
The Landscape and Therapeutic Targeting of BRCA1, BRCA2 and Other DNA Damage Response Genes in Pancreatic Cancer

Abstract
:1. Introduction
2. The Landscape of DDR Mutations in Pancreatic Cancer
3. DDR Role in Pancreatic Cancer
4. Platinum-Based Chemotherapy for BRCA1/BRCA2-Germline-Mutated Pancreatic Cancers
5. Clinical Trials of PARP Inhibitors in Pancreatic Cancers with Germline BRCA1/BRCA2 Mutations
6. PARP Inhibitors for Pancreatic Cancers with Somatic BRCA1/BRCA2 Mutations
7. PARP Inhibitors and Other Targeted Therapies for Pancreatic Cancers with Germline or Somatic Mutations in Other DDR-Associated Genes
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 72, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cabasag, C.J.; Ferlay, J.; Laversanne, M.; Vignat, J.; Weber, A.; Soerjomataram, I.; Bray, F. Pancreatic cancer: An increasing global public health concern. Gut 2022, 71, 1686–1687. [Google Scholar] [CrossRef] [PubMed]
- Zhen, D.B.; Coveler, A.; Zanon, S.; Reni, M.; Chiorean, E.G. Biomarker-driven and molecularly targeted therapies for pancreatic adenocarcinoma. Semin. Oncol. 2018, 45, 107–115. [Google Scholar] [CrossRef]
- Rémond, M.S.; Pellat, A.; Brezault, C.; Dhooge, M.; Coriat, R. Are targeted therapies or immunotherapies effective in metastatic pancreatic adenocarcinoma? ESMO Open 2022, 7, 100638. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.E.; El-Refai, S.M.; Sha, W.; Puccini, A.; Grothey, A.; George, T.J.; Hwang, J.J.; O’Neil, B.; Barrett, A.S.; Kadakia, K.C.; et al. Landscape of KRASG12C, Associated Genomic Alterations, and Interrelation with Immuno-Oncology Biomarkers in KRAS-Mutated Cancers. JCO Precis. Oncol. 2022, 6, e2100245. [Google Scholar] [CrossRef]
- McGarry, J.L.; Creavin, B.; Kelly, M.E.; Gallagher, T.K. Risk of pancreatic ductal adenocarcinoma associated with carriage of BRCA1 and/or BRCA2 mutation: A systematic review and meta-analysis. J. Surg. Oncol. 2022, 126, 1028–1037. [Google Scholar] [CrossRef]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients with Pancreatic Adenocarcinoma. J. Clin. Oncol. 2015, 33, 3124–3129. [Google Scholar] [CrossRef]
- Perkhofer, L.; Gout, J.; Roger, E.; de Almeida, F.K.; Simões, C.B.; Wiesmüller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2020, 70, 606–617. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. How do mutations affecting the breast cancer genes BRCA1 and BRCA2 cause cancer susceptibility? DNA Repair 2019, 81, 102668. [Google Scholar] [CrossRef] [PubMed]
- Chapin, W.J.; Reiss, K.A. PARPis and Other Novel, Targeted Therapeutics in Pancreatic Adenocarcinoma. Hematol. Oncol. Clin. N. Am. 2022, 36, 1019–1032. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Guo, Y.; Zeng, Y.; Song, Z.; Zhu, X.; Fan, N.; Zhang, Z.; Ren, G.; Zang, Y.; Rao, W. Clinically significant genomic alterations in the Chinese and Western patients with intrahepatic cholangiocarcinoma. BMC Cancer 2021, 21, 152. [Google Scholar] [CrossRef] [PubMed]
- The AACR Project GENIE Consortium; André, F.; Arnedos, M.; Baras, A.S.; Baselga, J.; Bedard, P.L.; Berger, M.F.; Bierkens, M.; Calvo, F.; Cerami, E.; et al. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, PO.17.00011. [Google Scholar] [CrossRef]
- Seeber, A.; Zimmer, K.; Kocher, F.; Puccini, A.; Xiu, J.; Nabhan, C.; Elliott, A.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; et al. Molecular characteristics of BRCA1/2 and PALB2 mutations in pancreatic ductal adenocarcinoma. ESMO Open 2020, 5, e000942. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Chittenden, A.B.; Morales-Oyarvide, V.; Rubinson, D.A.; Dunne, R.F.; Kozak, M.M.; Qian, Z.R.; Welch, M.W.; Brais, L.K.; Da Silva, A.; et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet. Med. 2019, 21, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef]
- Mohindroo, C.; De Jesus-Acosta, A.; Yurgelun, M.B.; Maitra, A.; Mork, M.; McAllister, F. The Evolving Paradigm of Germline Testing in Pancreatic Ductal Adenocarcinoma and Implications for Clinical Practice. Surg. Pathol. Clin. 2022, 15, 491–502. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Wattenberg, M.M.; Reiss, K.A. Determinants of Homologous Recombination Deficiency in Pancreatic Cancer. Cancers 2021, 13, 4716. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey-Jones, E.M.; Villaro, G.V.; Tutt, A.M. New Roles of Poly(ADP-Ribose) Polymerase Inhibitors in the Treatment of Breast Cancer. Cancer J. 2021, 27, 441–456. [Google Scholar] [CrossRef]
- Buisson, R.; Niraj, J.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.-L.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol. Cell 2017, 65, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [Green Version]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2010, 7, 98–111. [Google Scholar] [CrossRef]
- Kumar, R.J.; Chao, H.X.; A Simpson, D.; Feng, W.; Cho, M.-G.; Roberts, V.R.; Sullivan, A.R.; Shah, S.J.; Wozny, A.-S.; Fagan-Solis, K.; et al. Dual inhibition of DNA-PK and DNA polymerase theta overcomes radiation resistance induced by p53 deficiency. NAR Cancer 2020, 2, zcaa038. [Google Scholar] [CrossRef] [PubMed]
- Paculová, H.; Kohoutek, J. The emerging roles of CDK12 in tumorigenesis. Cell Div. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.; Leary, A.; Scott, C.; Serra, V.; Lord, C.; Bowtell, D.; Chang, D.; Garsed, D.; Jonkers, J.; Ledermann, J.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef] [Green Version]
- Westphalen, C.B.; Fine, A.D.; André, F.; Ganesan, S.; Heinemann, V.; Rouleau, E.; Turnbull, C.; Palacios, L.G.; Lopez, J.-A.; Sokol, E.S.; et al. Pan-cancer Analysis of Homologous Recombination Repair–associated Gene Alterations and Genome-wide Loss-of-Heterozygosity Score. Clin. Cancer Res. 2022, 28, 1412–1421. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Llop-Guevara, A.; Loibl, S.; Villacampa, G.; Vladimirova, V.; Schneeweiss, A.; Karn, T.; Zahm, D.-M.; Herencia-Ropero, A.; Jank, P.; van Mackelenbergh, M.; et al. Association of RAD51 with homologous recombination deficiency (HRD) and clinical outcomes in untreated triple-negative breast cancer (TNBC): Analysis of the GeparSixto randomized clinical trial. Ann. Oncol. 2021, 32, 1590–1596. [Google Scholar] [CrossRef]
- Shahda, S.; Timms, K.M.; Ibrahim, A.A.; Reid, J.E.; Cramer, H.M.; Radovich, M.; Ibrahim, S.; Allen, B.; O’Neil, B.H. Homologous Recombination Deficiency in Patients with Pancreatic Ductal Adenocarcinoma and Response to Chemotherapy. JCO Precis. Oncol. 2018, 2, 1–11. [Google Scholar] [CrossRef]
- Lohse, I.; Borgida, A.; Cao, P.; Cheung, M.; Pintilie, M.; Bianco, T.; Holter, S.; Ibrahimov, E.; Kumareswaran, R.; Bristow, R.G.; et al. BRCA1 and BRCA2 mutations sensitize to chemotherapy in patient-derived pancreatic cancer xenografts. Br. J. Cancer 2015, 113, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Park, J.Y.P.; Pacis, A.; Denroche, R.E.; Jang, G.H.; Zhang, A.; Cuggia, A.; Domecq, C.; Monlong, J.; Raitses-Gurevich, M.; et al. A Preclinical Trial and Molecularly Annotated Patient Cohort Identify Predictive Biomarkers in Homologous Recombination–deficient Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5462–5476. [Google Scholar] [CrossRef]
- Golan, T.; Barenboim, A.; Lahat, G.; Nachmany, I.; Goykhman, Y.; Shacham-Shmueli, E.; Halpern, N.; Brazowski, E.; Geva, R.; Wolf, I.; et al. Increased Rate of Complete Pathologic Response after Neoadjuvant FOLFIRINOX for BRCA Mutation Carriers with Borderline Resectable Pancreatic Cancer. Ann. Surg. Oncol. 2020, 27, 3963–3970. [Google Scholar] [CrossRef] [PubMed]
- Barenboim, A.; Lahat, G.; Geva, R.; Nachmany, I.; Nakache, R.; Goykhman, Y.; Brazowski, E.; Rosen, G.; Isakov, O.; Wolf, I.; et al. Neoadjuvant FOLFIRINOX for locally advanced and borderline resectable pancreatic cancer: An intention to treat analysis. Eur. J. Surg. Oncol. 2018, 44, 1619–1623. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.B.; Groot, V.P.; Gemenetzis, G.; Wei, J.; Cameron, J.L.; Weiss, M.J.; Goggins, M.; Wolfgang, C.L.; Yu, J.; He, J. BRCA1/BRCA2 Germline Mutation Carriers and Sporadic Pancreatic Ductal Adenocarcinoma. J. Am. Coll. Surg. 2018, 226, 630–637.e1. [Google Scholar] [CrossRef]
- Yu, S.; Agarwal, P.; Mamtani, R.; Symecko, H.; Spielman, K.; O’Hara, M.; O’Dwyer, P.J.; Schneider, C.; Teitelbaum, U.; Nathanson, K.L.; et al. Retrospective Survival Analysis of Patients with Resected Pancreatic Ductal Adenocarcinoma and a Germline BRCA or PALB2 Mutation. JCO Precis. Oncol. 2019, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Rahib, L.; Lyons, E.; De Arbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sohal, D.P.; et al. Outcomes in Patients with Pancreatic Adenocarcinoma with Genetic Mutations in DNA Damage Response Pathways: Results from the Know Your Tumor Program. JCO Precis. Oncol. 2019, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wattenberg, M.M.; Asch, D.; Yu, S.; O’Dwyer, P.J.; Domchek, S.M.; Nathanson, K.L.; Rosen, M.A.; Beatty, G.L.; Siegelman, E.S.; Reiss, K.A. Platinum response characteristics of patients with pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or PALB2 mutation. Br. J. Cancer 2019, 122, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Momtaz, P.; O’Connor, C.A.; Chou, J.F.; Capanu, M.; Park, W.; Bandlamudi, C.; Berger, M.F.; Kelsen, D.P.; Suehnholz, S.P.; Chakravarty, D.; et al. Pancreas cancer and BRCA: A critical subset of patients with improving therapeutic outcomes. Cancer 2021, 127, 4393–4402. [Google Scholar] [CrossRef]
- Reiss, K.A.; Yu, S.; Judy, R.; Symecko, H.; Nathanson, K.L.; Domchek, S.M. Retrospective Survival Analysis of Patients with Advanced Pancreatic Ductal Adenocarcinoma and Germline BRCA or PALB2 Mutations. JCO Precis. Oncol. 2018, 2, 1–9. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin with or without Veliparib in Patients with Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Botrus, G.; Roe, D.; Jameson, G.S.; Junior, P.L.S.U.; Korn, R.L.; Caldwell, L.; Bargenquast, T.; Miller, M.; Borazanci, E.H. Mitomycin C in Homologous Recombination Deficient Metastatic Pancreatic Cancer after Disease Progression on Platinum-Based Chemotherapy and Olaparib. Biomedicines 2022, 10, 2705. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Chung, S.Y.; Parakrama, R.; Fayyaz, F.; Jose, J.; Saif, M.W. The role of PARP inhibitors in BRCA mutated pancreatic cancer. Ther. Adv. Gastroenterol. 2021, 14, 1–11. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Kindler, H.L.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Overall Survival Results from the POLO Trial: A Phase III Study of Active Maintenance Olaparib Versus Placebo for Germline BRCA-Mutated Metastatic Pancreatic Cancer. J. Clin. Oncol. 2022, 40, 3929–3939. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib Monotherapy in Patients with Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Borazanci, E.; Korn, R.; Liang, W.S.; Guarnieri, C.; Haag, S.; Snyder, C.; Hendrickson, K.; Caldwell, L.; Von Hoff, D.; Jameson, G. An Analysis of Patients with DNA Repair Pathway Mutations Treated with a PARP Inhibitor. Oncologist 2019, 25, e60–e67. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, PO1700316. [Google Scholar] [CrossRef]
- Reiss, K.A.; Mick, R.; O’Hara, M.H.; Teitelbaum, U.; Karasic, T.B.; Schneider, C.; Cowden, S.; Southwell, T.; Romeo, J.; Izgur, N.; et al. Phase II Study of Maintenance Rucaparib in Patients With Platinum-Sensitive Advanced Pancreatic Cancer and a Pathogenic Germline or Somatic Variant in BRCA1, BRCA2, or PALBJ. Clin. Oncol. 2021, 39, 2497–2505. [Google Scholar] [CrossRef]
- Mohyuddin, G.R.; Aziz, M.; Britt, A.; Wade, L.; Sun, W.; Baranda, J.; Al-Rajabi, R.; Saeed, A.; Kasi, A. Similar response rates and survival with PARP inhibitors for patients with solid tumors harboring somatic versus Germline BRCA mutations: A Meta-analysis and systematic review. BMC Cancer 2020, 20, 507. [Google Scholar] [CrossRef]
- Binder, K.A.R.; Mick, R.; O’Hara, M.; Teitelbaum, U.; Karasic, T.; Schneider, C.; O’Dwyer, P.J.; Carpenter, E.; Pantel, A.; Makvandi, M.; et al. A phase II, single arm study of maintenance rucaparib in patients with plati-num-sensitive advanced pancreatic cancer and a pathogenic germline or somatic mutation in BRCA1, BRCA2 or PALB. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Park, D.; Bergin, S.M.; Jones, D.; Ru, P.; Koivisto, C.S.; Jeon, Y.-J.; Sizemore, G.M.; Kladney, R.D.; Hadjis, A.; Shakya, R.; et al. Ablation of the Brca1–Palb2 Interaction Phenocopies Fanconi Anemia in Mice. Cancer Res 2020, 80, 4172–4184. [Google Scholar] [CrossRef] [PubMed]
- Nacson, J.; Di Marcantonio, D.; Wang, Y.; Bernhardy, A.J.; Clausen, E.; Hua, X.; Cai, K.Q.; Martinez, E.; Feng, W.; Callén, E.; et al. BRCA1 Mutational Complementation Induces Synthetic Viability. Mol. Cell 2020, 78, 951–959.e6. [Google Scholar] [CrossRef]
- Her, J.; Bunting, S.F. BRCA1 and PALB2 in a Messy Breakup. Cancer Res 2020, 80, 4044–4045. [Google Scholar] [CrossRef] [PubMed]
- Sokol, E.S.; Jin, D.X.; Fine, A.; Trabucco, S.E.; Maund, S.; Frampton, G.; Molinero, L.; Antonarakis, E.S. PARP Inhibitor Insensitivity to BRCA1/2 Monoallelic Mutations in Microsatellite Instability-High Cancers. JCO Precis. Oncol. 2022, 6, e2100531. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, S.; Brown, J.; Yamaguchi, K.; Hamanishi, J.; Yamanoi, K.; Takaya, H.; Kaneyasu, T.; Mori, S.; Mandai, M.; Matsumura, N. Utility of Homologous Recombination Deficiency Biomarkers across Cancer Types. JCO Precis. Oncol. 2022, 6, e2200085. [Google Scholar] [CrossRef] [PubMed]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef] [Green Version]
- A Voutsadakis, I. Landscape of BRIP1 molecular lesions in gastrointestinal cancers from published genomic studies. World J. Gastroenterol. 2020, 26, 1197–1207. [Google Scholar] [CrossRef]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016, 6, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Kamphues, C.; Bova, R.; Bahra, M.; Klauschen, F.; Muckenhuber, A.; Sinn, B.V.; Warth, A.; Goeppert, B.; Endris, V.; Neuhaus, P.; et al. Ataxia-telangiectasia–Mutated Protein Kinase Levels Stratify Patients with Pancreatic Adenocarcinoma into Prognostic Subgroups with Loss Being a Strong Indicator of Poor Survival. Pancreas 2015, 44, 296–301. [Google Scholar] [CrossRef]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [Green Version]
- Jette, N.R.; Radhamani, S.; Arthur, G.; Ye, R.; Goutam, S.; Bolyos, A.; Petersen, L.F.; Bose, P.; Bebb, D.G.; Lees-Miller, S.P. Combined poly-ADP ribose polymerase and ataxia-telangiectasia mutated/Rad3-related inhibition targets ataxia-telangiectasia mutated-deficient lung cancer cells. Br. J. Cancer 2019, 121, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, C.R.; Wallez, Y.; Johnson, T.I.; Fernández, S.B.D.Q.; Durant, S.T.; Cadogan, E.B.; Lau, A.; Richards, F.M.; Jodrell, D.I. Complete loss of ATM function augments replication catastrophe induced by ATR inhibition and gemcitabine in pancreatic cancer models. Br. J. Cancer 2020, 123, 1424–1436. [Google Scholar] [CrossRef]
- Wallez, Y.; Dunlop, C.R.; Johnson, T.I.; Koh, S.-B.; Fornari, C.; Yates, J.W.; Fernández, S.B.D.Q.; Lau, A.; Richards, F.M.; Jodrell, D.I. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol. Cancer Ther. 2018, 17, 1670–1682. [Google Scholar] [CrossRef] [Green Version]
- Perkhofer, L.; Schmitt, A.; Carrasco, M.C.R.; Ihle, M.; Hampp, S.; Ruess, D.A.; Hessmann, E.; Russell, R.; Lechel, A.; Azoitei, N.; et al. ATM Deficiency Generating Genomic Instability Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Therapy-Induced DNA Damage. Cancer Res 2017, 77, 5576–5590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Güthle, M.; Zenke, M.; et al. Loss of ATM accelerates pancreatic cancer formation and epithelial–mesenchymal transition. Nat. Commun. 2015, 6, 7677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, V.; Wang, A.T.; Castroviejo-Bermejo, M.; Polanska, U.M.; Palafox, M.; Herencia-Ropero, A.; Jones, G.N.; Lai, Z.; Armenia, J.; Michopoulos, F.; et al. Identification of a Molecularly-Defined Subset of Breast and Ovarian Cancer Models that Respond to WEE1 or ATR Inhibition, Overcoming PARP Inhibitor Resistance. Clin. Cancer Res. 2022, 28, 4536–4550. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; MacCormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef]
- Seidel, P.; Rubarth, A.; Zodel, K.; Peighambari, A.; Neumann, F.; Federkiel, Y.; Huang, H.; Hoefflin, R.; Adlesic, M.; Witt, C.; et al. ATR represents a therapeutic vulnerability in clear cell renal cell carcinoma. J. Clin. Investig. 2022, 7, e156087. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Xu, R.-H.; Chin, K.; Lee, K.-W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.-A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- Martino, C.; Pandya, D.; Lee, R.; Levy, G.; Lo, T.; Lobo, S.; Frank, R.C. ATM-Mutated Pancreatic Cancer: Clinical and Molecular Re-sponse to Gemcitabine/Nab-Paclitaxel after Genome-Based Therapy Resistance. Pancreas 2020, 49, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Huang, T.-T.; Horibata, S.; Lee, J.-M. Cell cycle checkpoints and beyond: Exploiting the ATR/CHK1/WEE1 pathway for the treatment of PARP inhibitor–resistant cancer. Pharmacol. Res. 2022, 178, 106162. [Google Scholar] [CrossRef] [PubMed]
- Vakili-Samiani, S.; Khanghah, O.J.; Gholipour, E.; Najafi, F.; Zeinalzadeh, E.; Samadi, P.; Sarvarian, P.; Pourvahdani, S.; Kelaye, S.K.; Hamblin, M.R.; et al. Cell cycle involvement in cancer therapy; WEE1 kinase, a potential target as therapeutic strategy. Mutat. Res. Mol. Mech. Mutagen. 2022, 824, 111776. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Burkhart, R.A.; Beeharry, N.; Bhattacharjee, V.; Londin, E.R.; Cozzitorto, J.A.; Romeo, C.; Jimbo, M.; Norris, Z.A.; Yeo, C.J.; et al. HuR Posttranscriptionally Regulates WEE1: Implications for the DNA Damage Response in Pancreatic Cancer Cells. Cancer Res 2014, 74, 1128–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarts, M.; Sharpe, R.; Garcia-Murillas, I.; Gevensleben, H.; Hurd, M.S.; Shumway, S.D.; Toniatti, C.; Ashworth, A.; Turner, N.C. Forced Mitotic Entry of S-Phase Cells as a Therapeutic Strategy Induced by Inhibition of WEE1 Inhibition. Cancer Discov. 2012, 2, 524–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.H.; Nam, A.-R.; Park, J.E.; Bang, J.-H.; Bang, Y.-J.; Oh, D.-Y. Therapeutic Co-targeting of WEE1 and ATM Downregulates PD-L1 Expression in Pancreatic Cancer. Cancer Res. Treat. 2020, 52, 149–166. [Google Scholar] [CrossRef]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef] [Green Version]
- Saini, P.; Li, Y.; Dobbelstein, M. Wee1 is required to sustain ATR/Chk1 signaling upon replicative stress. Oncotarget 2015, 6, 13072–13087. [Google Scholar] [CrossRef] [Green Version]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination with Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef]
- Kausar, T.; Schreiber, J.S.; Karnak, D.; Parsels, L.A.; Parsels, J.D.; Davis, M.A.; Zhao, L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of Pancreatic Cancers to Gemcitabine Chemoradiation by WEE1 Kinase Inhibition Depends on Homologous Recombination Repair. Neoplasia 2015, 17, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Santisteban, I.; Llopis, A.; Krenning, L.; Vallejo-Rodríguez, J.; Broek, B.V.D.; Zubiaga, A.M.; Medema, R.H. Sustained CHK2 activity, but not ATM activity, is critical to maintain a G1 arrest after DNA damage in untransformed cells. BMC Biol. 2021, 19, 35. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.; Bin Hong, Y.; Kim, J.S.; Lee, H.; Yi, Y.W.; Kim, Y.J.; Wang, A.; Zhao, W.; Cho, C.H.; Seong, Y.; et al. Inhibition of checkpoint kinase 2 (CHK 2) enhances sensitivity of pancreatic adenocarcinoma cells to gemcitabine. J. Cell. Mol. Med. 2013, 17, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Takada, K.; Takeuchi, O.; Takagi, A.; Watanabe, K.; Hirohara, M.; Hamamoto, T.; Masuda, Y. Prexasertib increases the sensitivity of pancreatic cancer cells to gemcitabine and S-1. Oncol. Rep. 2019, 43, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Black, S.J.; Ozdemir, A.Y.; Kashkina, E.; Kent, T.; Rusanov, T.; Ristic, D.; Shin, Y.; Suma, A.; Hoang, T.; Chandramouly, G.; et al. Molecular basis of microhomology-mediated end-joining by purified full-length Polθ. Nat. Commun. 2019, 10, 4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, R.; Longley, M.J.; Sharief, F.S.; Hou, E.W.; Copeland, W.C.; Wilson, S.H. Human DNA polymerase theta possesses 5’-dRP lyase activity and functions in single-nucleotide base excision repair in vitro. Nucleic Acids Res. 2009, 37, 1868–1877. [Google Scholar] [CrossRef] [Green Version]
- Schrempf, A.; Bernardo, S.; Verge, E.A.A.; Otero, M.A.R.; Wilson, J.; Kirchhofer, D.; Timelthaler, G.; Ambros, A.M.; Kaya, A.; Wieder, M.; et al. POLθ processes ssDNA gaps and promotes replication fork progression in BRCA1-deficient cells. Cell Rep. 2022, 41, 111716. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Simpson, D.A.; Carvajal-Garcia, J.; Price, B.A.; Kumar, R.J.; Mose, L.E.; Wood, R.D.; Rashid, N.; Purvis, J.E.; Parker, J.S.; et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 2019, 10, 4286. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Färkkilä, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Koch, M.S.; Zdioruk, M.; Nowicki, M.O.; Griffith, A.M.; Aguilar-Cordova, E.; Aguilar, L.K.; Guzik, B.W.; Barone, F.; Tak, P.P.; Schregel, K.; et al. Perturbing DDR signaling enhances cytotoxic effects of local oncolytic virotherapy and modulates the immune environment in glioma. Mol. Ther. Oncolytics 2022, 26, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Botta, G.P.; Kato, S.; Patel, H.; Fanta, P.; Lee, S.; Okamura, R.; Kurzrock, R. SWI/SNF complex alterations as a biomarker of immunotherapy efficacy in pancreatic cancer. J. Clin. Investig. 2021, 6, e150453. [Google Scholar] [CrossRef] [PubMed]
- Hartman, S.J.; Bagby, S.M.; Yacob, B.W.; Simmons, D.M.; MacBeth, M.; Lieu, C.H.; Davis, S.L.; Leal, A.D.; Tentler, J.J.; Diamond, J.R.; et al. WEE1 Inhibition in Combination with Targeted Agents and Standard Chemotherapy in Preclinical Models of Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 642328. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene | TCGA All Alterations (n = 184 Profiled) | Amplifications | Mutations | (Likely) Oncogenic Mutations | MSK-IMPACT All Alterations (n = 383 Profiled) | Amplifications | Mutations | (Likely) Oncogenic Mutations |
|---|---|---|---|---|---|---|---|---|
| BRCA1 | 6 (3.3%) | 4 (2.2%) | 2 (1.1%) | 0 | 5 (1.3%) | 0 | 4 (1%) | 2 (0.5%) |
| BRCA2 | 2 (1.1%) | 0 | 2 (1.1%) | 2 (1.1%) | 13 (3.4%) | 0 | 13 (3.4%) | 10 (2.6%) |
| PALB2 | 1 (0.5%) | 0 | 1 (0.5%) | 0 | 2 (0.5%) | 0 | 2 (0.5%) | 1 (0.3%) |
| RAD51 | 1 (0.5%) | 1 (0.5%) | 0 | 0 | 0 | |||
| RAD51B | 0 | 0 | ||||||
| RAD51C | 1 (0.5%) | 0 | 1 (0.5%) | 1 (0.5%) | 0 | |||
| RAD51D | 4 (2.2%) | 4 (2.2%) | 0 | 0 | 1 (0.3%) | 0 | 0 | 0 |
| RAD50 | 3 (1.6%) | 2 (1.1%) | 0 | 0 | 3 (0.8%) | 0 | 2 (0.5%) | 0 |
| XRCC2 | 2 (1.1%) | 2 (1.1%) | 0 | 0 | 1 (0.3%) | 0 | 1 (0.3%) | 0 |
| ATM | 8 (4.3%) | 0 | 8 (4.3%) | 4 (2.2%) | 11 (2.9%) | 0 | 11 (2.9%) | 5 (1.3%) |
| ATR | 5 (2.7%) | 1 (0.5%) | 4 (2.2%) | 2 (1.1%) | 1 (0.3%) | 0 | 1 (0.3%) | 0 |
| BRIP1 | 5 (2.7%) | 4 (2.2%) | 1 (0.5%) | 0 | 4 (1%) | 2 (0.5%) | 2 (0.5%) | 1 (0.3%) |
| NBN | 8 (4.3%) | 8 (4.3%) | 0 | 0 | 3 (0.8%) | 2 (0.5%) | 1 (0.3%) | 0 |
| MRE11 | 1 (0.5%) | 0 | 1 (0.5%) | 0 | 0 | |||
| CHEK1 | 0 | 2 (0.5%) | 0 | 1 (0.3%) | 0 | |||
| CHEK2 | 1 (0.5%) | 0 | 1 (0.5%) | 0 | 0 | |||
| BARD1 | 5 (2.7%) | 3 (1.6%) | 2 (1.1%) | 0 | 3 (0.8%) | 1 (0.3%) | 3 (0.8%) | 2 (0.5%) |
| WEE1 | 1 (0.5%) | 1 (0.5%) | 0 | 0 | NA | |||
| PRKDC | 9 (4.9%) | 6 (3.3%) | 3 (2%) | 0 | NA | |||
| POLQ | 3 (1.6%) | 2 (1.1%) | 1 (0.5%) | 0 | NA | |||
| CDK12 | 9 (4.9%) | 8 (4.3%) | 1 (0.5%) | 0 | 8 (2.1%) | 6 (1.6%) | 2 (0.5%) | 1 (0.3%) |
| Total (non-overlapping) | 42 (22.8%) | 29 (15.8) | 15 (8.2%) | 9 (4.9) | 46 (12%) | 11 (2.9%) | 37 (9.7%) | 20 (5.2%) |
| Gene | Chinese All Alterations (n = 461 Profiled) | Amplifications | Mutations | (Likely) Oncogenic Mutations | Project GENIE All Alterations (n = 5874 Profiled) | Amplifications | Mutations | (Likely) Oncogenic Mutations |
|---|---|---|---|---|---|---|---|---|
| BRCA1 | 5 (1.1%) | 0 | 5 (1.1%) | 1 (0.2%) | 110 (2%) | 1 (0.02%) | 107 (1.9%) | 50 (1%) |
| BRCA2 | 8 (1.7%) | 0 | 8 (1.7%) | 5 (1.1%) | 242 (4.5%) | 7 (0.1%) | 237 (4.5%) | 143 (2.8%) |
| PALB2 | 7 (1.5%) | 0 | 7 (1.5%) | 3 (0.7%) | 75 (1.4%) | 1 (0.02%) | 72 (1.4%) | 35 (0.6%) |
| RAD51 | 1 (0.2%) | 0 | 0 | 7 (0.2%) | 0 | 7 (0.1%) | 2 (0.04%) | |
| RAD51B | 3 (0.7%) | 0 | 2 (0.4%) | 0 | 6 (0.2%) | 1 (0.02%) | 6 (0.1%) | 2 (0.04%) |
| RAD51C | 1 (0.2%) | 0 | 1 (0.2%) | 1 (0.2%) | 17 (0.4%) | 1 (0.02%) | 13 (0.2%) | 2 (0.04%) |
| RAD51D | 1 (0.2%) | 0 | 1 (0.2%) | 0 | 9 (0.2%) | 4 (0.08%) | 4 (0.08%) | 2 (0.04%) |
| RAD50 | 6 (1.3%) | 0 | 6 (1.3%) | 3 (0.7%) | 41 (0.9%) | 0 | 41 (0.9%) | 14 (0.3%) |
| XRCC2 | 2 (0.4%) | 0 | 2 (0.4%) | 1 (0.2%) | 15 (0.3%) | 0 | 15 (0.2%) | 3 (0.1%) |
| ATM | 21 (5%) | 0 | 21 (5%) | 16 (3.5%) | 251 (4.5%) | 0 | 271 (5.2%) | 171 (3.5%) |
| ATR | 6 (1.3%) | 0 | 6 (1.3%) | 3 (0.7%) | 97 (1.9%) | 1 (0.02%) | 96 (1.9%) | 28 (0.6%) |
| BRIP1 | 2 (0.4%) | 0 | 2 (0.4%) | 0 | 62 (1.2%) | 21 (0.4%) | 41 (0.8%) | 18 (0.3%) |
| NBN | 0 | 38 (0.7%) | 5 (0.1%) | 35 (0.7%) | 14 (0.3%) | |||
| MRE11 | 1 (0.2%) | 0 | 1 (0.2%) | 1 (0.2%) | 15 (0.3%) | 1 (0.02%) | 14 (0.3%) | 2 (0.04%) |
| CHEK1 | 1 (0.2%) | 0 | 1 (0.2%) | 0 | 15 (0.3%) | 0 | 15 (0.3%) | 3 (0.06%) |
| CHEK2 | 0 | 34 (0.7%) | 0 | 34 (0.6%) | 13 (0.2%) | |||
| BARD1 | 2 (0.4%) | 0 | 2 (0.4%) | 0 | 50 (1%) | 1 (0.02%) | 48 (1%) | 8 (0.1%) |
| WEE1 | 1 (0.2%) | 0 | 1 (0.2%) | 0 | NA | NA | NA | |
| PRKDC | 10 (2.2%) | 1 (0.2%) | 9 (2%) | 0 | 100 (4.1%) | 5 (0.1%) | 94 (4%) | NA |
| POLQ | 0 | 46 (2.9%) | 0 | 46 (2.9%) | NA | |||
| CDK12 | 5 (1.1%) | 5 (1.1%) | 0 | 79 (1.6%) | 34 (0.7%) | 41 (0.8%) | 8 (0.1%) | |
| Total non-overlapping | 76 (16.5) | 6 (1.3) | 71 (15.4) | 30 (6.5) | 1062 (18.1) | 61 (1.04) | 940 (16) | 336 (5.7) |
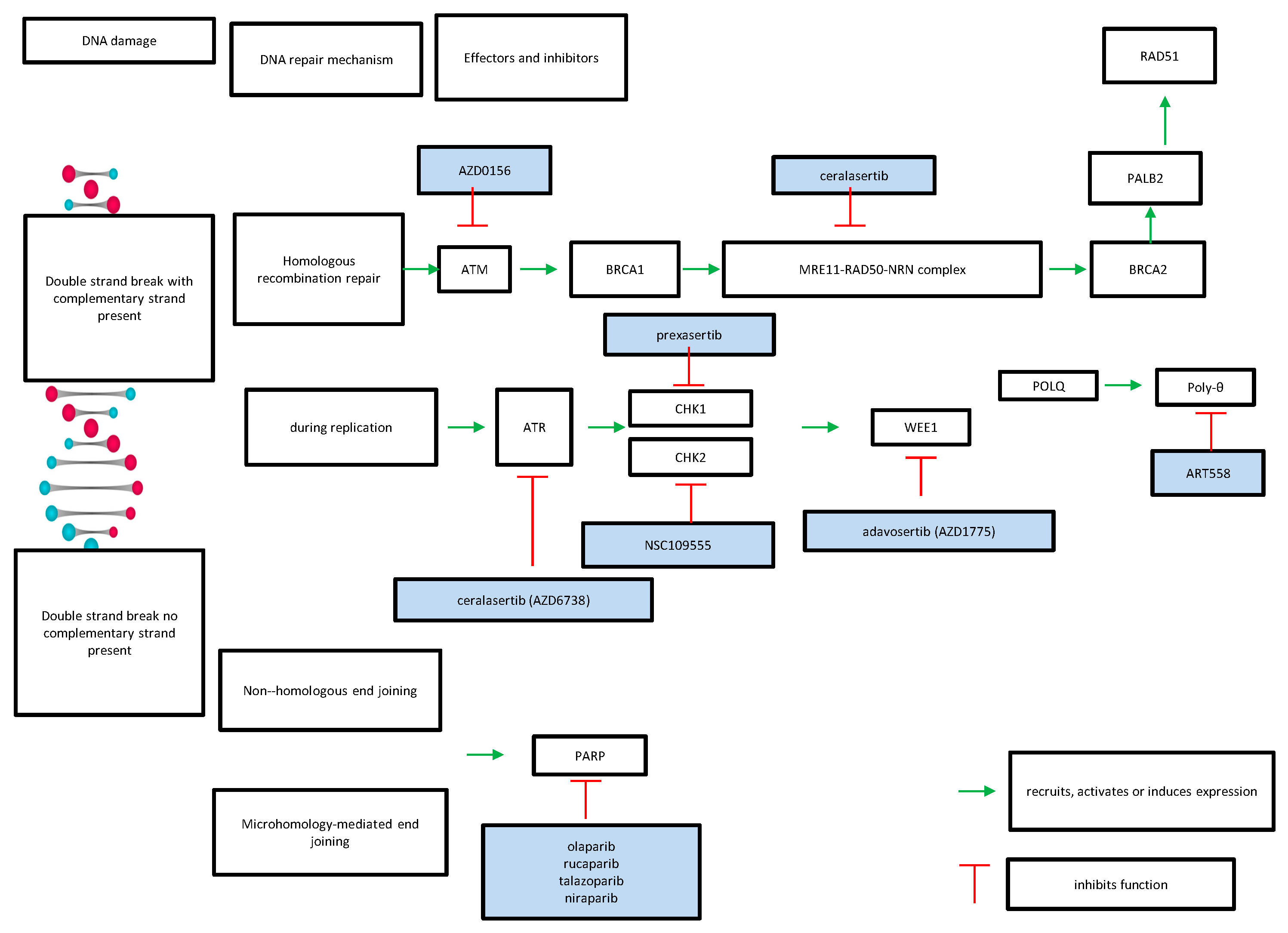
| Repair Pathway | Targeted Gene | Inhibitor |
|---|---|---|
| homologous recombination | ATR | ceralasertib |
| ATM | AZD0256 | |
| CHK2 | NSC109555 | |
| CHK1 | prexasertib | |
| WEE1 | adavosertib | |
| POLQ | ART558 | |
| non-homologous end-joining or microhomology-mediated end-joining | PARP | olaparib rucaparib talazoparib niraparib |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voutsadakis, I.A.; Digklia, A. The Landscape and Therapeutic Targeting of BRCA1, BRCA2 and Other DNA Damage Response Genes in Pancreatic Cancer. Curr. Issues Mol. Biol. 2023, 45, 2105-2120. https://doi.org/10.3390/cimb45030135
Voutsadakis IA, Digklia A. The Landscape and Therapeutic Targeting of BRCA1, BRCA2 and Other DNA Damage Response Genes in Pancreatic Cancer. Current Issues in Molecular Biology. 2023; 45(3):2105-2120. https://doi.org/10.3390/cimb45030135
Chicago/Turabian StyleVoutsadakis, Ioannis A., and Antonia Digklia. 2023. "The Landscape and Therapeutic Targeting of BRCA1, BRCA2 and Other DNA Damage Response Genes in Pancreatic Cancer" Current Issues in Molecular Biology 45, no. 3: 2105-2120. https://doi.org/10.3390/cimb45030135