
Synthesis, Molecular Docking, and Dynamic Simulation Targeting Main Protease (Mpro) of New, Thiazole Clubbed Pyridine Scaffolds as Potential COVID-19 Inhibitors

 ,
,  , , ,
, , ,  , ,
, ,  and
and 
Abstract
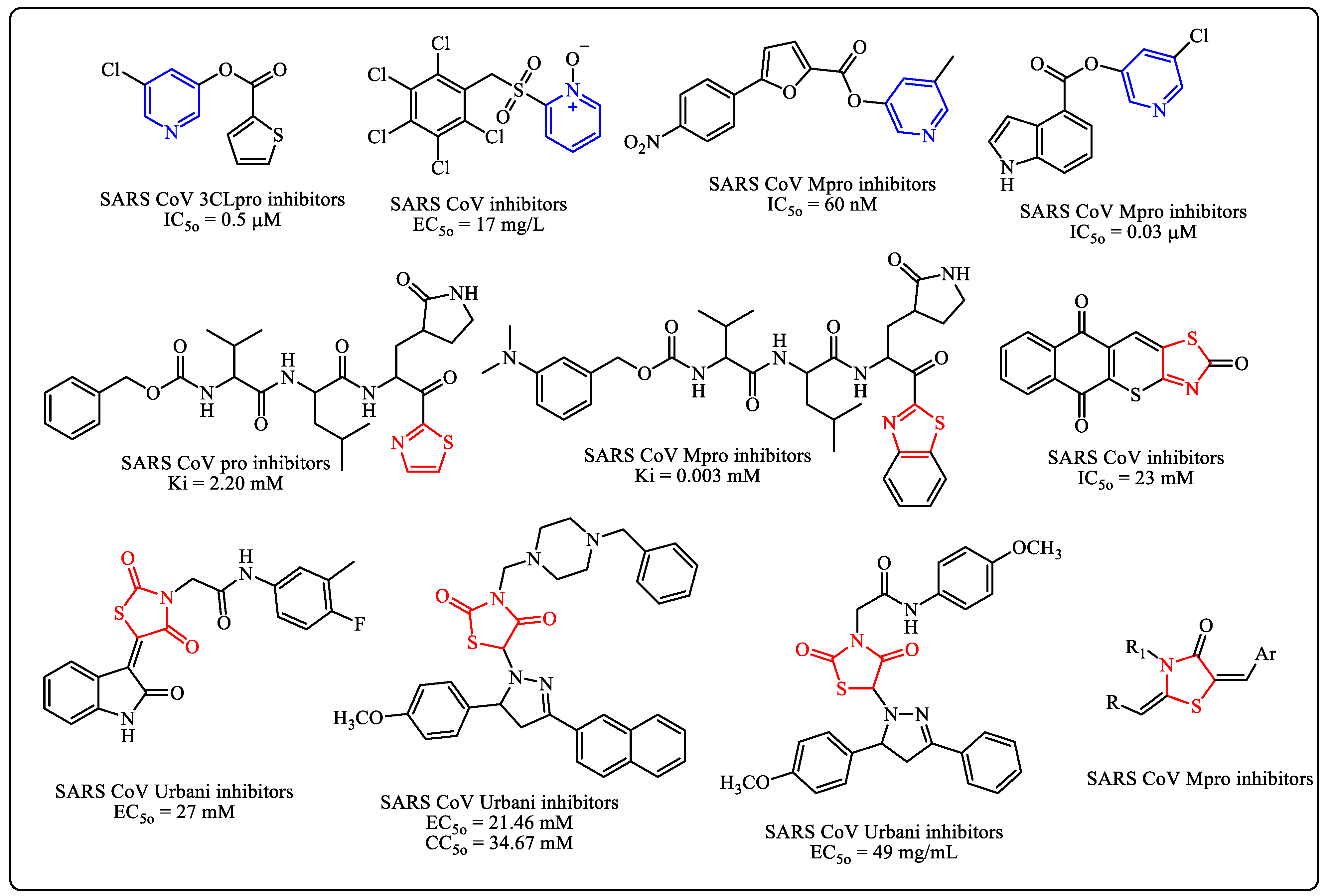
1. Introduction
2. Experimental
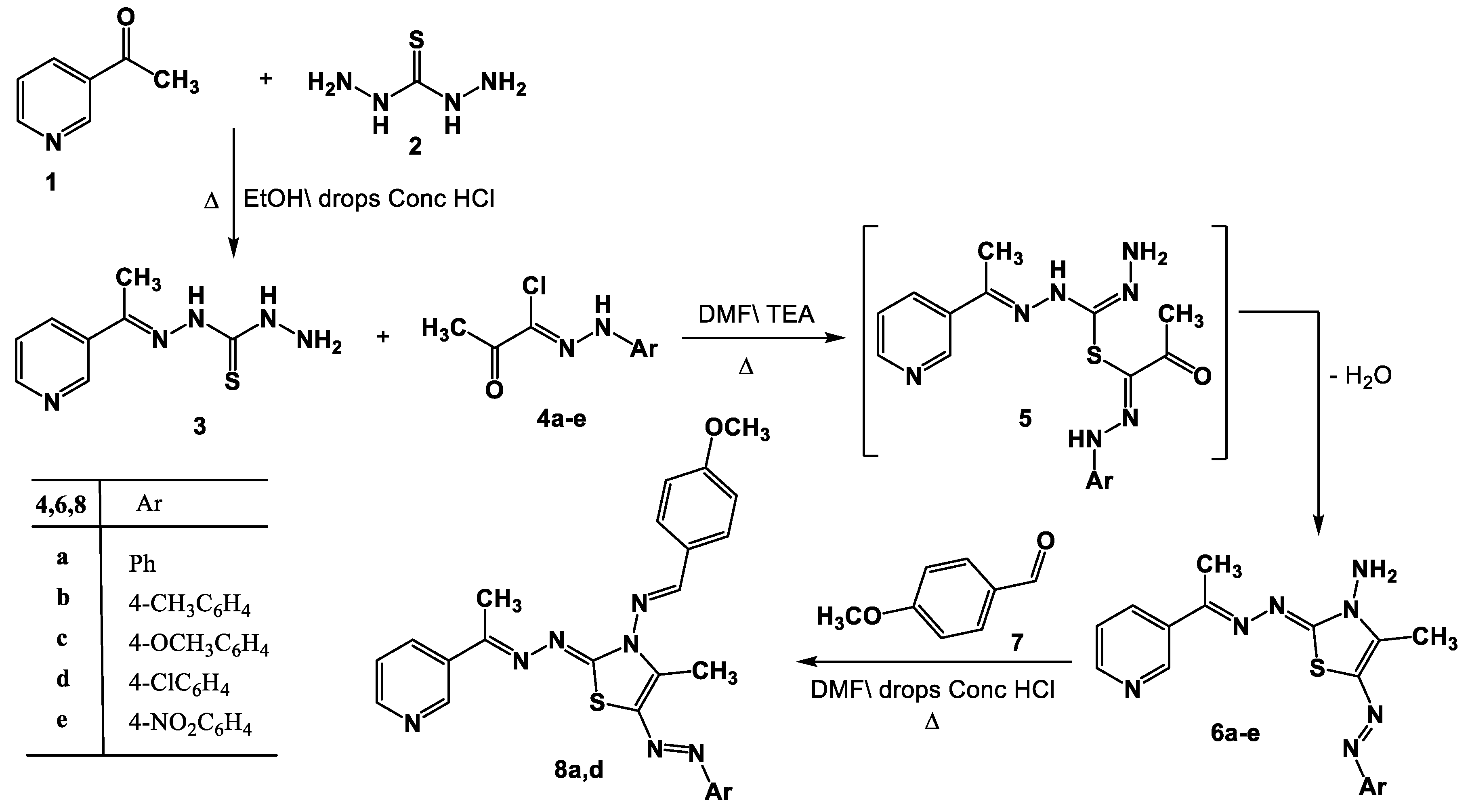
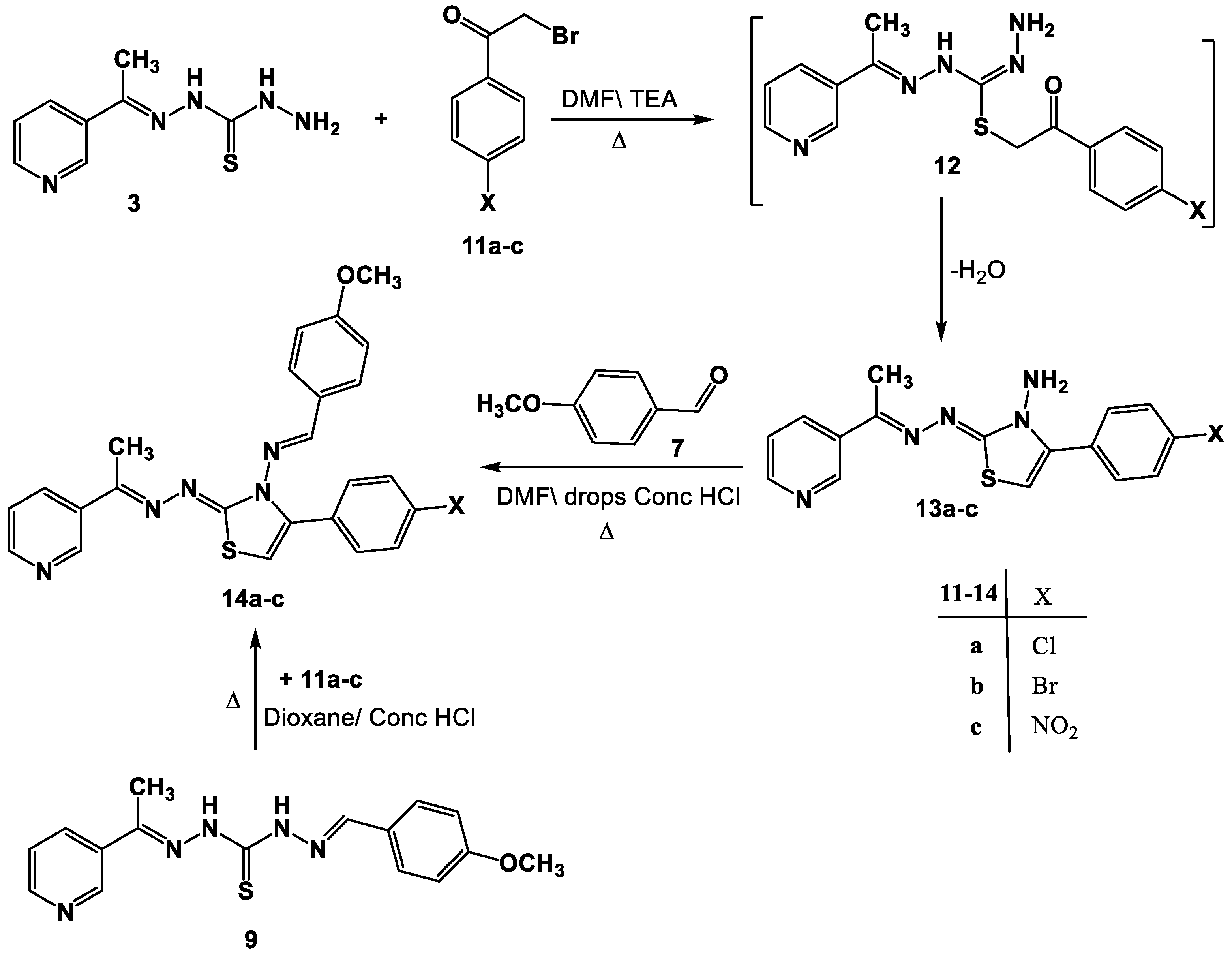
2.1. Chemistry
- Synthesis of thiazole derivatives 6a–e and 13a–c.
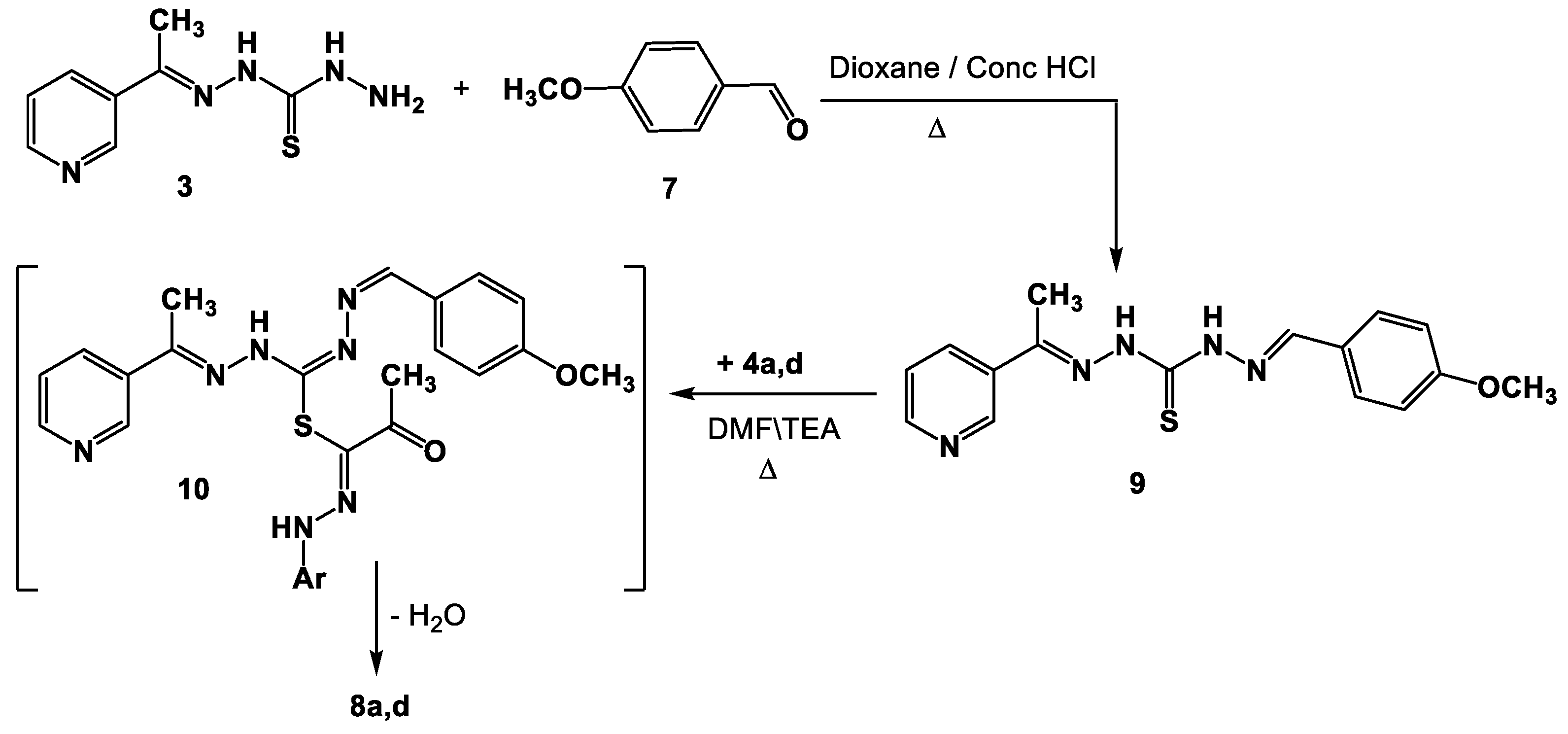
- Synthesis of Schiff bases 8a,d and 14a–c.
- Physical and spectral data of all synthesized compounds 6a–e, 13a–c, 8a,d and 14a–c are frond in the supporting information file.
2.2. Docking Method
2.2.1. Molecular Dynamics Simulation (MDs)
2.2.2. Binding Free Energy Analysis
- -
- ΔGbind specifies the binding free energy;
- -
- ΔGMM specifies the difference between the free energies of the ligand–macromolecule complex and the total energies of receptor and ligand in isolated forms;
- -
- ΔGSolv specifies the differences in the GSA solvation energies of the ligand–macromolecule complex and the sum of the solvation energies of the receptor and the ligand in the unbound state;
- -
- ΔGSA specifies the difference in the surface area energies for the receptor and the ligand.
3. Results and Discussion
3.1. Chemistry
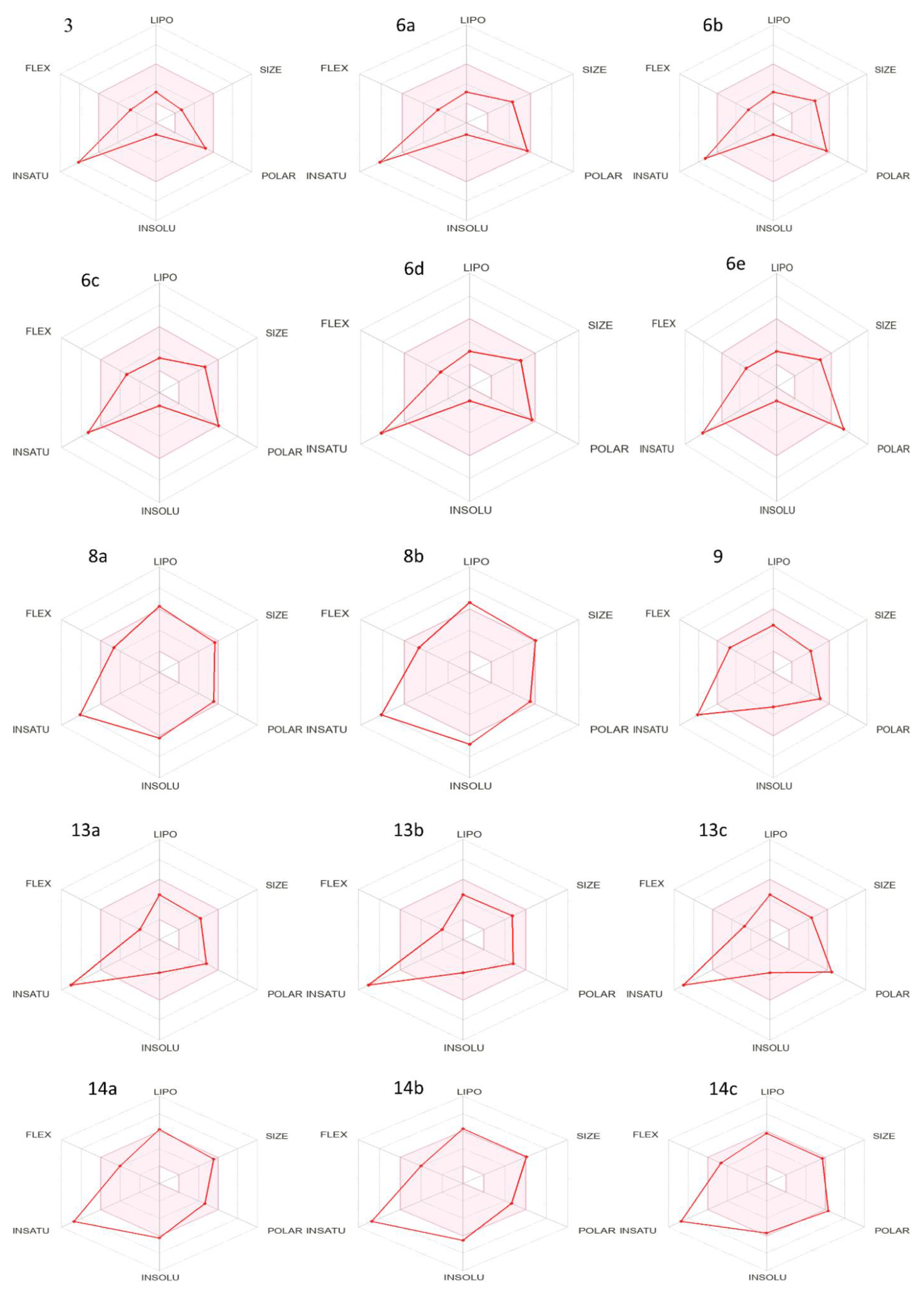
3.2. Physiochemical and Pharmacokinetics Profiling
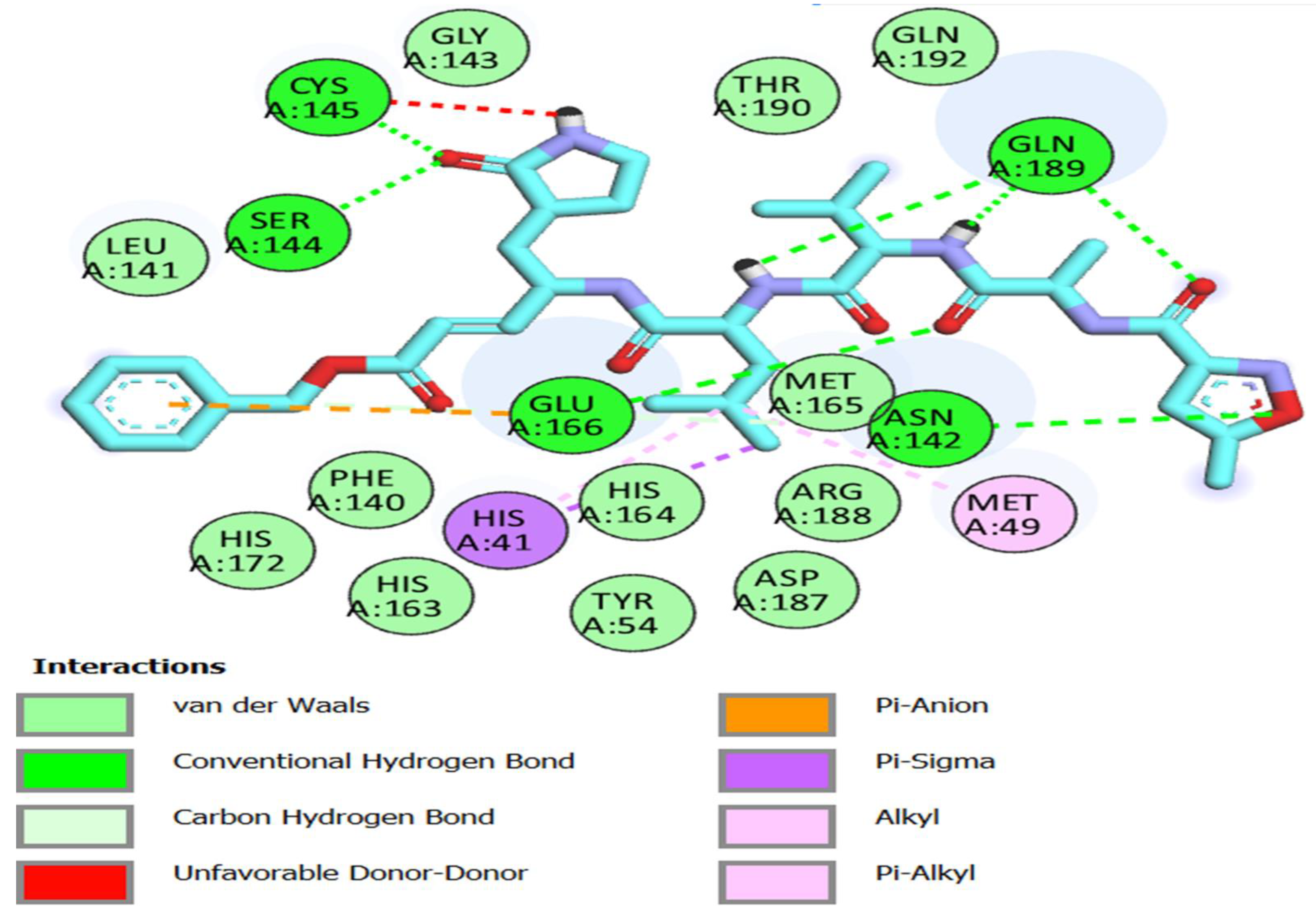
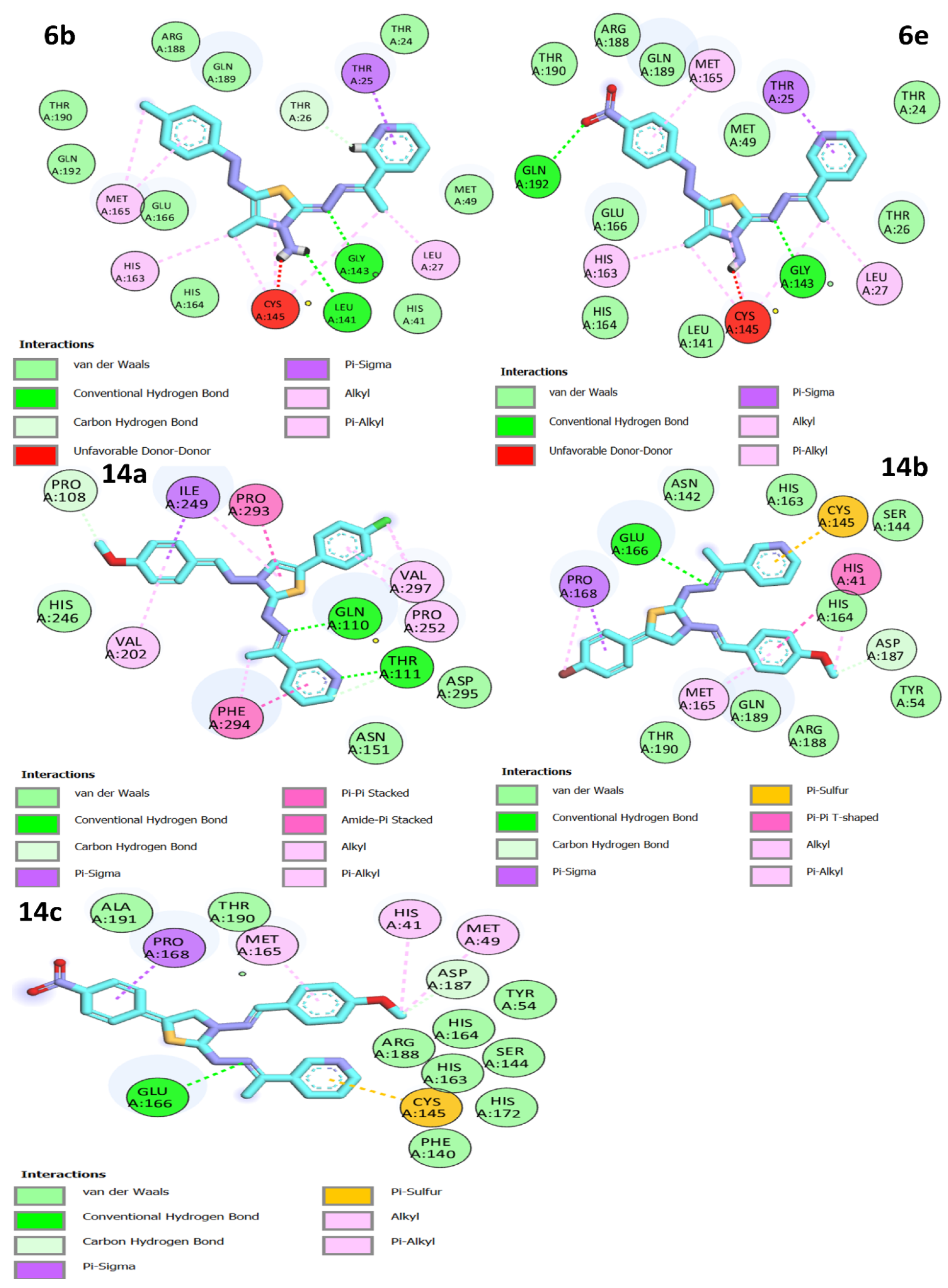
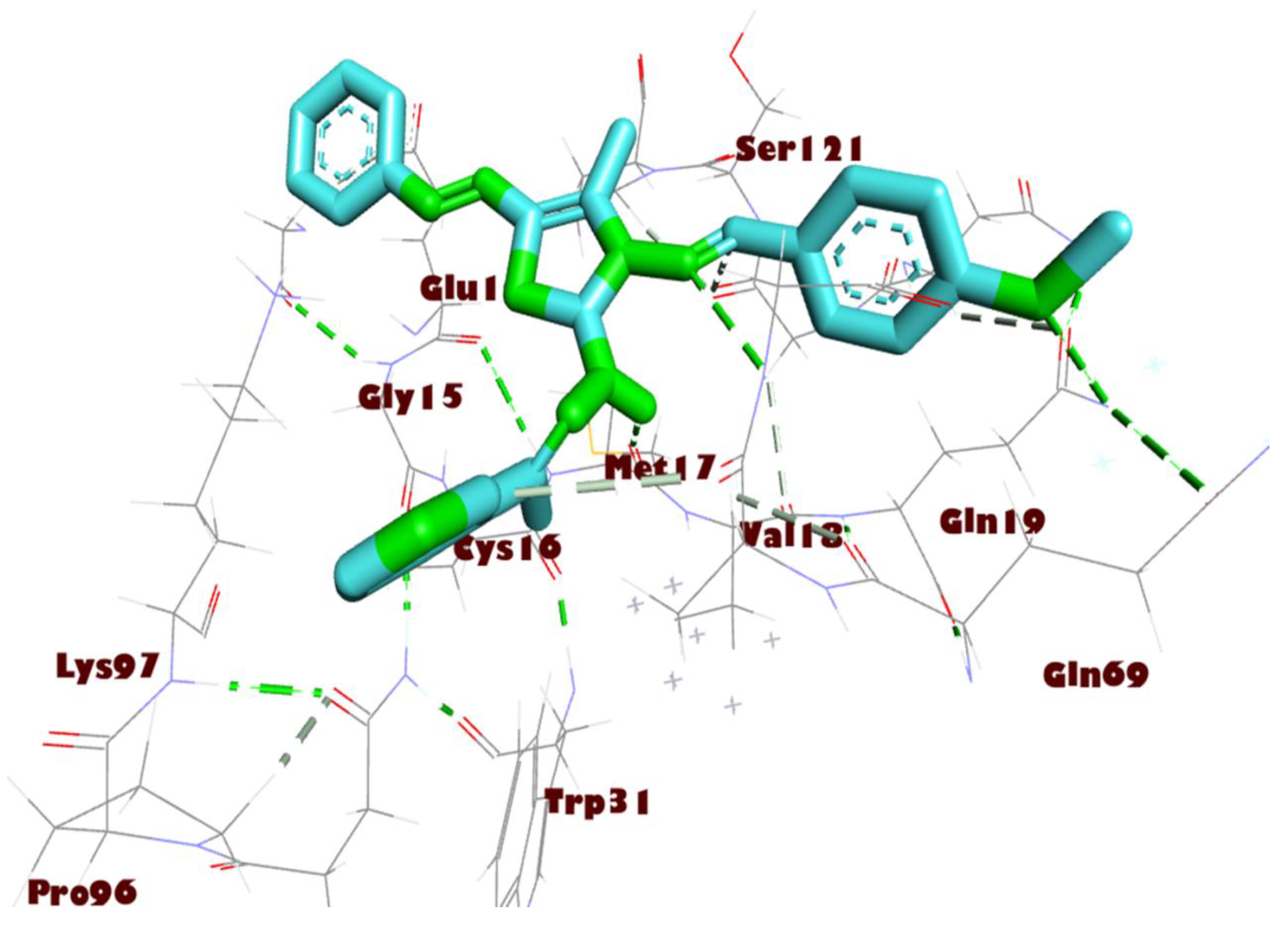
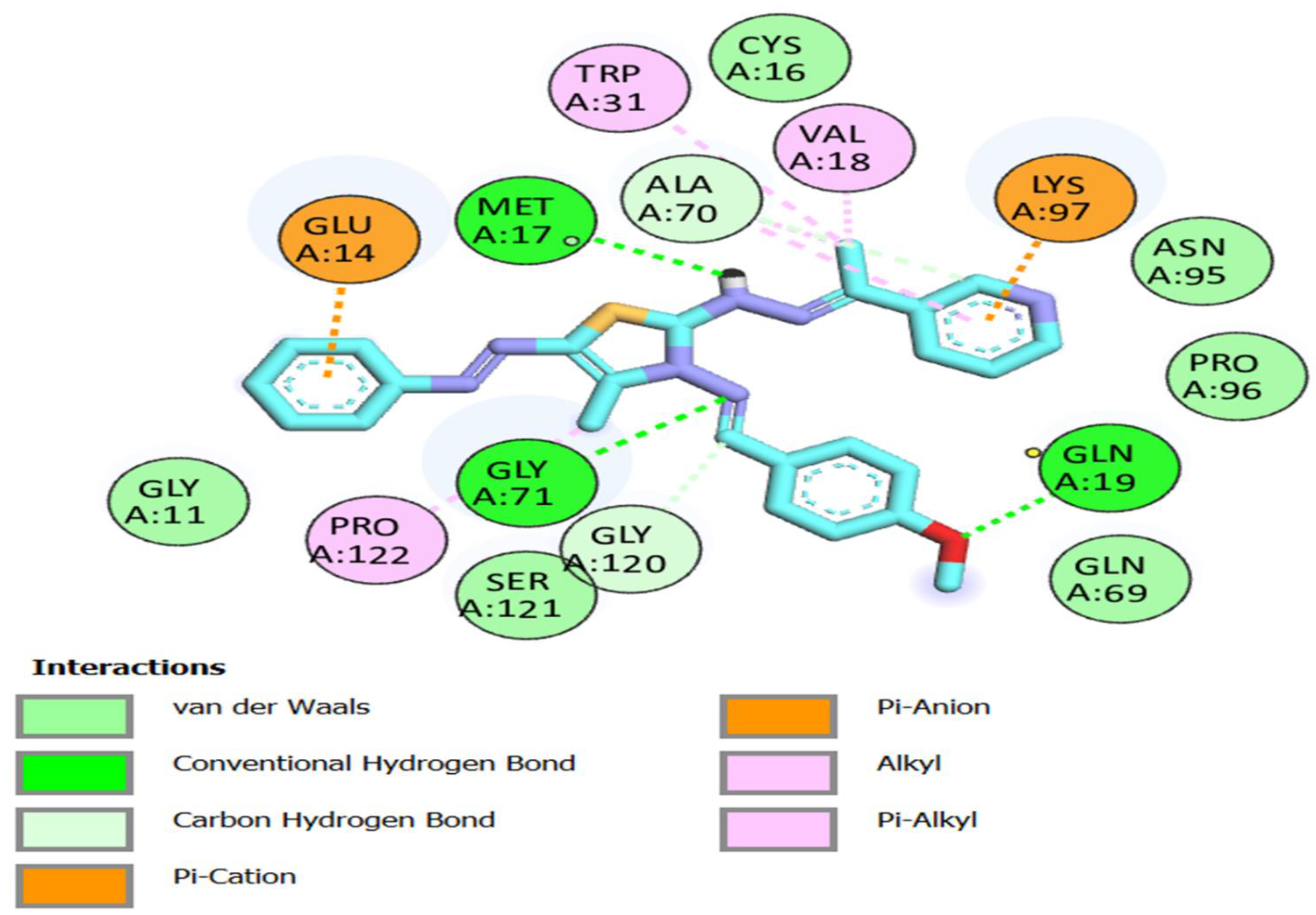
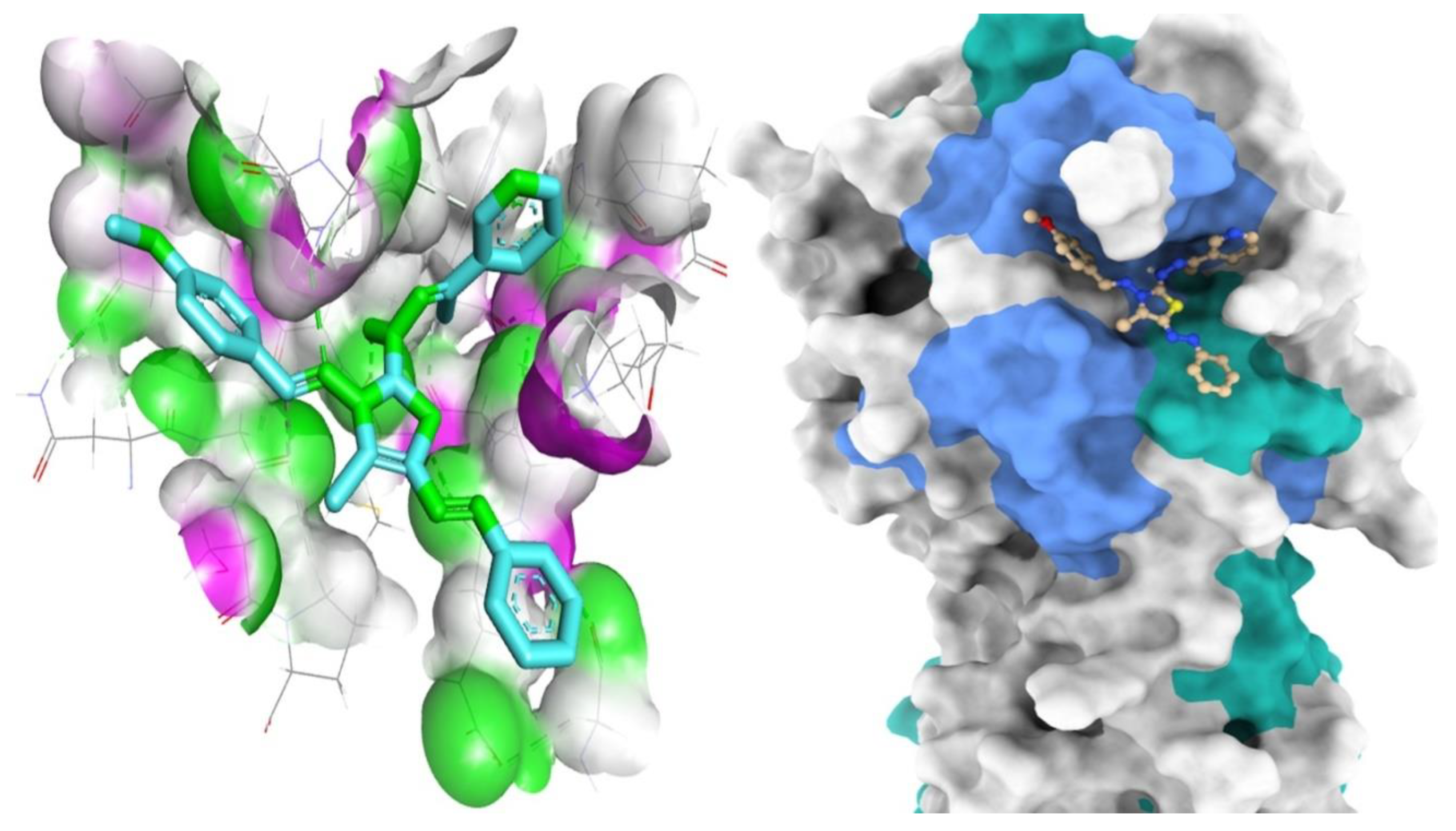
3.3. Molecular Docking Studies
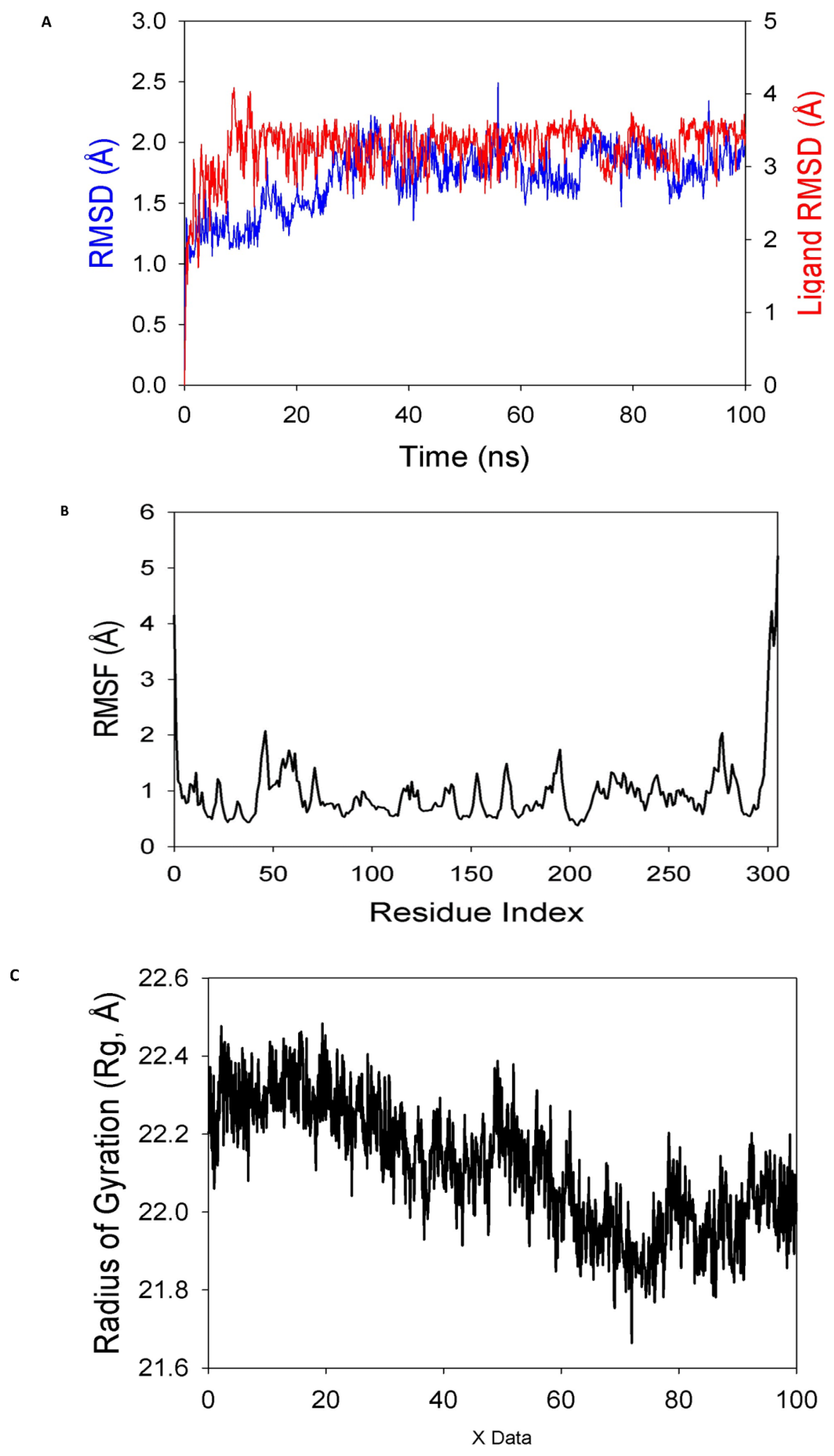
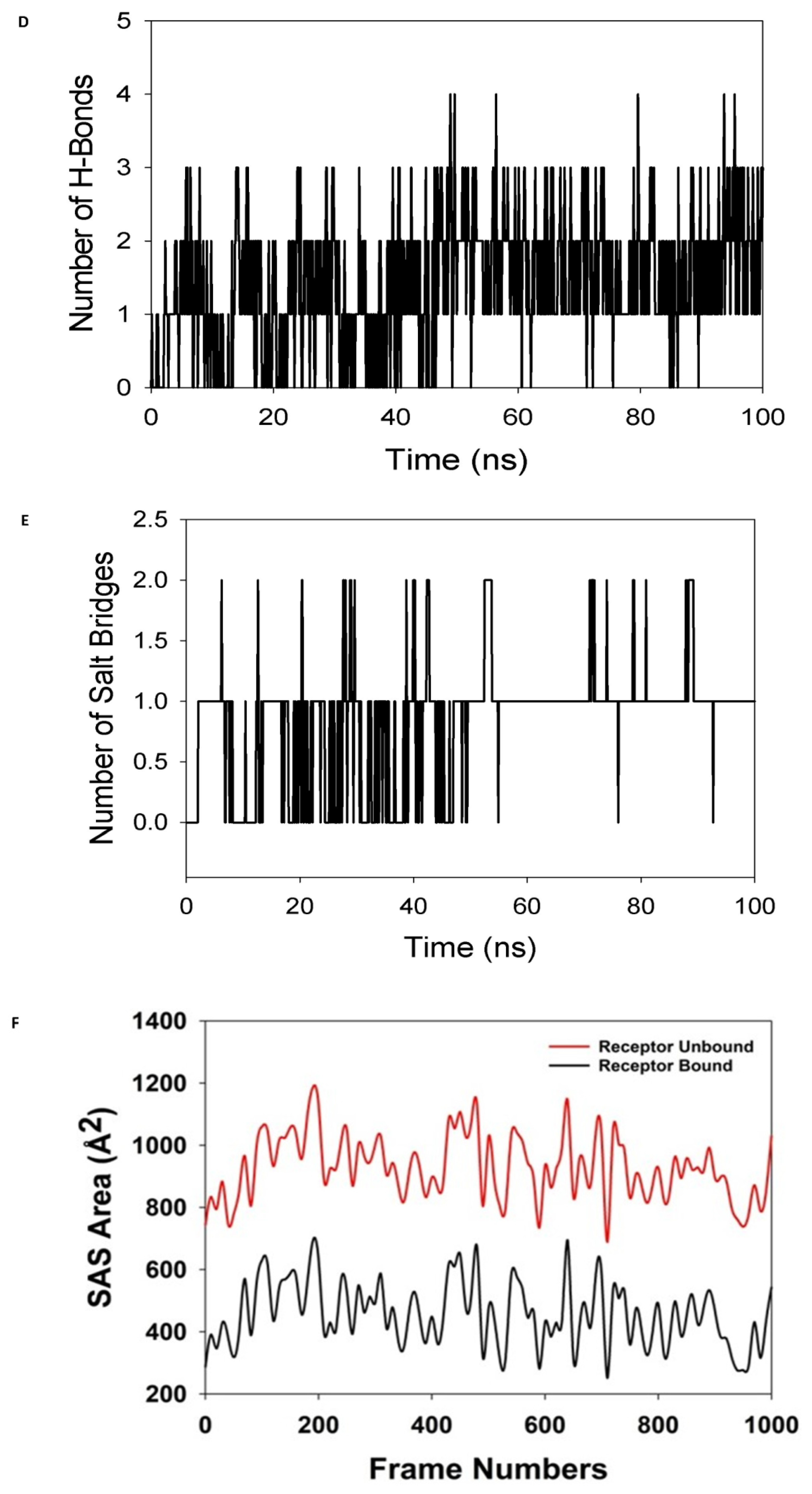
3.4. Simulation of Molecular Dynamics
3.4.1. Calculations of Molecular Mechanics Generalized Born Surface Area (MM-GBSA)
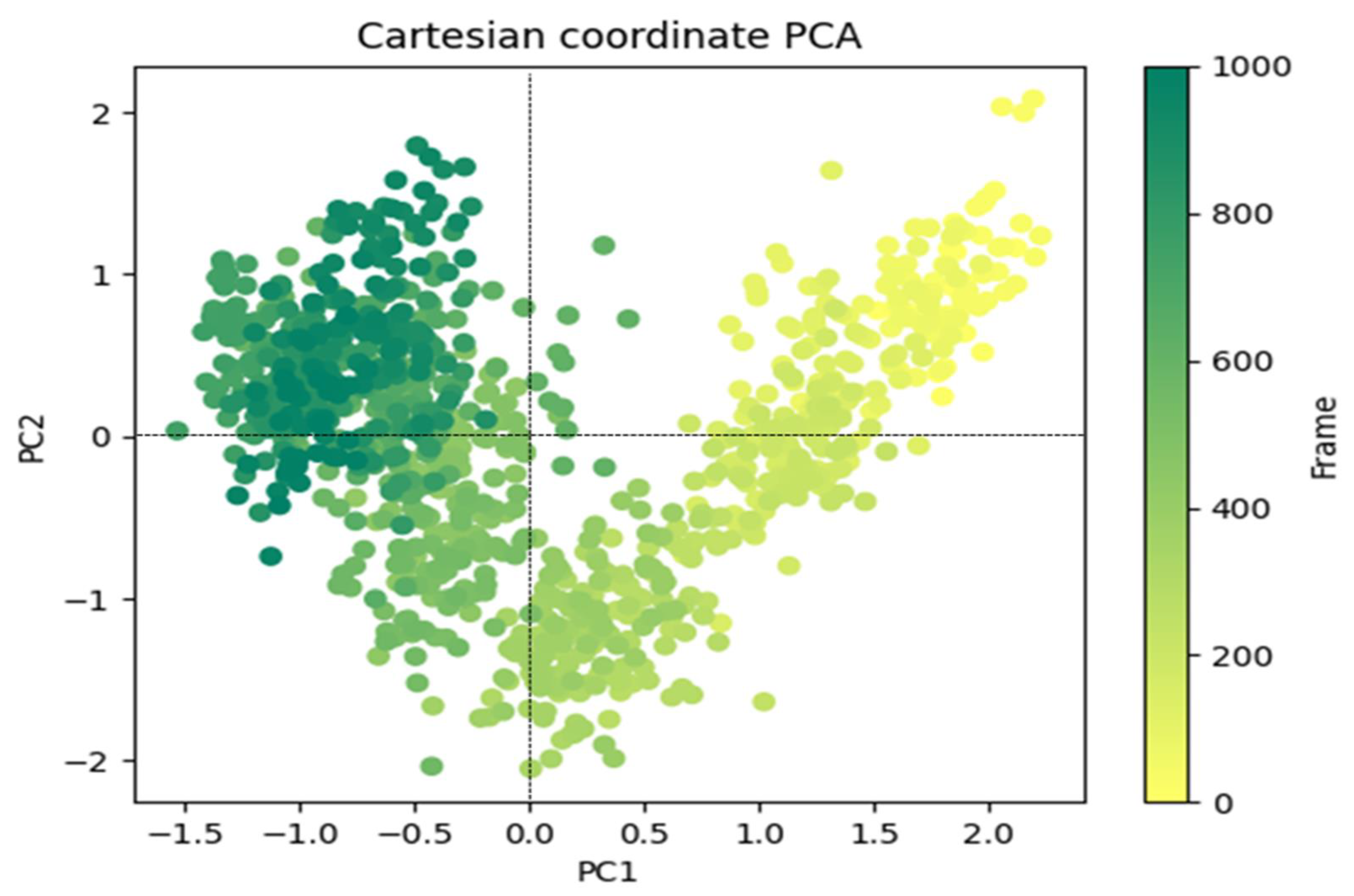
3.4.2. Principal Component Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef]
- Luo, C.M.; Wang, N.; Yang, X.L.; Liu, H.Z.; Zhang, W.; Li, B.; Hu, B.; Peng, C.; Geng, Q.B.; Zhu, G.J.; et al. Discovery of novel bat coronaviruses in South China that use the same receptor as Middle East respiratory syndrome coronavirus. J. Virol. 2018, 92, e00116. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, C.-M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.-H.; Sze, K.-H.; Yang, D.; Shuai, H.; et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef]
- Modjarrad, K.; Moorthy, V.S.; Ben Embarek, P.; Van Kerkhove, M.; Kim, J.; Kieny, M.-P. A roadmap for MERS-CoV research and product development: Report from a world health organization consultation. Nat. Med. 2016, 22, 701–705. [Google Scholar] [CrossRef]
- Magro, G. COVID-19: Review on latest available drugs and therapies against SARS-CoV-2. Coagulation and inflammation crosstalking. Virus Res. 2020, 286, 198070. [Google Scholar] [CrossRef]
- Samrat, S.K.; Tharappel, A.M.; Li, Z.; Li, H. Prospect of SARSCoV-2 spike protein: Potential role in vaccine and therapeutic development. Virus Res. 2020, 288, 198141. [Google Scholar] [CrossRef] [PubMed]
- Negi, M.; Chawla, P.A.; Faruk, A.; Chawla, V. Role of heterocyclic compounds in SARS and SARS CoV-2 pandemic. Bioorg. Chem. 2020, 104, 104315. [Google Scholar] [CrossRef] [PubMed]
- Abu-Melha, S.; Edrees, M.M.; Said, M.A.; Riyadh, S.M.; Al-Kaff, N.S.; Gomha, S.M. Potential COVID-19 drug candidates based on dia-zinyl-thiazol-imine moieties: Synthesis and greener pastures biological study. Molecules 2022, 27, 488. [Google Scholar] [CrossRef] [PubMed]
- Said, M.A.; Riyadh, S.M.; Al-Kaff, N.S.; Nayl, A.A.; Khalil, K.D.; Bräse, S.; Gomha, S.M. Synthesis and Greener Pastures Biological Study of Bis-thiadiazoles as Potential Covid-19 Drug Candidates. Arab. J. Chem. 2022, 15, 104101. [Google Scholar] [CrossRef]
- Gomha, S.M.; Riyadh, S.M.; Abdellattif, M.H.; Abolibda, T.Z.; Abdel-aziz, H.M.; Nayl, A.A.; Elgohary, A.M.; Elfiky, A.A. Synthesis and In Silico Study of Some New Bis-[1,3,4]Thiadiazolimines and Bis-Thiazolimines as Potential In-Hibitors for SARS-CoV-2 Main Protease. Curr. Issues Mol. Biol. 2022, in press. [Google Scholar] [CrossRef]
- Balzarini, J.; Stevens, M.; Andrei, G.; Snoeck, R.; Strunk, R.; Pierce, J.B.; Lacadie, J.A.; De Clercq, E.; Pannecouque, C. Pridine Oxide Derivatives: Structure-Activity Relationship for Inhibition of Human Immunodeficiency Virus and Cytomegalovirus Replication in Cell Culture. Helv. Chim. Acta 2002, 85, 2961–2974. [Google Scholar] [CrossRef]
- Balzarini, J.; Keyaert, E.; Vijgen, L.; Vandermeer, F.; Stevens, M.; De Clercq, E.; Egberink, H.; Van Ranst, M. Pyridine N-oxide derivatives are inhibitory to the human SARS and feline infectious peritonitis coronavirus in cell culture. J. Antimicrob. Chemother. 2006, 57, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Starčević, K.; Kralj, M.; Ester, K.; Sabol, I.; Grce, M.; Pavelić, K.; Karminski-Zamola, G. Synthesis, antiviral and antitumor activity of 2-substituted-5-amidino-benzimidazoles. Bioorg. Med. Chem. 2007, 15, 4419–4426. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.S.; Marzouk, M.I.; Ali, S.N.; Madkour, H.M.F. Synthesis, structure characterization and biological evaluation of new 6,8-dichloro-2-methyl-4H-chromen-4-one derivatives. Eur. J. Chem. 2012, 3, 220–227. [Google Scholar] [CrossRef]
- Niu, C.; Yin, J.; Zhang, J.Z.; Vederas, J.C.; James, M.N.G. Molecular docking identifies the binding of 3-chloropyridine moieties specifically to the S1pocket of SARS-CoV Mpro. Bioorrg. Med. Chem. 2008, 16, 293–302. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Sarkar, S.; Majee, A. Recent advances on heterocyclic compounds with antiviral properties. Chem. Heterocycl. Compd. 2021, 57, 410–416. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Raghavaiah, J.; Shahabi, D.; Yadav, M.; Anson, B.J.; Lendy, E.K.; Hattori, S.-I.; Higashi-Kuwata, N.; Mitsuya, H.; Mesecar, A.D. Indole Chloropyridinyl Ester-Derived SARS-CoV-2 3CLpro Inhibitors: Enzyme Inhibition, Antiviral Efficacy, Structure−Activity Relationship, and X-ray Structural Studies. J. Med. Chem. 2021, 64, 14702–14714. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic Dyad Residues His41 and Cys145 Impact the Catalytic Activity and Overall Conformational Fold of the Main SARS-CoV-2 Protease 3-Chymotrypsin-Like Protease. Front. Chem. 2021, 9, 692168. [Google Scholar] [CrossRef]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19). A Review. Clin. Rev. Educ. 2020, 3213, 1824–1836. [Google Scholar]
- Ghaleb, A.; Aouidate, A.; El Ayouchia, H.B.; Aarjane, M.; Anane, H.; Stiriba, S.E. In silico molecular investigations of pyridine N-Oxide compounds as potential inhibitors of SARS-CoV-2: 3D QSAR, molecular docking modeling, and ADMET screening. J. Biomol. Struct. Dyn. 2020, 40, 143–153. [Google Scholar] [CrossRef]
- Atamanyuk, D.; Zimenkovsky, B.; Atamanyuk, V.; Lesyk, R. 5-Ethoxymethylidene-4-thioxo-2-thiazolidinone as versatile building block for novel biorelevant small molecules with thiopyrano[2,3-d] [1,3]thiazole core. Synth. Commun. 2014, 44, 237–244. [Google Scholar] [CrossRef]
- Konno, S.; Thanigaimalai, P.; Yamamoto, T.; Nakada, K.; Kakiuchi, R.; Takayama, K.; Yamazaki, Y.; Yakushiji, F.; Akaji, K.; Kiso, Y.; et al. Design and synthesis of new tripeptide-type SARS-CoV 3CL protease inhibitors containing an electrophilic arylketone moiety. Bioorg. Med. Chem. 2013, 21, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Lesyk, R. Synthesis and anticancer and antiviral activities of new 2-pyrazoline-substituted 4-thiazolidinones. J. Heterocycl. Chem. 2013, 50, E55–E62. [Google Scholar] [CrossRef]
- Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Day, C.W.; Smee, D.F.; Grellier, P.; Lesyk, R. Synthesis and biological activity evaluation of 5-pyrazoline substituted 4-thiazolidinones. Eur. J. Med. Chem. 2013, 66, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Kaminskyy, D.V. Screening of the antiviral activity in the range of C5 and N3 substituted 4-thiazolidinone derivatives. J. Org. Pharm. Chem. 2015, 13, 64–69. [Google Scholar] [CrossRef]
- Gomha, S.M.; Abdelhady, H.A.; Hassain, D.Z.H.; Abdelmonsef, A.H.; El-Naggar, M.; Elaasser, M.M.; Mahmoud, H.K. Thiazole based thiosemicarbazones: Synthesis, cytotoxicity evaluation and molecular docking study. Drug Des. Dev. Ther. 2021, 15, 659–677. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.A.; Gomha, S.M.; Abdelaziz, M.R. Nany Serag. Synthesis and antiviral evaluation of some novel thiazoles and 1,3-thiazines substituted with pyrazole moiety against rabies virus. Turk. J. Chem. 2016, 40, 441–453. [Google Scholar] [CrossRef]
- Gomha, S.M.; Muhammad, Z.A.; Abdel-aziz, M.R.; Abdel-aziz, H.M.; Gaber, H.M.; Elaasser, M.M. One Pot Synthesis of new thiadiazolyl-pyridines as anticancer and antioxidant agents. J. Heterocycl. Chem. 2018, 55, 530–536. [Google Scholar] [CrossRef]
- Gomha, S.M.; Edrees, M.M.; Muhammad, Z.A.; El-Reedy, A.A.M. 5-(Thiophen-2-yl)-1,3,4-thiadiazole derivatives: Synthesis, molecular docking and in-vitro cytotoxicity evaluation as potential anticancer agents. Drug Des. Dev. Ther. 2018, 12, 1511–1523. [Google Scholar] [CrossRef]
- Edrees, M.M.; Abu-Melha, S.; Saad, A.M.; Kheder, N.A.; Gomha, S.M.; Muhammad, Z.A. Eco-friendly synthesis, characterization and biological evaluation of some new pyrazolines containing thiazole moiety as potential anticancer and antimicrobial agents. Molecules 2018, 23, 1970. [Google Scholar] [CrossRef]
- Gomha, S.M.; Abdelaziz, M.R.; Kheder, N.A.; Abdel-Aziz, H.M.; Alterary, S.; Mabkhot, Y.N. A Facile access and evaluation of some novel thiazole and 1,3,4-thiadiazole derivatives incorporating thiazole moiety as potent anticancer agents. Chem. Cent. J. 2017, 11, 105. [Google Scholar] [CrossRef] [PubMed]
- Gomha, S.M.; Abdel-Aziz, H.M.; El-Reedy, A.A.M. Facile synthesis of pyrazolo[3,4-c] pyrazoles bearing coumarine ring as anticancer agents. J. Heterocycl. Chem. 2018, 55, 1960–1965. [Google Scholar] [CrossRef]
- Abu-Melha, S.; Edrees, M.M.; Salem, H.H.; Kheder, N.A.; Gomha, S.M.; Abdelaziz, M.R. Synthesis and biological evaluation of some novel thiazole-based heterocycles as potential anticancer and antimicrobial agents. Molecules 2019, 24, 539. [Google Scholar] [CrossRef] [PubMed]
- Gomha, S.M.; Muhammad, Z.A.; Abdel-Aziz, H.M.; Matar, I.K.; El-Sayed, A.A. Green synthesis, molecular docking and anticancer activity of novel 1,4-dihydropyridine-3,5-dicarbohydrazones under grind-stone chemistry. Green Chem. Lett. Rev. 2020, 13, 6–17. [Google Scholar] [CrossRef]
- Sayed, A.R.; Abd El-lateef, H.M.; Gomha, S.M.; Abolibda, T.Z. L-Proline catalyzed green synthesis and anticancer evaluation of novel bioactive benzil bis-hydrazones under grinding technique. Green Chem. Lett. Rev. 2021, 14, 179–188. [Google Scholar] [CrossRef]
- Alshabanah, L.A.; Al-Mutabagani, L.A.; Gomha, S.M.; Ahmed, H.A. Three-component synthesis of some new coumarin derivatives as anti-cancer agents. Front. Chem. 2022, 9, 762248. [Google Scholar] [CrossRef]
- CCG. Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2016; Available online: https://scholar.google.com/scholar?cluster=7142026959131975597&hl=en&as_sdt=2005&sciodt=0,5 (accessed on 1 January 2023).
- El Gizawy, H.A.; Boshra, S.A.; Mostafa, A.; Mahmoud, S.H.; Ismail, M.I.; Alsfouk, A.A.; Taher, A.T.; Al-Karmalawy, A.A. Pimenta dioica (L.) Merr. bioactive constituents exert anti-SARS-CoV-2 and anti-inflammatory activities: Molecular docking and dynamics, in vitro, and in vivo studies. Molecules 2021, 26, 5844. [Google Scholar] [CrossRef]
- El-Shershaby, M.H.; El-Gamal, K.M.; Bayoumi, A.H.; El-Adl, K.; Alswah, M.; Ahmed, H.E.A.; Al-Karmalamy, A.A.; Abulkhair, H.S. The antimicrobial potential and pharmacokinetic profiles of novel quinoline-based scaffolds: Synthesis and in silico mechanistic studies as dual DNA gyrase and DHFR inhibitors. New J. Chem. 2021, 45, 13986–14004. [Google Scholar] [CrossRef]
- Wang, K.Y.; Liu, F.; Jiang, R.; Yang, X.; You, T.; Liu, X.; Xiao, C.Q.; Shi, Z.; Jiang, H.; Rao, Z. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar]
- Amin, E.; Abdel-Bakky, M.S.; Mohammed, H.A.; Hassan, M.H.A. Chemical Profiling and Molecular Docking Study of Agathophora alopecuroides. Life 2022, 12, 1852. [Google Scholar] [CrossRef]
- Khalilullah, H.; Agarwal, D.K.; Ahsan, M.J.; Jadav, S.S.; Mohammed, H.A.; Khan, M.A.; Mohammed, S.A.A.; Khan, R. Synthesis and Anti-Cancer Activity of New Pyrazolinyl-Indole Derivatives: Pharmacophoric Interactions and Docking Studies for Identifying New EGFR Inhibitors. Int. J. Mol. Sci. 2022, 23, 6548. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.; Mutahir, S.; Humayun, M.; Khan, M.A.; Al-Humaidi, J.Y.; Refat, M.S.; Abouzied, A.S. Facile Synthesis of ZIF-67 for the Adsorption of Methyl Green from Wastewater: Integrating Molecular Models and Experimental Evidence to Comprehend the Removal Mechanism. Molecules 2022, 27, 8385. [Google Scholar] [CrossRef] [PubMed]
- Alesawy, M.S.; Al-Karmalawy, A.A.; Elkaeed, E.B.; Alswah, M.; Belal, A.; Taghour, M.S.; Eissa, I.H. Design and discovery of new 1, 2, 4-triazolo [4, 3-c] quinazolines as potential DNA intercalators and topoisomerase II inhibitors. Arch. Der Pharm. 2021, 354, 2000237. [Google Scholar] [CrossRef] [PubMed]
- Eliaa, S.G.; Al-Karmalawy, A.A.; Saleh, R.M.; Elshal, M.F. Empagliflozin and doxorubicin synergistically inhibit the survival of triple-negative breast cancer cells via interfering with the mTOR pathway and inhibition of calmodulin: In vitro and molecular docking studies. ACS Pharmacol. Transl. Sci. 2020, 3, 1330–1338. [Google Scholar] [CrossRef]
- El-Shershaby, M.H.; Ghiaty, A.; Bayoumi, A.H.; Al-Karmalawy, A.A.; Husseiny, E.M.; El-Zoghbi, M.S.; Abulkhair, H.S. From triazolophthalazines to triazoloquinazolines: A bioisosterism-guided approach toward the identification of novel PCAF inhibitors with potential anticancer activity. Bioorg. Med. Chem. 2021, 42, 116266. [Google Scholar] [CrossRef]
- Soltan, M.A.; Elbassiouny, N.; Gamal, H.; Elkaeed, E.B.; Eid, R.A.; Eldeen, M.A.; Al-Karmalawy, A.A. In silico prediction of a multitope vaccine against Moraxella catarrhalis: Reverse vaccinology and immunoinformatics. Vaccines 2021, 9, 669. [Google Scholar] [CrossRef]
- McConkey, B.J.; Sobolev, V.; Edelman, M. The performance of current methods in ligand–protein docking. Curr. Sci. 2002, 83, 845–856. [Google Scholar]
- Abdallah, A.E.; Alesawy, M.S.; Eissa, S.I.; El-Fakharany, E.M.; Kalaba, M.H.; Sharaf, M.H.; Shama, N.M.A.; Mahmoud, S.H.; Mostafa, A.; Al-Karmalawy, A.A. Design and synthesis of new 4-(2-nitrophenoxy) benzamide derivatives as potential antiviral agents: Molecular modeling and in vitro antiviral screening. New J. Chem. 2021, 45, 6557–16571. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC′06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Chow, E.; Rendleman, C.A.; Bowers, K.J.; Dror, R.O.; Hughes, D.H.; Gullingsrud, J.; Sacerdoti, F.D.; Shaw, D.E. Desmond Performance on a Cluster of Multicore Processors; DE Shaw Research Technical Report DESRES/TR-2008-01; DE Shaw Research: New York, NY, USA, 2008. [Google Scholar]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nose-Hoover chains-the canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Toukmaji, A.Y.; Board, J.A. Ewald summation techniques in perspective: A survey. Comput. Phys. Commun. 1996, 95, 73–92. [Google Scholar] [CrossRef]
- Kagami, L.P.; das Neves, G.M.; Timmers, L.F.S.M.; Caceres, R.A.; Eifler-Lima, V.L. Geo-Measures: A PyMOL plugin for protein structure ensembles analysis. Comput. Biol. Chem. 2020, 87, 107322. [Google Scholar] [CrossRef]
- Abbas, I.M.; Riyadh, S.M.; Abdallah, M.A.; Gomha, S.M. A novel route to tetracyclic fused tetrazines and thiadiazines. J. Heterocycl. Chem. 2006, 43, 935–942. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. Swiss ADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Abouzied, A.S.; Abd-Rabo, M.M.; Huwaimel, B.; Almahmoud, S.A.; Almarshdi, A.A.; Alharbi, F.M.; Alenzi, S.S.; Albsher, B.N.; Alafnan, A. In Silico Pharmacokinetic Profiling of the Identified Bioactive Metabolites of Pergularia tomentosa L. Latex Extract and In Vitro Cytotoxic Activity via the Induction of Caspase-Dependent Apoptosis with S-Phase Arrest. Pharmaceuticals 2022, 15, 1132. [Google Scholar] [CrossRef]
- Waring, M.J. Defining optimum lipophilicity and molecular weight ranges for drug candidates—Molecular weight dependent lower log D limits based on permeability. Bioorg. Med. Chem. Lett. 2009, 19, 2844–2851. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef]
- Bosshard, H.R.; Marti, D.N.; Jelesarov, I. Protein stabilization by salt bridges: Concepts, experimental approaches and clarification of some misunderstandings. J. Mol. Recognit. 2004, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | |||||
|---|---|---|---|---|---|
| Properties | Compounds | ||||
| 3 | 6a | 6b | 6c | 6d | |
| Formula | C8H11N5S | C17H17N7S | C18H19N7S | C18H19N7OS | C17H16ClN7S |
| Molecular weight | 209.07 g/mol | 351.43 g/mol | 365.46 g/mol | 381.45 g/mol | 385.87 g/mol |
| N. Heavy atoms | 14 | 25 | 26 | 27 | 26 |
| Number of aromatic heavy atoms | 6 | 18 | 17 | 16 | 16 |
| Number of Rotatable Bonds | 4 | 4 | 4 | 5 | 4 |
| HBA | 5 | 5 | 5 | 6 | 5 |
| HBD | 4 | 1 | 1 | 1 | 1 |
| Molar Refractivity | 58.6 | 100.24 | 105.20 | 106.73 | 105.25 |
| TPSA | 107.42 Å2 | 121.52 Å2 | 121.52 Å2 | 130.75 Å2 | 121.52 Å2 |
| Log P | 1.52 | 1.52 | 1.52 | 1.52 | 1.52 |
| Log S | −1.82 Very soluble | −1.20 Very soluble | −1.20 Very soluble | −1.20 Very soluble | −1.20 Very soluble |
| (GI absorption) | High | High | High | High | High |
| BBB | Nil | Nil | Nil | No | No |
| CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4 | No | No | No | No | No |
| No | No | No | No | No | |
| No | No | No | No | No | |
| No | No | No | No | No | |
| No | No | No | No | No | |
| Druglikeness (Lipinski) | Yes 0 violations | Yes 0 violations | Yes 0 violations | Yes 0 violations | Yes 0 violations |
| b | |||||
| Properties | Compounds | ||||
| 6e | 8a | 8d | 9 | 13a | |
| Formula | C17H16N8O2S | C25H23N7OS | C25H22ClN7OS | C16H17N5OS | C16H14ClN5S |
| Molecular weight | 396.43 g/mol | 469.56 g/mol | 504.01 g/mol | 327.40 g/mol | 343.83 g/mol |
| Number of aromatic heavy atoms | 28 | 34 | 35 | 23 | 23 |
| Number of Rotatable Bonds | 17 | 23 | 23 | 12 | 17 |
| HBA | 5 | 7 | 7 | 7 | 3 |
| HBD | 7 | 7 | 7 | 4 | 3 |
| Molar Refractivity | 1 | 0 | 0 | 2 | 1 |
| TPSA | 109.06 | 136.02 | 141.03 | 95.37 | 95.10 |
| Log P | 167.34 Å2 | 117.09 Å2 | 117.09 Å2 | 102.99 Å2 | 96.80 Å2 |
| Log S | 1.52 | 4.81 | 4.95 | 2.67 | 2.67 |
| (GI absorption) | −1.20 Very soluble | −6.24 Poorly soluble | −6.83 Poorly soluble | −3.28 Moderately soluble | −3.28 Moderately soluble |
| BBB | High | Low | Low | High | High |
| Number of aromatic heavy atoms | No | No | No | No | No |
| CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4 | No | No | No | No | No |
| No | No | No | No | No | |
| No | No | Yes | Yes | Yes | |
| No | No | No | No | No | |
| No | No | No | Yes | Yes | |
| Druglikeness (Lipinski) | Yes 0 violations | Yes 0 violations | Yes 1 violation: MW > 500 | Yes 0 violations | Yes 0 violations |
| c | |||||
| Properties | Compounds | ||||
| 13b | 13c | 14a | 14b | 14c | |
| Formula | C16H14BrN5S | C16H14N6O2S | C24H20ClN5OS | C24H20BrN5OS | C24H20N6O3S |
| Molecular weight | 388.28 g/mol | 354.39 g/mol | 461.97 g/mol | 506.42 g/mol | 472.52 g/mol |
| N. Heavy atoms | 23 | 25 | 32 | 32 | 34 |
| Number of aromatic heavy atoms | 17 | 17 | 23 | 23 | 23 |
| Number of Rotatable Bonds | 3 | 4 | 6 | 6 | 7 |
| HBA | 3 | 5 | 5 | 5 | 7 |
| HBD | 1 | 1 | 0 | 0 | 0 |
| Molar Refractivity | 97.79 | 98.91 | 130.88 | 133.57 | 134.69 |
| TPSA | 96.80 Å2 | 142.62 Å2 | 92.37 Å2 | 92.37 Å2 | 138.19 Å2 |
| Log P | 2.67 | 2.67 | 4.40 | 4.46 | 3.66 |
| Log S | −3.28 Moderately soluble | −3.28 Moderately soluble | −6.22 Poorly soluble | −6.54 Poorly soluble | −5.69 Moderately soluble |
| (GI absorption) | High | High | High | High | Low |
| BBB | No | No | No | No | No |
| CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4 | No | No | No | No | No |
| No | No | Yes | Yes | Yes | |
| Yes | Yes | Yes | Yes | Yes | |
| No | No | No | No | No | |
| Yes | Yes | Yes | No | Yes | |
| Druglikeness (Lipinski) | Yes 0 violations | Yes 0 violations | Yes 0 violations | Yes 1 violation: MW > 500 | Yes 0 violations |
| Compounds | Binding Energy (kcal/mol) | Hydrogen Bond Interactions | Distance (Å) | Hydrophobic Interactions | Distance (Å) |
|---|---|---|---|---|---|
| 3 | −5.8 | GLN 110 THR 111 SER 158 THR 292 ASP 295 | 2.75 2.25 2.32 2.16 2.49 | VAL 104 ILE 106 GLN 110 | 3.82 3.93 3.67 |
| 6a | −7.8 | GLY 143 SER 144 CYS 145 | 2.07 2.29 2.72 | THR 25 ASN 142 MET 165 GLN 189 | 3.85 3.61 3.43 3.35 |
| 6b | −8.3 | LEU 141 GLY 143 SER 144 CYS 145 | 1.91 1.78 2.65 2.92 | THR 25 LEU 27 MET 165 GLN 189 | 3.94 3.80 3.32 3.54 |
| 6c | −7.9 | GLY 143 SER 144 CYS145 HIS 164 THR 190 | 1.66 3.29 3.42 4.10 3.96 | THR 25 LEU 27 MET 165 GLN 189 | 3.98 3.95 3.34 3.38 |
| 6d | −7.3 | LEU 141 GLY 143 SER 144 CYS 145 | 2.42 1.63 2.51 2.79 | THR 25 LEU 27 MET 165 GLN 189 | 3.82 3.97 3.42 3.38 |
| 6e | −8.4 | GLN 192 GLY 143 SER 144 CYS 145 | 2.35 1.72 2.61 2.92 | LEU 27 MET 165 HIS 163 | 3.90 3.41 3.43 |
| 8a | −8.6 | GLY 71 GLN 19 MET 17 | 3.41 2.99 3.52 | GLU 14 GLY 120 ALA 70 LYS 97 VAL 18 TRP 31 | 2.67 3.91 3.32 3.78 2.98 3.53 |
| 8d | −7.8 | GLY 143 GLU 166 | 3.47 2.76, 2.86 | MET 165 | 3.91 |
| 9 | −7.2 | MET 17 GLN 19 GLY 71 | 2.58 2.28 2.48 | VAL 18 GLN 19 TRP 31 GLN 69 PRO 96 | 3.69 3.72 3.68, 3.73 3.73 3.99 |
| 13a | −6.8 | GLU 14 GLY 15 | 3.14 2.24,3.65 | ALA 70 VAL 73 PRO 96 | 3.38 3.94 3.55 |
| 13b | −7.2 | GLY 15 | 2.5 | GLU 14 ALA 70 PRO 96 | 3.92 3.46 3.55 |
| 13c | −6.7 | GLU 166 | 3.36 | PRO 168 GLN 189 | 3.74 3.56 |
| 14a | −8.4 | GLN 110 THR 111 | 2.39 | PHE 294 VAL 202 PRO 252 PRO 292 ILE 249 | 3.64 3.68 3.72, 3.65 3.45 3.49 |
| 14b | −8.3 | GLU 166 | 2.86 | MET 165 GLU 166 PRO 168 GLN 189 | 3.76 3.90 3.73 3.74 |
| 14c | −8.1 | GLU 166 | 2.32 | MET 165 GLN 189 PRO 168 CYS 145 | 3.53 3.48 3.44 3.84 |
| Inhibitor N3 | −8 | ASN 142 GLY 143 GLU 166 GLN 189 SER 144 CYS 145 | 2.81 2.38 3.27 2.01,2.72,2.58 2.67 2.72 | HIS 41 MET 49 ASP 187 GLN 189 HIS 163 HIS 172 | 3.68 3.64 3.83 3.69 5.02 5.40 |
| Energies (kcal/mol) | 6LU7 + 8a |
|---|---|
| ΔGbind | −56.81 ± 6.79 |
| ΔGbindLipo | −18.08 ± 1.04 |
| ΔGbindvdW | −48.49 ± 2.18 |
| ΔGbindCoulomb | −25.47 ± 6.20 |
| ΔGbindHbond | −1.73 ± 0.34 |
| ΔGbindSolvGB | 31.74 ± 3.34 |
| ΔGbindCovalent | 5.23 ± 4.41 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alghamdi, A.; Abouzied, A.S.; Alamri, A.; Anwar, S.; Ansari, M.; Khadra, I.; Zaki, Y.H.; Gomha, S.M. Synthesis, Molecular Docking, and Dynamic Simulation Targeting Main Protease (Mpro) of New, Thiazole Clubbed Pyridine Scaffolds as Potential COVID-19 Inhibitors. Curr. Issues Mol. Biol. 2023, 45, 1422-1442. https://doi.org/10.3390/cimb45020093
Alghamdi A, Abouzied AS, Alamri A, Anwar S, Ansari M, Khadra I, Zaki YH, Gomha SM. Synthesis, Molecular Docking, and Dynamic Simulation Targeting Main Protease (Mpro) of New, Thiazole Clubbed Pyridine Scaffolds as Potential COVID-19 Inhibitors. Current Issues in Molecular Biology. 2023; 45(2):1422-1442. https://doi.org/10.3390/cimb45020093
Chicago/Turabian StyleAlghamdi, Adel, Amr S. Abouzied, Abdulwahab Alamri, Sirajudheen Anwar, Mukhtar Ansari, Ibrahim Khadra, Yasser H. Zaki, and Sobhi M. Gomha. 2023. "Synthesis, Molecular Docking, and Dynamic Simulation Targeting Main Protease (Mpro) of New, Thiazole Clubbed Pyridine Scaffolds as Potential COVID-19 Inhibitors" Current Issues in Molecular Biology 45, no. 2: 1422-1442. https://doi.org/10.3390/cimb45020093
APA StyleAlghamdi, A., Abouzied, A. S., Alamri, A., Anwar, S., Ansari, M., Khadra, I., Zaki, Y. H., & Gomha, S. M. (2023). Synthesis, Molecular Docking, and Dynamic Simulation Targeting Main Protease (Mpro) of New, Thiazole Clubbed Pyridine Scaffolds as Potential COVID-19 Inhibitors. Current Issues in Molecular Biology, 45(2), 1422-1442. https://doi.org/10.3390/cimb45020093