-Pathway-Mediated Autophagy. Antioxidants 2022, 11, 908. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, H.; Chen, C.; Wang, X.; Qu, F.; Wang, H.; Yang, K.; Wang, Q.; Zhao, N.; Meng, J.; et al. Ivermectin induces autophagy-mediated cell death through the AKT/mTOR signaling pathway in glioma cells. Biosci. Rep. 2019, 39, BSR20192489. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Li, W.; Xu, H.; Liu, J.; Ren, L.; Yang, Y.; Li, S.; Wang, J.; Ji, T.; Du, G. Sinomenine ester derivative inhibits glioblastoma by inducing mitochondria-dependent apoptosis and autophagy by PI3K/AKT/mTOR and AMPK/mTOR pathway. Acta. Pharm. Sin. B 2021, 11, 3465–3480. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, F.; Liao, W.; Yu, L.; Hu, Z.; Li, M.; Xia, H. Curcumin suppresses glioblastoma cell proliferation by p-AKT/mTOR pathway and increases the PTEN expression. Arch. Biochem. Biophys. 2020, 689, 108412. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Roy, T.; Uddin, M.B.; Banang-Mbeumi, S.; Chamcheu, R.N.; Walker, A.L.; Liu, Y.; Huang, S. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells 2019, 8, 803. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Feng, L.; Ye, L.; Wang, L.; Xiao, B.; Tao, X.; Chang, Q. Ganoderic Acid A Metabolites and Their Metabolic Kinetics. Front. Pharmacol. 2017, 8, 101. [Google Scholar] [CrossRef]
- Cheng, Y.; Xie, P. Ganoderic acid A holds promising cytotoxicity on human glioblastoma mediated by incurring apoptosis and autophagy and inactivating PI3K/AKT signaling pathway. J. Biochem. Mol. Toxicol. 2019, 33, e22392. [Google Scholar] [CrossRef]
- Wu, G.; Lu, J.; Guo, J.; Li, Y.; Tan, W.; Dang, Y.; Zhong, Z.; Xu, Z.; Chen, X.; Wang, Y. Ganoderic acid DM, a natural triterpenoid, induces DNA damage, G1 cell cycle arrest and apoptosis in human breast cancer cells. Fitoterapia 2012, 83, 408–414. [Google Scholar] [CrossRef]
- Feng, Z.; Wu, R.; Lin, M.; Hu, W. Tumor suppressor p53: New functions of an old protein. Front. Biol. 2011, 6, 58–68. [Google Scholar] [CrossRef]
- Liang, C.; Tian, D.; Liu, Y.; Li, H.; Zhu, J.; Li, M.; Xin, M.; Xia, J. Review of the molecular mechanisms of Ganoderma lucidum triterpenoids: Ganoderic acids A, C2, D, F, DM, X and Y. Eur. J. Med. Chem. 2019, 174, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast APG8P, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Kalamida, D.; Giatromanolaki, A.; Zois, C.E.; Sivridis, E.; Pouliliou, S.; Mitrakas, A.; Gatter, K.C.; Harris, A.L. Autophagosome Proteins LC3A, LC3B and LC3C Have Distinct Subcellular Distribution Kinetics and Expression in Cancer Cell Lines. PLoS ONE 2015, 10, e0137675. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Fayos, A.C.; G-García, M.E.; Pérez-Gómez, J.M.; Montero-Hidalgo, A.J.; Martín-Colom, J.; Doval-Rosa, C.; Blanco-Acevedo, C.; Torres, E.; Toledano-Delgado, Á.; Sánchez-Sánchez, R.; et al. Metformin and simvastatin exert additive antitumour effects in glioblastoma via senescence-state: Clinical and translational evidence. eBioMedicine 2023, 90, 104484. [Google Scholar] [CrossRef]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Kulesza, B.; Jonak, K.; Baj, J.; Grochowski, C. Metformin as Potential Therapy for High-Grade Glioma. Cancers 2020, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Guarnaccia, L.; Navone, S.E.; Masseroli, M.M.; Balsamo, M.; Caroli, M.; Valtorta, S.; Moresco, R.M.; Campanella, R.; Schisano, L.; Fiore, G.; et al. Effects of Metformin as Add-On Therapy against Glioblastoma: An Old Medicine for Novel Oncology Therapeutics. Cancers 2022, 14, 1412. [Google Scholar] [CrossRef]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.; Lemarié, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Cohen-Jonathan Moyal, E.; et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS ONE 2015, 10, e0123721. [Google Scholar] [CrossRef]
- Scotland, S.; Saland, E.; Skuli, N.; de Toni, F.; Boutzen, H.; Micklow, E.; Sénégas, I.; Peyraud, R.; Peyriga, L.; Théodoro, F.; et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia 2013, 27, 2129–2138. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Tanti, J.; Bost, F. The combination of metformin and 2-deoxyglucose inhibits autophagy and induces AMPK-dependent apoptosis in prostate cancer cells. Autophagy 2010, 6, 670–671. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Huang, F.; Wilson, K.A.; Tan-Wilson, A. Protein storage vacuole acidification as a control of storage protein mobilization in soybeans. J. Exp. Bot. 2007, 58, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Sooro, M.A.; Zhang, P. Autophagic Regulation of p62 is Critical for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Xiao, D.; Wang, L.; Dong, L.; Yan, Z.; Shen, Z.; Chen, S.; Chen, Y.; Zhao, W. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012, 3, e275. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chhipa, R.R.; Pooya, S.; Wortman, M.; Yachyshin, S.; Chow, L.M.L.; Kumar, A.; Zhou, X.; Sun, Y.; Quinn, B.; et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. USA 2014, 111, 435. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Morales, L.D.; Zhang, Y.; Mito, S.; Tsin, A. Niclosamide induces protein ubiquitination and inhibits multiple pro-survival signaling pathways in the human glioblastoma U-87 MG cell line. PLoS ONE 2017, 12, e0184324. [Google Scholar] [CrossRef]
- Cheng, B.; Wei, Y.; Guerra, L.; Shirvani-Arani, R.; Balderas, S.; Valdez, L.; Tsin, A.; Fang, X. Anti-cancer effect of Cissus quadrangularis on human glioblastoma cells. RPS Pharm. Pharmacol. Rep. 2023, 2, rqad014. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Rodriguez, A.S.; Morales, M.A.; Fang, X. Autophagy Modulation and Its Implications on Glioblastoma Treatment. Curr. Issues Mol. Biol. 2023, 45, 8687-8703. https://doi.org/10.3390/cimb45110546
Chen J, Rodriguez AS, Morales MA, Fang X. Autophagy Modulation and Its Implications on Glioblastoma Treatment. Current Issues in Molecular Biology. 2023; 45(11):8687-8703. https://doi.org/10.3390/cimb45110546
Chicago/Turabian StyleChen, Johnny, Andrea Salinas Rodriguez, Maximiliano Arath Morales, and Xiaoqian Fang. 2023. "Autophagy Modulation and Its Implications on Glioblastoma Treatment" Current Issues in Molecular Biology 45, no. 11: 8687-8703. https://doi.org/10.3390/cimb45110546