
Role of Oxidative Stress on the Etiology and Pathophysiology of Amyotrophic Lateral Sclerosis (ALS) and Its Relation with the Enteric Nervous System

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Relevant Clinical Features of ALS
2. Etiology of Amyotrophic Lateral Sclerosis
2.1. Familiar Form
2.2. Sporadic Form
3. Pathophysiological Mechanisms of the Disease
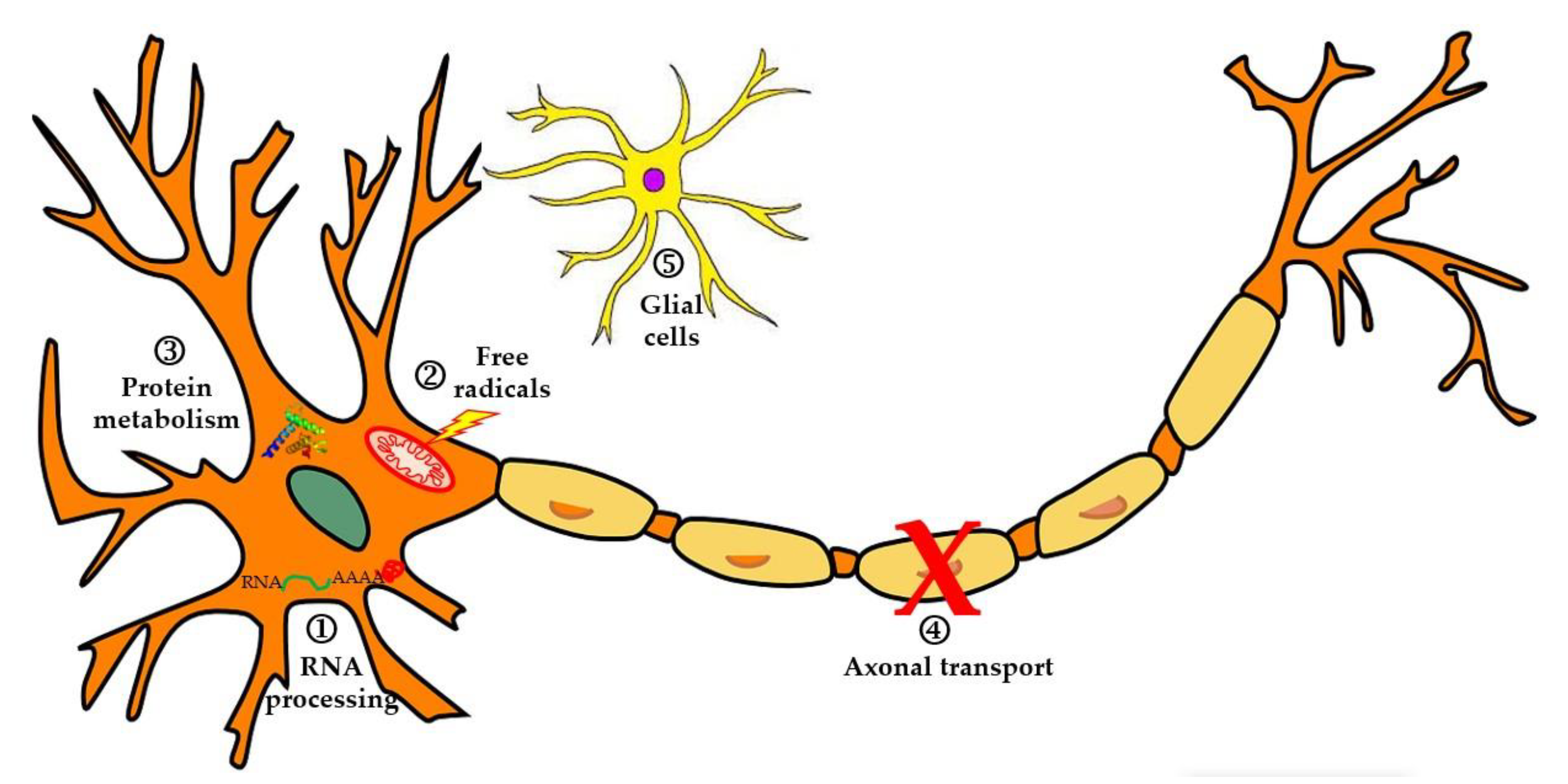
3.1. Alterations in RNA and Pathologic Cytoplasmic Aggregates
3.2. Changes in the Protein Degradation System
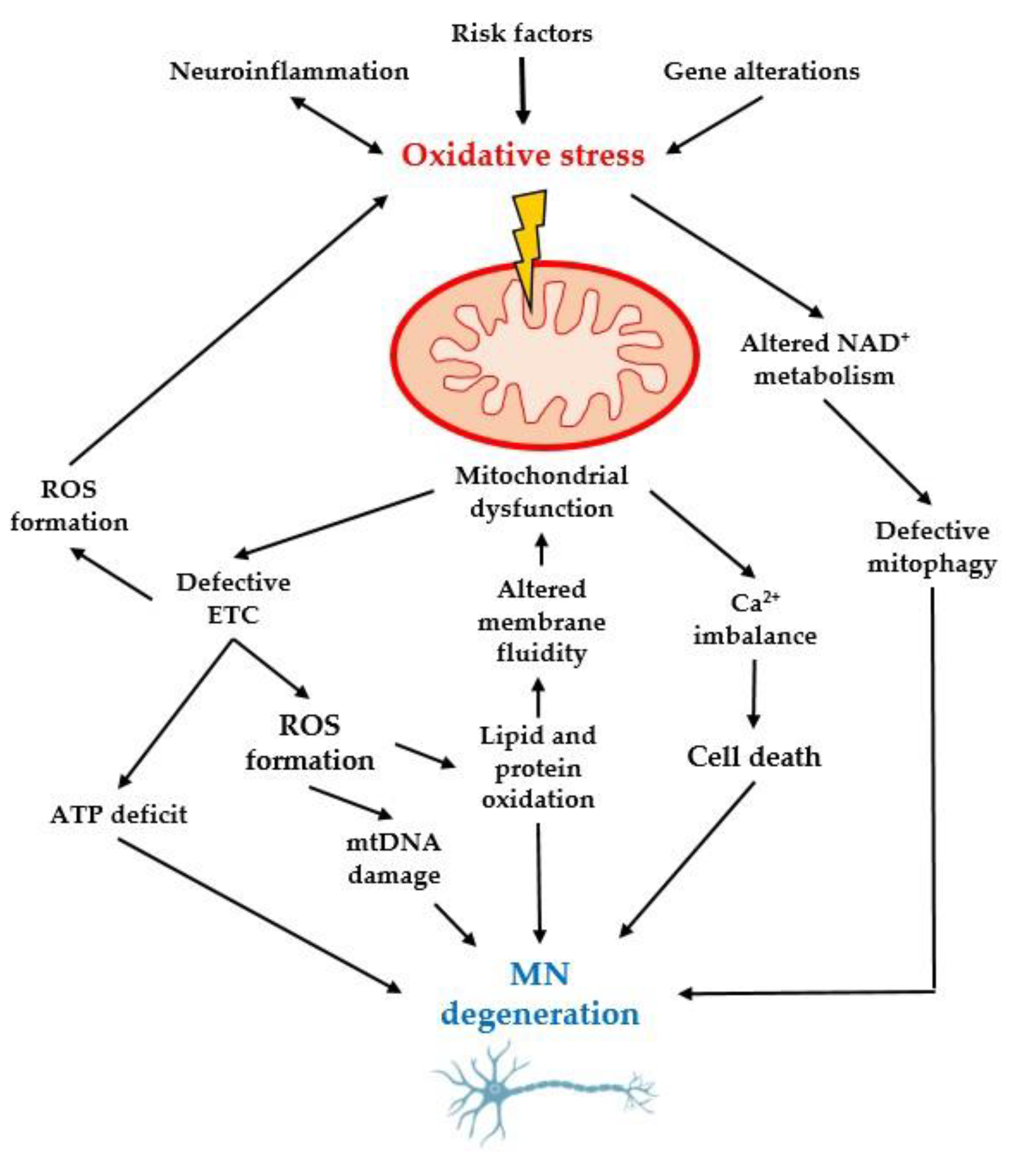
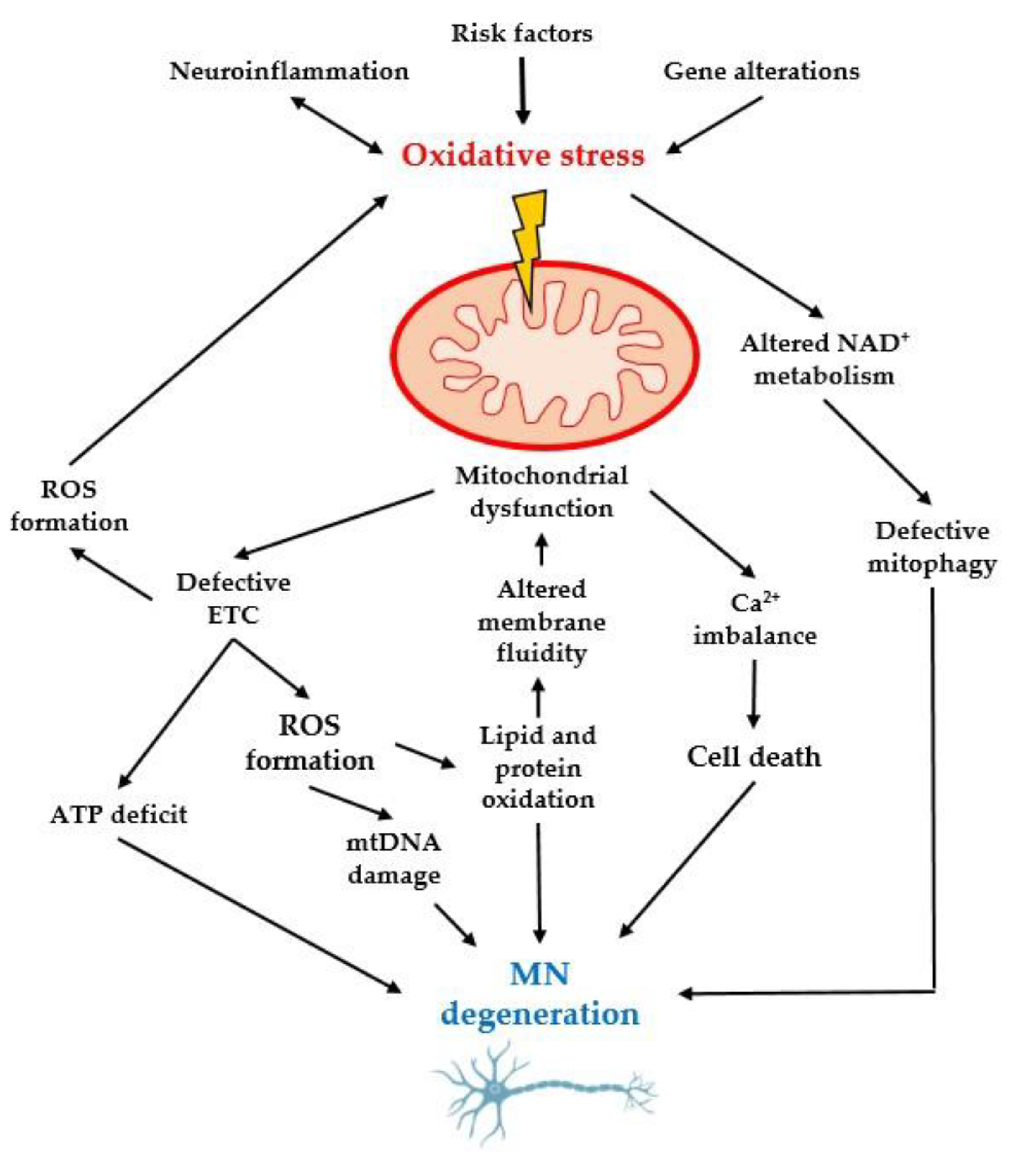
3.3. Importance of Oxidative Stress and Mitochondrial Dysfunction
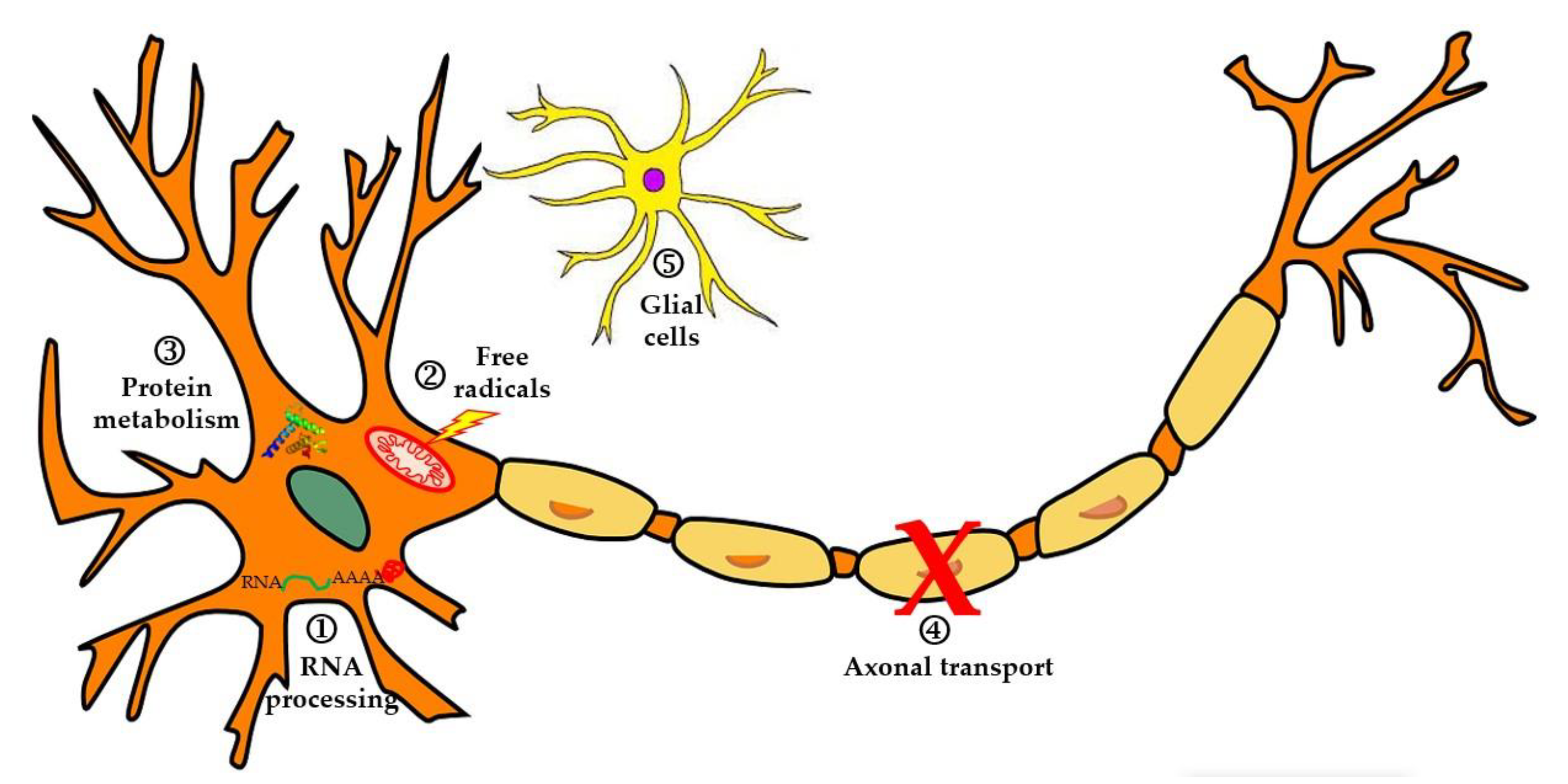
3.4. Axonal Transport Defects in Motor Neurons
3.5. Glutamate Excitotoxicity and Its Relation to Apoptosis
3.6. Roles of Microglia and Neuroinflammation in ALS
4. Pathogenic Risk Factors Involved in ALS
4.1. Neurodegeneration
4.2. Relationship of Physical Activity Intensity and Muscle Metabolism
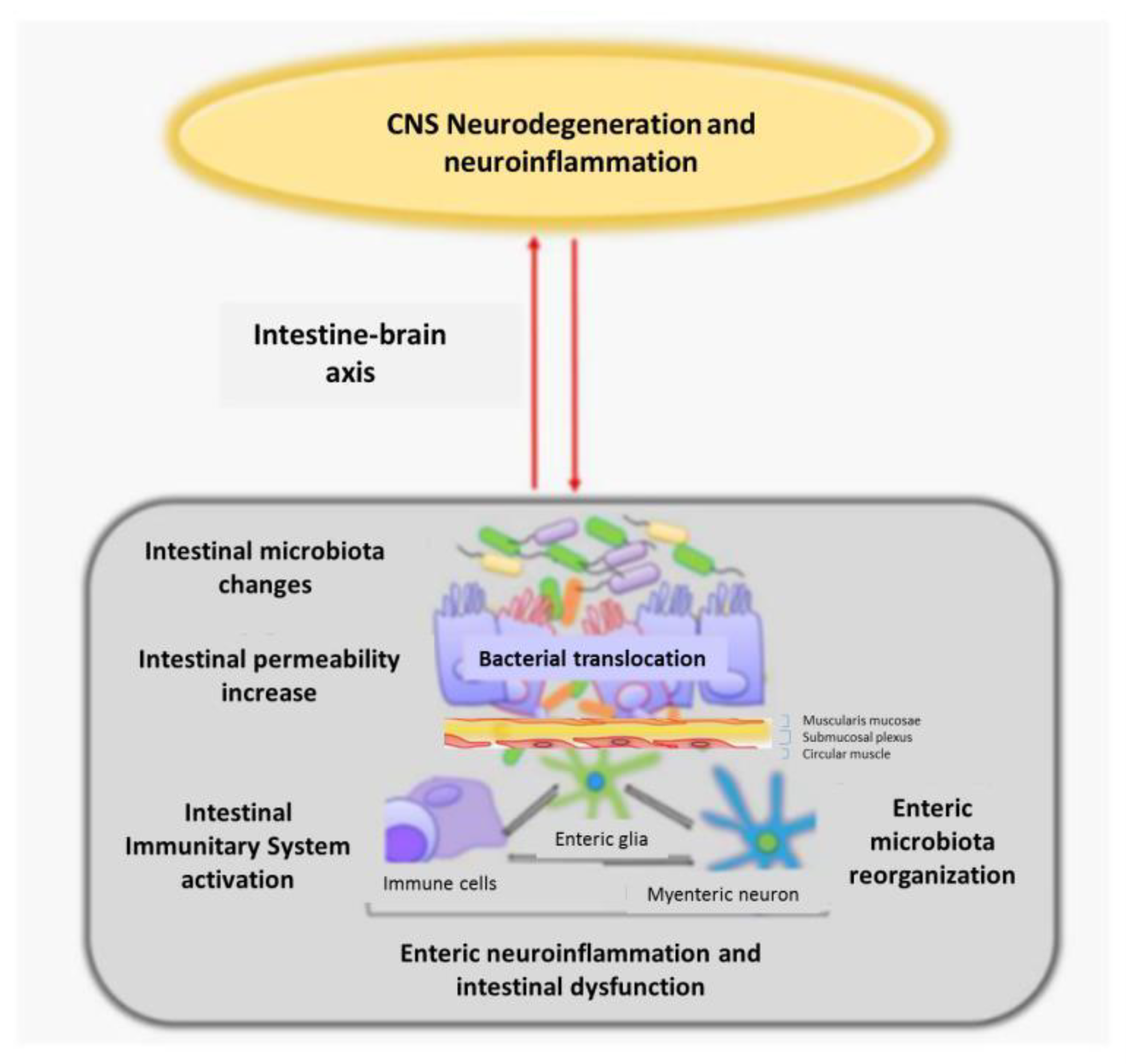
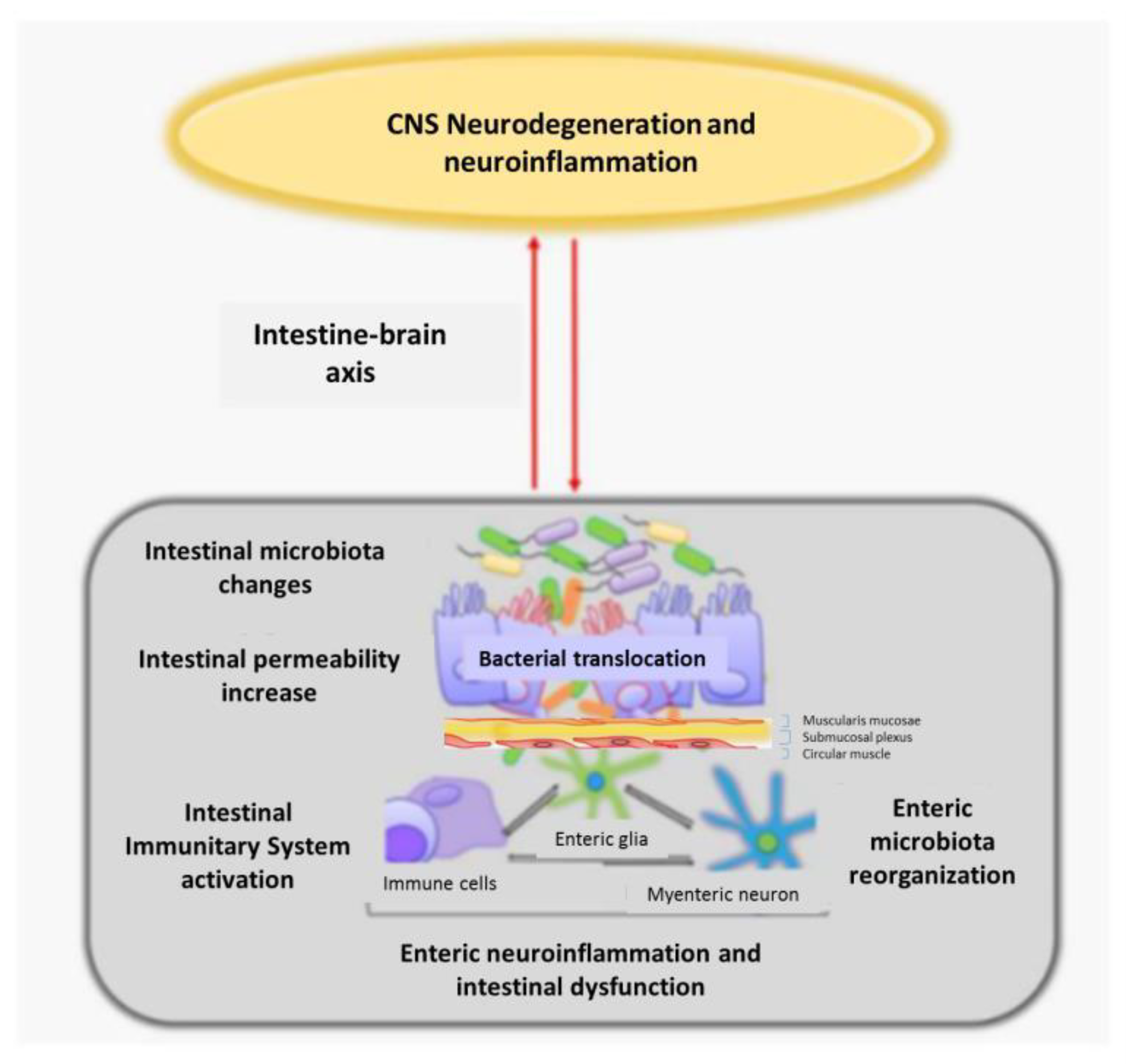
4.3. Intestinal Dysbiosis with Enteric Nervous System Involvement
5. Conclusions and Future Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H., Jr. Amyotrophic Lateral Sclerosis and Other Motor Neuron Diseases. In Harrison’s Principles of Internal Medicine, 20th ed.; Fauci, A.S., Kasper, D.L., Longo, D.L., Braunwald, E., Hauser, S.L., Jameson, J.L., Eds.; McGraw-Hill: New York, NY, USA, 2018; Volume 2, pp. 3141–3147. [Google Scholar]
- Conde, B.; Winck, J.C.; Azevedo, L.F. Estimating Amyotrophic Lateral Sclerosis and Motor Neuron Disease Prevalence in Portugal Using a Pharmaco-Epidemiological Approach and a Bayesian Multiparameter Evidence Synthesis Model. Neuroepidemiology 2019, 53, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Castro-Rodríguez, E.; Azagra, R.; Gómez-Batiste, X.; Povedano, M. Amyotrophic lateral sclerosis (ALS) from Primary Care. Epidemiology and clinical-care characteristics. Aten. Primaria 2021, 53, 102158. [Google Scholar] [CrossRef]
- Jericó, I.; Elizalde-Beiras, I.; Pagola, I.; Torné, L.; Galbete, A.; Delfrade-Osinaga, J.; Vicente, E. Clinical features and incidence trends of amyotrophic lateral sclerosis in Navarre, Spain, 2007–2018: A population-based study. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 401–409. [Google Scholar] [CrossRef]
- Nowicka, N.; Juranek, J.; Juranek, J.K.; Wojtkiewicz, J. Risk Factors and Emerging Therapies in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2019, 20, 2616. [Google Scholar] [CrossRef] [Green Version]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Maragakis, N.J.; Galvez-Jimenez, N. Epidemiology and Pathogenesis of Amyotrophic Lateral Sclerosis; UpToDate Inc.: Waltham, MA, USA, 2021. [Google Scholar]
- Grad, L.I.; Rouleau, G.A.; Ravits, J.; Cashman, N.R. Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harb. Perspect. Med. 2017, 7, a024117. [Google Scholar] [CrossRef] [Green Version]
- Elman, L.B.; McCluskey, L. Clinical Features of Amyotrophic Lateral Sclerosis and Other Forms of Motor Neuron Disease; UpToDate Inc.: Waltham, MA, USA, 2021; Volume 31, p. 1. [Google Scholar]
- Couratier, P.; Lautrette, G.; Luna, J.A.; Corcia, P. Phenotypic variability in amyotrophic lateral sclerosis. Rev. Neurol. 2021, 177, 536–543. [Google Scholar] [CrossRef]
- Elman, L.; McClusckey, L. Diagnosis of Amyotrophic Lateral Sclerosis and Other Forms of Motor Neuron Disease; UpToDate Inc.: Waltham, MA, USA, 2021. [Google Scholar]
- Chiò, A.; Mazzini, L.; Mora, G. Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology 2021, 167, 107986. [Google Scholar] [CrossRef]
- Sever, B.; Ciftci, H.; DeMirci, H.; Sever, H.; Ocak, F.; Yulug, B.; Tateishi, H.; Tateishi, T.; Otsuka, M.; Fujita, M.; et al. Comprehensive Research on Past and Future Therapeutic Strategies Devoted to Treatment of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 23, 2400. [Google Scholar] [CrossRef] [PubMed]
- Carrera-Juliá, S.; Moreno, M.L.; Barrios, C.; de la Rubia Ortí, J.E.; Drehmer, E. Antioxidant Alternatives in the Treatment of Amyotrophic Lateral Sclerosis: A Comprehensive Review. Front. Physiol. 2020, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Ng, L.; Khan, F.; Young, C.A.; Galea, M. Symptomatic treatments for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst. Rev. 2017, 1, CD011776. [Google Scholar] [CrossRef] [PubMed]
- Danel-Brunaud, V.; Touzet, L.; Chevalier, L.; Moreau, C.; Devos, D.; Vandolaeghe, S.; Defebvre, L. Ethical considerations and palliative care in patients with amyotrophic lateral sclerosis: A review. Rev. Neurol. 2017, 173, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Ingre, C.; Roos, P.M.; Piehl, F.; Kamel, F.; Fang, F. Risk factors for amyotrophic lateral sclerosis. Clin. Epidemiol. 2015, 7, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Riancho, J.; Gonzalo, I.; Ruiz-Soto, M.; Berciano, J. Why do motor neurons degenerate? Actualization in the pathogenesis of amyotrophic lateral sclerosis. Neurologia 2019, 34, 27–37. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Ravits, J.M.; La Spada, A.R. ALS motor phenotype heterogeneity, focality, and spread: Deconstructing motor neuron degeneration. Neurology 2009, 73, 805. [Google Scholar] [CrossRef] [Green Version]
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2018, 390, 2084–2098. [Google Scholar] [CrossRef]
- Lacorte, E.; Ferrigno, L.; Leoncini, E.; Corbo, M.; Boccia, S.; Vanacore, N. Physical activity, and physical activity related to sports, leisure and occupational activity as risk factors for ALS: A systematic review. Neurosci. Biobehav. Rev. 2016, 66, 61–79. [Google Scholar] [CrossRef]
- Ibba, G.; Piu, C.; Uleri, E.; Serra, C.; Dolei, A. Disruption by SaCas9 Endonuclease of HERV-Kenv, a Retroviral Gene with Oncogenic and Neuropathogenic Potential, Inhibits Molecules Involved in Cancer and Amyotrophic Lateral Sclerosis. Viruses 2018, 10, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obrenovich, M.; Jaworski, H.; Tadimalla, T.; Mistry, A.; Sykes, L.; Perry, G.; Bonomo, R.A. The Role of the Microbiota-Gut-Brain Axis and Antibiotics in ALS and Neurodegenerative Diseases. Microorganisms 2020, 8, 784. [Google Scholar] [CrossRef] [PubMed]
- Furtado Bastos, A.; Orsini, M.; Machado, D.; Mello, M.P.; Nader, S.; Silva, J.G.; da Silva Catharino, A.M.; de Freitas, M.R. Amyotrophic lateral sclerosis: One or multiple causes? Neurol. Int. 2011, 3, e4. [Google Scholar] [CrossRef] [Green Version]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.P.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Williams, K.L.; Warraich, S.T.; Yang, S.; Solski, J.A.; Fernando, R.; Rouleau, G.A.; Nicholson, G.A.; Blair, I.P. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2527.e3–2527.e10. [Google Scholar] [CrossRef] [PubMed]
- Weishaupt, J.H.; Bartels, C.; Pölking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Sperling, S.; Bohn, M.; Hüther, G.; et al. Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J. Pineal Res. 2006, 41, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Homma, K.; Ichijo, H. SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv. Biol. Regul. 2016, 60, 95–104. [Google Scholar] [CrossRef]
- Pandi-Perumal, S.R.; BaHammam, A.S.; Brown, G.M.; Spence, D.W.; Bharti, V.K.; Kaur, C.; Hardeland, R.; Cardinali, D.P. Melatonin antioxidative defense: Therapeutical implications for aging and neurodegenerative processes. Neurotox. Res. 2013, 23, 267–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Landers, J.E. Mutations in the Profilin 1 Gene Cause Familial Amyotrophic Lateral Sclerosis. Nature 2012, 488, 499. [Google Scholar] [CrossRef] [Green Version]
- Ikenaka, K.; Katsuno, M.; Kawai, K.; Ishigaki, S.; Tanaka, F.; Sobue, G. Disruption of axonal transport in motor neuron diseases. Int. J. Mol. Sci. 2012, 13, 1225–1238. [Google Scholar] [CrossRef]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; Estrela, J.M.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef]
- Cheah, B.C.; Vucic, S.; Krishnan, A.; Kiernan, M.C. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr. Med. Chem. 2010, 17, 1942–1959. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Guo, Y.S.; Liu, Y.L.; Wu, S.Y.; Yang, C.; Wu, D.X.; Wu, H.R.; Zhang, Y.S.; Li, C.Y. Oxidative stress in immune-mediated motoneuron destruction. Brain Res. 2009, 1302, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Reyes, N.A.; Fisher, J.K.; Austgen, K.; Vandenberg, S.; Huang, E.J.; Oakes, S.A. Blocking the mitochondrial apoptotic pathway preserves motor neuron viability and function in a mouse model of amyotrophic lateral sclerosis. J. Clin. Investig. 2010, 120, 3673–3679. [Google Scholar] [CrossRef] [Green Version]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from Familial and Sporadic ALS Patients are Toxic to Motor Neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visser, A.E.; Rooney, J.P.K.; D’Ovidio, F.; Westeneng, H.J.; Vermeulen, R.C.H.; Beghi, E.; Chiò, A.; Logroscino, G.; Hardiman, O.; Euro-Motor Consortium; et al. Multicentre, cross-cultural, population-based, case-control study of physical activity as risk factor for amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 797–803. [Google Scholar] [CrossRef]
- Swash, M. Physical activity as a risk factor in ALS. J. Neurol. Neurosurg. Psychiatry 2018, 89, 793. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zaken, S.; Nefussy, B.; Meckel, Y.; Eliakim, A.; Nemet, D.; Gotkine, M.; Lorber, D.; Zeev, A.; Drory, V.E. Common genetic basis of ALS patients and soccer players may contribute to disease risk. Neurol. Sci. 2022, 43, 4231–4238. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.D.; Kamel, F. Military service, deployments, and exposures in relation to amyotrophic lateral sclerosis etiology and survival. Epidemiol. Rev. 2015, 37, 55–70. [Google Scholar] [CrossRef] [Green Version]
- Huisman, M.H.; Seelen, M.; de Jong, S.W.; Dorresteijn, K.R.; van Doormaal, P.T.; van der Kooi, A.J.; de Visser, M.; Schelhaas, H.J.; van den Berg, L.H.; Veldink, J.H. Lifetime physical activity and the risk of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxidative Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef]
- Harwood, C.A.; Westgate, K.; Gunstone, S.; Brage, S.; Wareham, N.J.; McDermott, C.J.; Shaw, P.J. Long-term physical activity: An exogenous risk factor for sporadic amyotrophic lateral sclerosis? Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 377. [Google Scholar] [CrossRef] [Green Version]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863. [Google Scholar] [CrossRef] [Green Version]
- Pehar, M.; Vargas, M.R.; Cassina, P.; Barbeito, A.G.; Beckman, J.S.; Barbeito, L. Complexity of astrocyte-motor neuron interactions in amyotrophic lateral sclerosis. Neurodegener. Dis. 2005, 2, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Kazmi, S.A.; Jameson, K.G.; Hsiao, E.Y. The Microbiome as a Modifier of Neurodegenerative Disease Risk. Cell Host Microbe 2020, 28, 201. [Google Scholar] [CrossRef]
- Mittal, R.; Debs, L.H.; Patel, A.P.; Nguyen, D.; Patel, K.; O’Connor, G.; Grati, M.; Mittal, J.; Yan, D.; Eshraghi, A.A.; et al. Neurotransmitters: The critical modulators regulating gut-brain axis. J. Cell. Physiol. 2017, 232, 2359. [Google Scholar] [CrossRef] [Green Version]
- Biagi, E.; Franceschi, C.; Rampelli, S.; Severgnini, M.; Ostan, R.; Turroni, S.; Consolandi, C.; Quercia, S.; Scurti, M.; Monti, D.; et al. Gut Microbiota and Extreme Longevity. Curr. Biol. 2016, 26, 1480–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iatsenko, I.; Boquete, J.P.; Lemaitre, B. Microbiota-Derived Lactate Activates Production of Reactive Oxygen Species by the Intestinal NADPH Oxidase Nox and Shortens Drosophila Lifespan. Immunity 2018, 49, 929–942.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stebegg, M.; Silva-Cayetano, A.; Innocentin, S.; Jenkins, T.P.; Cantacessi, C.; Gilbert, C.; Linterman, M.A. Heterochronic faecal transplantation boosts gut germinal centres in aged mice. Nat. Commun. 2019, 10, 2443. [Google Scholar] [CrossRef]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef]
- Gotkine, M.; Kviatcovsky, D.; Elinav, E. Amyotrophic lateral sclerosis and intestinal microbiota—Toward establishing cause and effect. Gut Microbes 2020, 11, 1833. [Google Scholar] [CrossRef]
- Zhang, Y.; Ogbu, D.; Garrett, S.; Xia, Y.; Sun, J. Aberrant enteric neuromuscular system and dysbiosis in amyotrophic lateral sclerosis. Gut Microbes 2021, 13, 1996848. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Kwong, L.K.; Sampathu, D.M.; Trojanowski, J.Q.; Lee, V.M. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: Protein misfolding diseases without amyloidosis. Arch. Neurol. 2007, 64, 1388–1394. [Google Scholar] [CrossRef] [Green Version]
- Sorarú, G.; Orsetti, V.; Buratti, E.; Baralle, F.; Cima, V.; Volpe, M.; D’ascenzo, C.; Palmieri, A.; Koutsikos, K.; Pegoraro, E.; et al. TDP-43 in skeletal muscle of patients affected with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 240–243. [Google Scholar] [CrossRef]
- Riva, N.; Gentile, F.; Cerri, F.; Gallia, F.; Podini, P.; Dina, G.; Falzone, Y.M.; Fazio, R.; Lunetta, C.; Calvo, A.; et al. Phosphorylated TDP-43 aggregates in peripheral motor nerves of patients with amyotrophic lateral sclerosis. Brain 2022, 145, 276–284. [Google Scholar] [CrossRef]
- Pattle, S.B.; O’Shaughnessy, J.; Kantelberg, O.; Rifai, O.M.; Pate, J.; Nellany, K.; Hays, N.; Arends, M.J.; Horrocks, M.H.; Waldron, F.M.; et al. pTDP-43 aggregates accumulate in non-central nervous system tissues prior to symptom onset in amyotrophic lateral sclerosis: A case series linking archival surgical biopsies with clinical phenotypic data. J. Pathol. Clin. Res. 2023, 9, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Fricker, R.A.; Green, E.L.; Jenkins, S.I.; Griffin, S.M. The Influence of Nicotinamide on Health and Disease in the Central Nervous System. Int. J. Tryptophan Res. 2018, 11, 117864691877665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145. [Google Scholar] [CrossRef]
- Martin, S.; Battistini, C.; Sun, J. A Gut Feeling in Amyotrophic Lateral Sclerosis: Microbiome of Mice and Men. Front. Cell. Infect. Microbiol. 2022, 12, 839526. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, C.; Antonioli, L.; Colucci, R.; Blandizzi, C.; Fornai, M. Interplay among gut microbiota, intestinal mucosal barrier and enteric neuro-immune system: A common path to neurodegenerative diseases? Acta Neuropathol. 2018, 136, 345–361. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Pingarrón, L.; Almeida, H.; Soria-Aznar, M.; Reyes-Gonzales, M.C.; Terrón, M.P.; García, J.J. Role of Oxidative Stress on the Etiology and Pathophysiology of Amyotrophic Lateral Sclerosis (ALS) and Its Relation with the Enteric Nervous System. Curr. Issues Mol. Biol. 2023, 45, 3315-3332. https://doi.org/10.3390/cimb45040217
López-Pingarrón L, Almeida H, Soria-Aznar M, Reyes-Gonzales MC, Terrón MP, García JJ. Role of Oxidative Stress on the Etiology and Pathophysiology of Amyotrophic Lateral Sclerosis (ALS) and Its Relation with the Enteric Nervous System. Current Issues in Molecular Biology. 2023; 45(4):3315-3332. https://doi.org/10.3390/cimb45040217
Chicago/Turabian StyleLópez-Pingarrón, Laura, Henrique Almeida, Marisol Soria-Aznar, Marcos C. Reyes-Gonzales, María Pilar Terrón, and Joaquín J. García. 2023. "Role of Oxidative Stress on the Etiology and Pathophysiology of Amyotrophic Lateral Sclerosis (ALS) and Its Relation with the Enteric Nervous System" Current Issues in Molecular Biology 45, no. 4: 3315-3332. https://doi.org/10.3390/cimb45040217
APA StyleLópez-Pingarrón, L., Almeida, H., Soria-Aznar, M., Reyes-Gonzales, M. C., Terrón, M. P., & García, J. J. (2023). Role of Oxidative Stress on the Etiology and Pathophysiology of Amyotrophic Lateral Sclerosis (ALS) and Its Relation with the Enteric Nervous System. Current Issues in Molecular Biology, 45(4), 3315-3332. https://doi.org/10.3390/cimb45040217