Abstract
Elucidation of the tertiary structure of proteins is an important task for biological and medical studies. AlphaFold, a modern deep-learning algorithm, enables the prediction of protein structure to a high level of accuracy. It has been applied in numerous studies in various areas of biology and medicine. Viruses are biological entities infecting eukaryotic and procaryotic organisms. They can pose a danger for humans and economically significant animals and plants, but they can also be useful for biological control, suppressing populations of pests and pathogens. AlphaFold can be used for studies of molecular mechanisms of viral infection to facilitate several activities, including drug design. Computational prediction and analysis of the structure of bacteriophage receptor-binding proteins can contribute to more efficient phage therapy. In addition, AlphaFold predictions can be used for the discovery of enzymes of bacteriophage origin that are able to degrade the cell wall of bacterial pathogens. The use of AlphaFold can assist fundamental viral research, including evolutionary studies. The ongoing development and improvement of AlphaFold can ensure that its contribution to the study of viral proteins will be significant in the future.
1. Introduction
Proteins play a crucial role both in building biological structures and in managing biochemical processes in living organisms. Proteins are linear unbranched polymers of amino acid residues. To possess biological activity, proteins adopt unique three-dimensional structures (folds), which is known as the “native state” [1,2]. The folded structure is determined by the amino acid sequence of the protein (“primary structure”) [3,4], and the formation of the folded native conformation (“tertiary structure”) starts with rapid folding into a “secondary structure”, which is a local spatial conformation of the polypeptide backbone, stabilised by intramolecular hydrogen bonds [5]. The most common elements of the secondary structure are α-helices and β-sheets. The so-called “quaternary structure” is the result of assembly of the folded proteins or protein subunits into protein complexes of fully functional protein [6]. Thus, the protein structure can be described using four levels of organisation: a primary, secondary, tertiary and, for some proteins, quaternary structure (Figure 1).
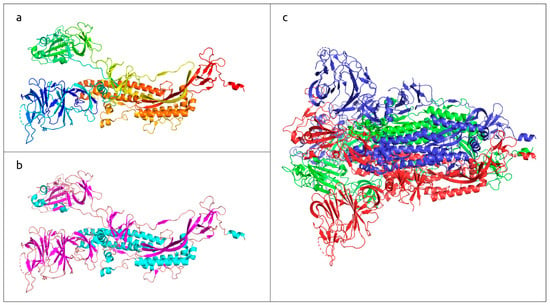
Figure 1.
Three-dimensional structure of SARS-CoV-2 trimeric spike glycoprotein, determined with electron microscopy (PDB code #7DF3 [7]). (a) Monomeric subunit coloured based on a rainbow gradient scheme, where the N-terminus of the polypeptide chain is coloured blue, and the C-terminus is coloured red. (b) Monomeric subunit coloured based on the secondary structure, where α-helices are coloured cyan, β-sheets are coloured magenta, and loops are coloured wheat. (c) Quaternary structure of functional trimer, where each monomer is coloured in a different colour.
Knowledge of the three-dimensional structure of proteins is important for understanding their functions. A detailed knowledge of three-dimensional structure is crucial for protein structure-based drug design [8]. The main techniques for determining protein structures are X-ray crystallography [9], NMR spectroscopy [10] and Cryoelectron microscopy [11]. Experimentally determined protein structures are stored in databases, the largest of them being the publicly available Protein Data Bank (PDB) (https://www.rcsb.org/, accessed on 1 March 2023). As of March 2023, the PDB database contained about 202,000 experimentally determined structures, most of which belonged to proteins. This is, however, just a small fraction of all proteins for which the primary sequences are known. The UniProtKB/TrEMBL database alone contains over 200 million sequence records, (database release 2022_05 of 14 December 2022 contained 229,580,745 sequence entries, https://www.ebi.ac.uk/uniprot/TrEMBLstats, accessed on 1 March 2023). Thus, the prediction of the three-dimensional structure of a protein is an urgent problem that aims to fill the gap between the large known number of primary sequences and the relatively small number of known structures.
Prediction of the three-dimensional structure of proteins is a difficult task. For a long time, the main prediction methods included comparative modelling (homology modelling), threading and ab initio and machine-learning approaches [12,13]. The development of end-to-end machine-learning approaches in recent years has resulted in the emergence of new techniques that can often outperform other methods [2,14]. Moreover, recent progress associated with deep-learning methods enables speculation about a revolution in protein-structure prediction [15]. One of the most popular deep-learning techniques is Alphabet–Google DeepMind’s neural network-based end-to-end solution AlphaFold2 (AlphaFold, AF2), which was presented in the CASP14 competition [16], the second iteration of the AlphaFold system entered in CASP13 [17]. AlphaFold employs a deep-learning approach and a conventional neural network. This technique is able to predict the distance and torsion distribution of proteins, using training schemes of experimentally determined PDB structures, protein primary sequences and the multiple sequence alignment (MSA) of proteins. In CASP14, AlphaFold2 structures had a median backbone accuracy of 0.96 Å RMSD95 (Cα root-mean-square deviation at 95% residue coverage) and an all-atom accuracy of 1.5 Å RMSD95. The corresponding values for the prediction of the best alternative method were 2.8 Å and 3.5 Å [16]. The high level of accuracy of AlphaFold2 predictions boosted the popularity of this technique. One might even talk about “AlphaFold mania”, given the astonishing increase in the number of journal articles and preprints citing AlphaFold2 AI software [18]. As of the beginning of March 2023, the original paper [16] published in July 2021, which described AlphaFold2′s release, with its source code, was accessed about a million times and, according to the Web of Science metric, was cited about 5000 times (https://www.nature.com/articles/s41586-021-03819-2/metrics, accessed on 1 March 2023).
The updated version of AlphaFold2, called AlphaFold-Multimer, also developed by DeepMind, was released several months after AlphaFold2 [19]. AlphaFold-Multimer was designed to predict the three-dimensional structure of protein complexes. AlphaFold-Multimer was benchmarked on a large dataset of 4446 protein complexes, successfully predicting the interface in 70% of cases of heteromeric interfaces and in 72% of cases of homomeric interfaces. A high level of predictive accuracy was demonstrated in 26% of cases of heteromeric interfaces and 36% of cases of homomeric interfaces.
The level of accuracy of AlphaFold (and other AI protein-folding methods, such as RoseTTAFold [20]) makes it tempting to use AlphaFold predictions in various fields of biological and medical research. In particular, virology, the importance of which has become especially evident in the light of the recent COVID-19 pandemic, has received a new tool that can solve a number of problems requiring the knowledge of three-dimensional protein structures. Virology studies viruses, probably the most widespread entities on Earth [21]. Viruses infect various cellular organisms, including eukaryotes, archaea and bacteria. In the latter case, they are called “bacteriophages”, or “phages”. Phages and their proteins that are harmful to bacteria can be used to fight bacterial infection in humans, animals and plants [22,23]. So-called “phage therapy”, or the use of bacteriophages to treat bacterial infections, can assist in the context of the rise of antimicrobial resistance [24]. This review describes different cases of the use of AlphaFold for the purposes of viral research. It summarizes the results of the studies involving AlphaFold predictions, analyses the possible advantages and disadvantages of AlphaFold for predictions of viral proteins and discusses corresponding studies (Table 1).

Table 1.
Summary of the studies discussed.
2. Application of AF2 for Research on Eukaryotic Viruses
2.1. Application of AlphaFold for SARS-CoV-2 Research
The outbreak of severe acute respiratory syndrome caused by coronavirus 2 (SARS-CoV-2, realm Ribozyviria, class Pisoniviricetes, order Nidovirales, family Coronaviridae, genus Betacoronavirus) and the spread of associated infection boosted research on coronaviruses. The structure of SARS-CoV-2 spike (S) glycoprotein, the main target of antibodies, has been determined by cryo-electron microscopy and was used in the development of vaccines and inhibitors [82,83]. S glycoprotein promotes entry into the cell. Another target of drug design is main protease cutting the initial translated propeptide into functional viral proteins. The crystal structure of the SARS-CoV-2 main protease was also obtained experimentally [84].
To assist the solution of tasks related to general research and drug design, different structure prediction techniques, including AlphaFold, were used for prediction of SARS-CoV-2 proteins [25,26,27,28,29,85]. The main task was probably the investigation of the mechanism of interaction of the SARS-CoV-2 receptor-binding protein (RBP), which is the SARS-CoV-2 spike, and the angiotensin-converting enzyme 2 (ACE2) receptor. AF2 predictions enabled clarification of the structural features of monomeric and multimeric formulations of the vaccine and suggested that monomeric formulation presents more antigenic epitopes [27]. The emergence of new immune-escaping variants of SARS-CoV-2, such as Omicron BA1, made it important to study potential mutation sites that do not yet exist in nature but could increase the binding affinity of RBD and the receptor [29]. AF2 predictions were successfully used to find an explanation for the observed reduction in the neutralisation of SARS-CoV-2 variants of concern compared with other variants [28]. AF2 predictions can be combined with molecular dynamics simulations to improve modelling accuracy [86] and to predict the physical properties of proteins. Such models can be used for studies of both qualitative and quantitative aspects of the formation of the quaternary structure of proteins [85]. AlphaFold models are useful for revealing possible ligand binding sites. Together with virtual screening and in silico validation, these approaches provide the basis for the biological testing of new drugs and for the repurposing of natural products [25].
The accuracy of predicted structures can be assessed using computational techniques [87] and via experimental methods, e.g., optical spectroscopy or measurement of solution residual dipolar couplings data (RDCs) [30,88]. A meticulous evaluation of the concordance of AF2 models of the SARS-CoV-2 homodimeric 3C-like protease (Mpro) with residual dipolar couplings (RDCs) measured in solution for 15N–1HN and 13C′–1HN atom pairs indicated the close agreement of AlphaFold predictions with experimental data (Figure 2) [30].
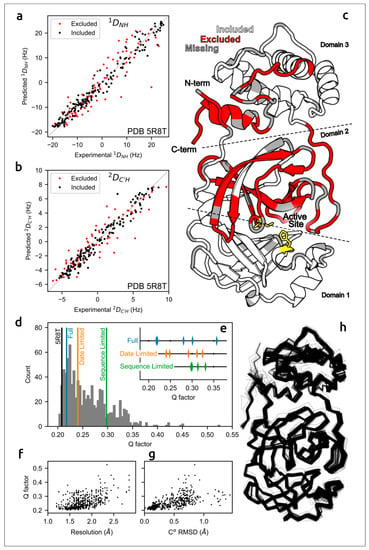
Figure 2.
Agreement between measured Mpro RDCs and values predicted by AF2-derived models. (a) 1DNH and (b) 2DC′H experimental couplings vs. those predicted from X-ray structure 5R8T. (c) Excluded residues (red) illustrated on a ribbon diagram (PDB code 5R8T; only a single chain is shown, for clarity); residues with missing RDCs are shown in grey and the catalytic dyad is shown in yellow. (d) Q-factors from SVD fits of 1DNH and 2DC′H RDCs to the included region of all available Mpro X-ray structures, plotted as a histogram, with the top-ranked (Amber-relaxed) AF2 models obtained using full, date-limited and sequence-limited implementations marked. (e) Q-factors of all Amber-relaxed models. (f) X-ray structure resolution vs. Q-factor and (g) Cα RMSD (relative to 5R8T) vs. Q-factor. (h) Cα wireframe of all 352 PDB structures. Images courtesy of Dr. Adriaan Bax. Reprinted/adapted with permission from Ref. [30]. Not subject to U.S. Copyright.
Interestingly, the high level of accuracy of AF2 predictions makes it possible to use AlphaFold predictions to determine a macromolecular structure from crystallographic diffraction experiments. It has been shown that a template-free AF2 model, generated by the AlphaFold2 group, was of sufficient quality to phase the native SARS-CoV-2 ORF8 dataset by molecular replacement, overcoming the limitations of the crystallographic phasing problem [26]. However, a comparison of RMSD (root mean square deviation of atomic positions) values of SARS-CoV-2 spike RBD, the laboratory-derived structure with both trRosetta-generated models [89] and models generated by AlphaFold v2.1.0, indicated the high level of accuracy of both methods, but the better results were obtained with trRosetta.
2.2. Application of AlphaFold to Study Eukaryotic Viruses
AlphaFold is widely used in research on other eukaryotic viruses, including monkeypox virus (MPXV) [31,32,33,34], herpes simplex virus [35,36], hepatitis E virus (HEV) [37] and other viral pathogens of humans and economically significant animals and plants [38,39,40,41,42,43]. Monkeypox virus (MPXV) represents a new serious threat to human health. MPXV has spread to 110 countries (https://www.cdc.gov/poxvirus/mpox/response/2022/world-map.html, accessed on 1 March 2023). As of 1 March 2023, there were 86,231 confirmed cases worldwide, of which 84,858 cases occurred in locations that had not previously reported MPXV cases. Monkeypox virus is classified as a member of realm Varidnaviria, class Pokkesviricetes, order Chitovirales, family Poxviridae, genus Orthopoxvirus and is evolutionarily close to vaccinia virus (VACV), the smallpox virus. AlphaFold-derived structures of the recombinantly expressed MPXV antigen truncations to their VACV homologues have indicated that MPXV and VACV antigens are likely to achieve similar conformations [34]. The World Health Organisation (WHO) has recommended the current anti-smallpox drugs tecovirimat, brincidofovir and cidofovir for the treatment of monkeypox [90]. Brincidofovir and cidofovir inhibit DNA polymerase (DNAP), while tecovirimat is an inhibitor for poxvirus phospholipase D (protein F13) [91], but specific antiviral treatment requires new drugs.
MPXV DNA polymerase (DNAP) is a very important antiviral drug target. The laboratory-derived structure of MPXV DNAP was deposited in the RCSB PDB database (PDB code 8HG1) in mid-November 2022, and a paper describing this structure was published in January 2023 [92]. Before that, the AF2-derived structure was obtained and used in the search and design of new inhibitors of MPXV DNAP. The molecules found were predicted to bind to the MPXV DNAP with a binding energy comparable to that of brincidofovir and cidofovir. New MPXV DNAP inhibitors are important in the context of possible drug resistance, which can arise due to mutations in proteins of the DNA replication complex (RC). Studies of the effect of mutations in MPXV RC using AF2-generated models have suggested similar mechanisms of drug resistance to cidofovir in monkeypox and vaccinia viruses [32]. It appears that the use of highly accurate AlphaFold predictions can assist the forecasting of the emergence of drug-resistant variants of concern to improve preparedness for them.
The molecular mechanism of interaction of tecovirimat with the monkeypox phospholipase D (F13) was studied using AlphaFold models and molecular dynamics simulations [33]. The results suggested a detailed mechanism of inhibition of F13 by tecovirimat (Figure 3) and supported the efficacy of tecovirimat against monkeypox virus, emphasising the importance of the availability of precise modelling for revealing molecular mechanisms of drug action.
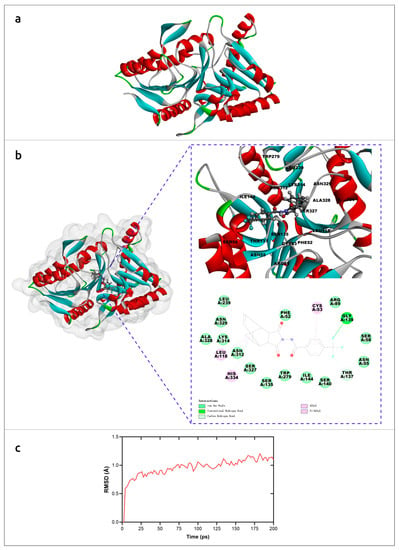
Figure 3.
Molecular simulation analysis of tecovirimat with F13 from monkeypox virus. (a) Overview of the F13 protein structure from monkeypox virus generated by AlphaFold. (b) The minimum free energy poses with of F13 protein and tecovirimat and corresponding interactions plots. (c) RMSD of monkeypox virus F13-tecovirimat complex during the production stage of molecular dynamics. Images courtesy of Dr. Leiliang Zhang. Reprinted/adapted with permission from Ref. [33]. © 2022 The British Infection Association.
The development of new drugs is barely possible without an understanding of the mechanisms of viral infection. This knowledge can often require robust structural analysis, which can make use of modern deep-learning structure prediction methods. AlphaFold can facilitate the elucidation of the functionality of viral proteins.
Herpesviruses constitute an important group of pathogens that infect animals, including humans. Herpesviruses infect most vertebrates, causing a lifelong latent infection [93]. Herpesviruses belong to the realm Duplodnaviria, class Herviviricetes, order Herpesvirales, and comprise the families Alloherpesviridae, Herpesviridae and Malacoherpesviridae [94]. Human herpesviruses belong to the family Herpesviridae. Herpes simplex virus 1 (HSV-1) (genus Alphaherpesviruse), residing in sensory neurons or sympathetic neurons, has been shown to severely modify infected cells and to remodel the composition and architecture of cellular membranes [35,95,96]. One of the HSV-1 proteins, phosphatase adaptor UL21, mediates dephosphorylation and accelerates the rate of ceramide to sphingomyelin conversion, altering cell membranes and influencing viral replication [35]. AlphaFold-Multimer modelling has revealed the details of the interaction of UL21 and viral protein UL16 and has enabled the suggestion of the functionality of domains of the latter protein using its structural features. Specific protein–protein interactions have been shown to be essential for lipid metabolism [35]. The use of AlphaFold has also shown that another HSV-1 protein, the tegument protein UL37, interacts with the cytoplasmic surface of the lipid membrane, suggesting that UL37 can be a peripheral membrane protein [36]. AlphaFold predictions have suggested the domain organisation of UL37, and assisted experimental studies and molecular dynamics simulation have clarified the structural features and molecular mechanisms of UL37 interactions.
Fundamentally similar tasks concerning research on other viral pathogens of animals, including humans, and plants can be made easier by the use of AlphaFold predictions. These tasks include mechanisms that are crucial for viral attachment, penetration, replication, release and other steps in the viral infection cycle. They can include the investigation of viral proteins and membranes [38,41,43], viral proteins and DNA [39] and studies of viral proteins, glycoproteins and their mutations [37,40,42]. It is noteworthy that AlphaFold predictions are often used as part of an integrated approach, making the planning of experiments easier and improving understanding of the results obtained.
3. Application of AlphaFold for Research on Bacteriophages
Bacteriophages (a.k.a. phages) are viruses that infect and replicate in bacterial cells alone. Bacteriophages are ubiquitous—they can be found in water, soil and various living organisms [97]. The total number of bacteriophages can be estimated at 1031 viral particles, which is 10–100 times the number of cells [98]. The total mass of these particles is about a trillion tons [99]. Phages are also members of plant and animal microbiomes, including humans. For example, the human gastrointestinal tract contains more than 1012 phage virions [100]. The ability of bacteriophages to destroy the cells of pathogenic bacteria attracted the attention of scientists as early as the beginning of the 20th century. In recent decades, interest in bacteriophage therapy has begun to grow, primarily due to the spread of antibiotic resistance. Phage therapy has important advantages [101], including sustained bactericidal activity and “autodosing”, wherein the number of phages positively correlates with the number of host bacteria. Furthermore, phages have low intrinsic toxicity, and phage therapy is characterised by minimal disruption of normal flora and the lack of cross-resistance with antibiotics.
The practical use of phages for phage therapy requires an understanding of the structural bases of interactions of the host receptor and phage receptor-binding proteins (RBPs); the latter can include tail fibre and tail spike proteins (TFP and TSP). In addition, phage RBPs, as well as endolysins and ectolysins, the proteins that cause cell lysis, can be used as antibacterial agents by themselves [45,102]. The analysis of the structural features of phage RBPs and lysins can use modern deep-learning techniques, including AlphaFold. Together with experimental studies, AlphaFold predictions can be used to elucidate the domain organisation of TFP, TSP and cell-wall degrading enzymes, to reveal the sites of phage particle binding and enzymatic domains (Figure 4) [45,46,47,52].
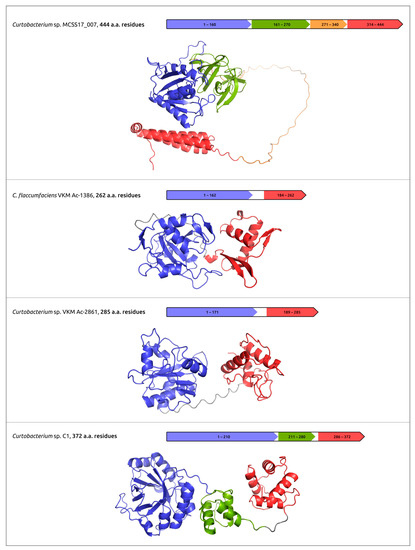
Figure 4.
Predicted domain architecture and AlphaFold models of putative endolysins encoded in the prophage-derived regions. Reprinted/adapted with permission from Ref. [47]. © 2023 by the authors.
As well as in the case of eukaryotic viruses mentioned above, AlphaFold predictions can contribute to building the model of the viral particle [48,103] or the virion parts, including the attachment apparatus [46,50] and phage egress machinery [51]. All the steps of phage infection are accompanied by macromolecular interactions that include proteins, so AlphaFold’s highly accurate structural predictions can assist in the elucidation of the mechanisms of the formation of the phage nucleus [49], lysogeny maintenance [53] or anti-phage defence [44,54]. AlphaFold can also be useful in the trivial but relevant task of phage genome annotation, assisting the prediction of genes’ functionality. As of January 2023, 19,499 GenBank sequences, assigned to class Caudoviricetes, contained 1,731,815 coding regions, 67% of which were annotated as hypothetical proteins. In some cases, BLAST search and HMM-HMM motif comparisons fail to assign a function to proteins encoded in phage genomes, but analysis of fold of AF2-derived structures can assist to clarify this function [55].
It seems that no large-scale studies have been published on the accuracy of modelling using AF2 compared with the predictions of other algorithms. However, comparing the predicted average local distance difference test (lDDT) score of the 54 AF2-derived models of the major capsid protein and ATPase subunit of phage terminase indicated an impressive level of accuracy of the predictions [55]. Interestingly, structural predictions of more conserved terminase were more accurate than those of major capsid protein, (terminase lDDT mean: 0.988, median: 0.996; major capsid protein lDDT mean: 0.907, median: 0.929). The average lDDT of the ATPase domains extracted from the ATPase subunit of phage terminase models was even higher (mean: 0.998, median: 0.999). An evaluation of models of the same major capsid proteins, carried out using a different deep-learning algorithm, RoseTTAFold, showed a lower accuracy of prediction (lDDT mean: 0.634, median: 0.649) than with the AlphaFold models (Figure 5).
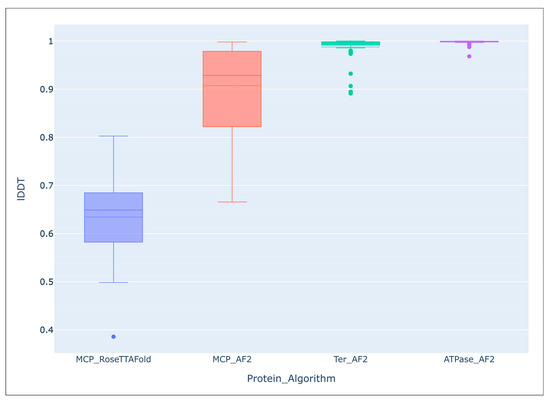
Figure 5.
Comparison of the overall accuracy of predictions made with the Local Distance Difference Test (lDDT), using the DeepAccNet accuracy predictor. MCP_RoseTTAFlold–RoseTTAFlold models of the MCP, MCP_AF2–AlphaFold models of the MCP, Ter_AF2–terminase ATPase subunits’ models predicted with AlphaFold, ATPase_AF2–ATPase domain of terminase ATPase subunits’ models predicted with AlphaFold. Reprinted/adapted with permission from Ref. [55]. © 2023 by the authors.
4. Application of AlphaFold for Evolutionary and Taxonomic Studies
Comparing structural similarity and specific structural features can clarify the evolutionary relationships between proteins. Furthermore, the emergence of new high-precision algorithms for predicting the structure of proteins, including AlphaFold, can enable the identification of evolutionary relationships between highly divergent discovered proteins, using the results of structural modelling. The evolution of proteins may be accompanied by the appearance of new domains, and comparative analysis of AF2-derived structures can help reveal patterns of protein evolution. Studies of bacteriophage tail sheath proteins, an important part of phages’ contractile injection system, have enabled the identification of the common core domain, including both N-terminal and C-terminal parts. The remaining variable parts consisting of one or more moderately conserved domains have, presumably, been added during phage evolution (Figure 6) [58].
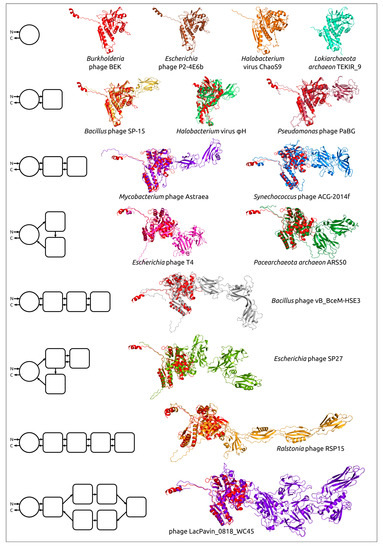
Figure 6.
Examples of the structural architecture of AF2-derived contractile phage sheath proteins [58]. Proteins consisting of two and more domains are superimposed with the modelled structure of the Burkholderia phage BEK tail sheath protein, depicted in the red colour. The schemes on the left show the structural architecture of proteins. The main domain is depicted as a circle, with additional domains represented as squares with rounded corners. The direction of the polypeptide chain, from the N- to the C-termini, is shown with arrows. Reprinted/adapted with permission from Ref. [58]. © 2022 by the authors.
Structural similarity is widely used to evaluate evolutionary relationships between proteins whose amino acid sequence homology level is low or cannot be determined at all [104,105]. The structural similarity between two proteins can be assessed using root-mean-square deviation (RMSD) or other metrics such as template modelling score (TM-score) and DALI Z-score; the latter two metrics have a number of advantages over RMSD [84,106]. Clustering of experimentally determined structures of major capsid proteins using the DALI Z-score has already been used to illustrate the common origin of some viral groups and to cluster prokaryotic viruses [56,104]. Integrated use of both experimental structures and AF2-derived structures can be used for elucidation of evolutionary relationships and taxonomic classification of bacteriophages and eukaryotic viruses [57,59,107]. AlphaFold modelling and subsequent clustering have been used in taxonomic studies of archaeal viruses [56]. Clustering using AlphaFold showed interesting and often biologically meaningful results [55]. Clustering using structures predicted by AlphaFold showed interesting and often biologically meaningful results (Figure 7). It should also be noted that the native state of viral proteins can change according to the state of the viral particle (e.g., empty, full, expanded capsids) and according to the stage of viral particle assembly [108,109,110,111]. The correlation between structural similarity and sequence identity is not absolute due to conformational plasticity, solvent effects and ligand binding [112]. Most of these limitations apply to studies that involve experimentally determined structures, but, hypothetically, they could be exacerbated by structural prediction errors. Therefore, predicting the effectiveness of using AlphaFold for the analysis of structural similarity and evolutionary history, based only on the similarity of the predicted structures, seems to be a difficult task [55].
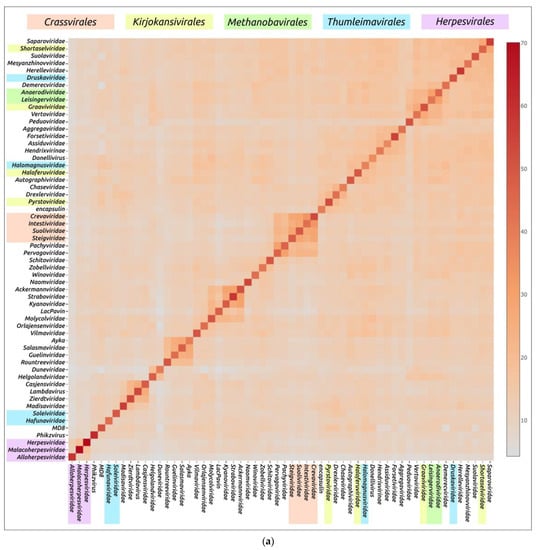
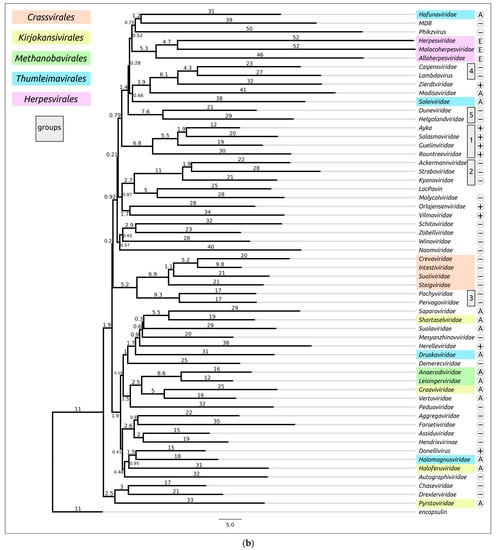
Figure 7.
Heatmap (a) and dendrogram (b) based on the pairwise Z-score comparisons of 57 major capsid proteins and encapsulin AF models, using DALI. The branch lengths are measured using the DALI Z-score, and the tree was rooted to encapsulin. “A”—archaeal viruses, “E”—eukaryotic viruses, “+”—phages infecting Gram-positive bacteria, and “−”—phages infecting Gram-negative bacteria. Groups correspond to clusters found as a result of structural comparison. Reprinted/adapted with permission from Ref. [55]. © 2023 by the authors.
5. Further Development of AlphaFold and Machine Learning Techniques
5.1. AlphaFold-Multimer and Prediction of Multi-Chain Protein Complexes
Originally, AF2 was designed to predict monomeric protein structures. Consequently, interactions between different proteins, subunits and domains in multimers were not described in the AlphaFold database [61]. As a result, some large multi-domain protein complexes may not have been modelled accurately enough. Several publications have, however, explored how AF2 could be used for predicting both homo- and heteromeric complexes [62,63,64]. Moreover, it has been pointed out that an AI system outperforms standard docking methods in as much as it does not require starting protein structures [62].
In addition, a number of approaches have been developed to make AlphaFold work well for complicated protein structures with multiple bindings. Recent versions of AF2, such as those incorporated into ColabFold, enable multimer structures to be uploaded [63]. They include AlphaFold-Multimer, the extension developed by the DeepMind team, which significantly improves the accuracy of predicting multimeric interactions [19]. This new instrument is an AlphaFold algorithm that is specially modified to use multimeric data and trained on oligomeric proteins. However, there is evidence that this multimeric modification has not succeeded in predicting the key features of some protein complexes [65]. Currently, AlphaFold-Multimer does not include the self-distillation of multimer predictions, so the authors believe there is potential for future accuracy enhancements.
To overcome the limitation described above, combining AF2 with experimental methods, e.g., cryo-electron tomography and/or other computer-based tools such as RoseTTAFold, provides more robust results [64,66]. Other authors have suggested combining AlphaFold models of protein complexes with differential covalent labelling mass spectrometry data by applying RosettaDock [67]. The use of cryo-electron microscopy maps, integrated with AlphaFold, for multi-chain protein complex prediction also encourages the creation of accurate and reliable models [68].
Other approaches include the use of optimised multiple sequence alignment together with AF2 [69] and the application of a Monte Carlo tree search [70]. The latter works well but only with symmetric protein complexes and when the stoichiometry of the subcomponents is known.
5.2. AlphaFill
A study from Massachusetts Institute of Technology, which mainly focused on the limitations of AF2 in the drug industry [74], showed that the use of AF2 together with molecular docking simulations to predict protein-ligand bindings demonstrated poor performance that, in some cases, was comparable to pure chance. At the same time, this study indicated how prediction accuracy might be improved with the integration of machine-learning-based approaches. The authors of the study expected their research to encourage the development of machine-learning methods that would complement AlphaFold.
AlphaFill is a new tool that has been developed to solve the problem with ligands and cofactors in the AlphaFold protein structure database [75]. AlphaFill uses an algorithm that employs sequence and structure similarity analysis to graft missing molecules and ions from experimental data into predicted protein structures. The algorithm has been successfully validated against experimental structures.
6. Critique of AlphaFold
AlphaFold has probably revolutionised the determination of protein molecular structure. Today, AF2 is a state-of-the-art deep-learning tool that demonstrates an accuracy in predicting protein folding that was previously unattainable using computational tools. The quality of its predictions is, however, not consistent. Furthermore, in some cases, Artificial Intelligence (AI) systems are unable to provide highly accurate results. As reported by the EMBL’s European Bioinformatics Institute, 35% of the more than 214 million AF2 predictions have been found to be very accurate [60], which indicates that its predictions are often not inferior to those obtained experimentally. It should also be pointed out that 45% of these predictions still could be used for some applications, in spite of their accuracy being inferior to that of experimentally retrieved structures. Therefore, although AF2 is an outstanding tool, it is important to consider its limitations to ensure that investigations provide reliable results.
6.1. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions
When AlphaFold encounters difficulties with obtaining highly accurate predictions, the problem very often relates to intrinsically disordered proteins (IDPs) or intrinsically disordered protein regions (IDRs) [71]. AI systems perform excellently when predicting well-folded proteins, but about a third of eukaryotic proteins are intrinsically disordered or contain disordered regions [72]. Moreover, IDPs play an important role in physiological functions, such as in protein signalling networks.
The reason for AlphaFold encountering difficulties when predicting IDRs may be that these proteins and regions are often not solved by X-ray crystallography; AF2 is mainly designed to use X-ray data [62]. There is a database, DisProt, that contains consolidated information on IDPs [72]. If AF2 or another AI system could be tailored so that it can extract conformational features from DisProt or some other experiment-based databases, then this might enable prediction of IDPs/IDRs in the future.
6.2. Protein Interactions with Metal Ions, DNA, RNA, Cofactors, Ligands and Post-Translational Modifications
Many proteins can physiologically function only in the form of complexes with various ions and molecules, such as hemoglobin. Such interactions are especially crucial for drug discovery. It is to be expected, therefore, that much of AlphaFold’s criticism is related to the fact that it omits protein-ligand interactions in its predictions [18,73].
AlphaFold is not designed for the prediction of post-translational modifications (PTMs) of proteins, such as protein glycosylation. This fact has attracted the attention of the scientific community, with recent studies demonstrating the relevance and importance of glycosylation in the SARS-CoV-2 spike protein or in human proteins. According to research, between 50% and 70% of the 20,000 predicted human proteins are thought to be glycosylated [113]. Bagdonas et al. suggested that the use of sequence- and structure-based studies might address not only the ligand and cofactor interactions problem but also issues related to PTMs [76]. The authors presented an example of glycosylation to demonstrate the potential of their proposed approach, developing an algorithm integrated into Privateer software. This tool ‘transfers’ protein glycosylation from a library of structurally balanced glycan blocks to the protein folding from AlphaFold.
6.3. Protein Conformations
Proteins are not static; they take on various structures, depending on their surroundings or the stage in the functional cycle. Conformational changes in proteins are closely related to their functions and regulations. They can be caused by binding to other molecules, by PTMs or by changes at the pH and temperature levels, for example. AlphaFold provides a static picture of protein folding and does not incorporate information about its dynamics [77]. There is also no clarity as to which conformation of the protein will be predicted by AlphaFold [61]. Consequently, this AI system offers only partial information about the key features of the relationship between protein structure and function.
The situation is complicated by the fact that data on these conformations obtained under experimental conditions also have limitations. Nevertheless, at the moment, it seems that predictions of conformations and the dynamics of protein structures are only possible using experimental methods, such as time-resolved crystallography and structural distributions from cryo-EM data [78].
6.4. Mutations
According to some studies, it appears that AF2 is unable to predict defects in protein structures caused by mutations [79]. One investigation showed that differences between mutated and wild-type structures predicted by AlphaFold were extremely small [80]. Other researchers have found that it is impossible to obtain a reliable evaluation of the impact of mutation on protein stability with the direct application of AI predictions [81]. Thus, predicting the effect of mutations on protein stability should be carried out as a specific task, although this will be hampered by the limited amount of data available for training deep-learning models.
6.5. Database Loopholes
As a deep neural network, AF2 cannot correctly predict absolutely unknown structures on which it was not trained. It is based on MSA and experimentally obtained structures stored in the database. Similarly, AF2 also lacks predictive accuracy where fewer sequences are available for alignment [65]. Accordingly, the AI’s quality performance will depend on how much experimental and previous computational data have been collected and stored in databases. This is not really a limitation, since it may be considered as an opportunity, given that the more data that are collected, the more accurate predictions will become.
7. Conclusions
Protein structure modelling is an important task that helps fundamental and applied research in the field of virology. The AlphaFold deep-learning algorithm, which has been proven to be a highly accurate prediction method, can be used in the design of new drugs and in studies of viral pathogens and mechanisms of viral infection. In bacteriophage research, AlphaFold predictions can also be used to model receptor-binding proteins and glycopolymer-degrading enzymes, helping to develop new antibacterials and biocontrol agents.
Author Contributions
Conceptualisation, P.E.; methodology, P.E.; formal analysis, D.G. and P.E.; investigation, D.G. and P.E.; writing—original draft preparation, P.E., D.G., K.M. and M.S.; writing—review and editing, K.M., M.S. and P.E.; visualisation, D.G. and P.E.; supervision, P.E.; project administration, K.M. and P.E.; funding acquisition, K.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Russian Science Foundation, grant #21-16-00047.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are grateful to the staff of the Laboratory of Aquatic Microbiology of the Limnological Institute of the Siberian Branch of the Russian Academy of Sciences for valuable consultations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Creighton, T.E. Protein Folding. Biochem. J. 1990, 270, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Marcu, Ş.-B.; Tăbîrcă, S.; Tangney, M. An Overview of Alphafold’s Breakthrough. Front. Artif. Intell. 2022, 5, 875587. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. The Formation and Stabilization of Protein Structure. Biochem. J. 1972, 128, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.S. The Anatomy and Taxonomy of Protein Structure. In Advances in Protein Chemistry; Anfinsen, C.B., Edsall, J.T., Richards, F.M., Eds.; Academic Press: Cambridge, USA, 1981; Volume 34, pp. 167–339. [Google Scholar]
- Rose, G.D.; Fleming, P.J.; Banavar, J.R.; Maritan, A. A Backbone-Based Theory of Protein Folding. Proc. Natl. Acad. Sci. USA 2006, 103, 16623–16633. [Google Scholar] [CrossRef]
- Janin, J.; Bahadur, R.P.; Chakrabarti, P. Protein–Protein Interaction and Quaternary Structure. Q. Rev. Biophys. 2008, 41, 133–180. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational Dynamics of SARS-CoV-2 Trimeric Spike Glycoprotein in Complex with Receptor ACE2 Revealed by Cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef]
- Śledź, P.; Caflisch, A. Protein Structure-Based Drug Design: From Docking to Molecular Dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef]
- Smyth, M.S.; Martin, J.H.J. X Ray Crystallography. Mol. Pathol. 2000, 53, 8. [Google Scholar] [CrossRef]
- Klukowski, P.; Riek, R.; Güntert, P. Rapid Protein Assignments and Structures from Raw NMR Spectra with the Deep Learning Technique ARTINA. Nat. Commun. 2022, 13, 6151. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Chiu, W.; Dai, W.; Flatt, J.W.; Hudson, B.P.; Kaelber, J.T.; Khare, S.D.; Kulczyk, A.W.; Lawson, C.L.; et al. Electron Microscopy Holdings of the Protein Data Bank: The Impact of the Resolution Revolution, New Validation Tools, and Implications for the Future. Biophys. Rev. 2022, 14, 1281–1301. [Google Scholar] [CrossRef]
- Agnihotry, S.; Pathak, R.K.; Singh, D.B.; Tiwari, A.; Hussain, I. Chapter 11—Protein Structure Prediction. In Bioinformatics; Singh, D.B., Pathak, R.K., Eds.; Academic Press: Cambridge, USA, 2022; pp. 177–188. ISBN 978-0-323-89775-4. [Google Scholar]
- Kuhlman, B.; Bradley, P. Advances in Protein Structure Prediction and Design. Nat. Rev. Mol. Cell. Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, S.; Sowdhamini, R.; Cadet, F.; Offmann, B. A Glance into the Evolution of Template-Free Protein Structure Prediction Methodologies. Biochimie 2020, 175, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Bouatta, N.; AlQuraishi, M. Structural Biology at the Scale of Proteomes. Nat. Struct. Mol. Biol. 2023, 30, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- AlQuraishi, M. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef]
- Callaway, E. What’s next for AlphaFold and the AI Protein-Folding Revolution. Nature 2022, 604, 234–238. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein Complex Prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Antonelli, G.; Pistello, M. Virology: A Scientific Discipline Facing New Challenges. Clin. Microbiol. Infect. 2019, 25, 133–135. [Google Scholar] [CrossRef]
- Summers, W.C. The Strange History of Phage Therapy. Bacteriophage 2012, 2, 130–133. [Google Scholar] [CrossRef]
- Miroshnikov, K.A.; Evseev, P.V.; Lukianova, A.A.; Ignatov, A.N. Tailed Lytic Bacteriophages of Soft Rot Pectobacteriaceae. Microorganisms 2021, 9, 1819. [Google Scholar] [CrossRef]
- Brives, C.; Pourraz, J. Phage Therapy as a Potential Solution in the Fight against AMR: Obstacles and Possible Futures. Palgrave Commun. 2020, 6, 100. [Google Scholar] [CrossRef]
- Abdelkader, A.; Elzemrany, A.A.; El-Nadi, M.; Elsabbagh, S.A.; Shehata, M.A.; Eldehna, W.M.; El-Hadidi, M.; Ibrahim, T.M. In-Silico Targeting of SARS-CoV-2 NSP6 for Drug and Natural Products Repurposing. Virology 2022, 573, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Flower, T.G.; Hurley, J.H. Crystallographic Molecular Replacement Using an in Silico-Generated Search Model of SARS-CoV-2 ORF8. Protein Sci. 2021, 30, 728–734. [Google Scholar] [CrossRef]
- Jansen van Vuren, P.; McAuley, A.J.; Kuiper, M.J.; Singanallur, N.B.; Bruce, M.P.; Riddell, S.; Goldie, S.; Mangalaganesh, S.; Chahal, S.; Drew, T.W.; et al. Highly Thermotolerant SARS-CoV-2 Vaccine Elicits Neutralising Antibodies against Delta and Omicron in Mice. Viruses 2022, 14, 800. [Google Scholar] [CrossRef]
- Singanallur, N.B.; van Vuren, P.J.; McAuley, A.J.; Bruce, M.P.; Kuiper, M.J.; Gwini, S.M.; Riddell, S.; Goldie, S.; Drew, T.W.; Blasdell, K.R.; et al. At Least Three Doses of Leading Vaccines Essential for Neutralisation of SARS-CoV-2 Omicron Variant. Front. Immunol. 2022, 13, 883612. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, S.; Jing, T.; Wang, W.; Zhang, E.Y.; Zhang, F.; Yang, Y. In Silico Protein Folding Prediction of COVID-19 Mutations and Variants. Biomolecules 2022, 12, 1665. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.J.; Courtney, J.M.; Shen, Y.; Ying, J.; Bax, A. Concordance of X-Ray and AlphaFold2 Models of SARS-CoV-2 Main Protease with Residual Dipolar Couplings Measured in Solution. J. Am. Chem. Soc. 2021, 143, 19306–19310. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Chakraborty, S.; Ahmad, M.; Kumar, V.; Tailor, P.B.; Biswal, B.K. Identification of Probable Inhibitors for the DNA Polymerase of the Monkeypox Virus through the Virtual Screening Approach. Int. J. Biol. Macromol. 2023, 229, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.R.; Sachdev, S.; Reddy, A.S.; Kandasamy, S.L.; Byrareddy, S.N.; Lorson, C.L.; Singh, K. Mutations in the Monkeypox Virus Replication Complex: Potential Contributing Factors to the 2022 Outbreak. J. Autoimmun. 2022, 133, 102928. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, Y.; Li, K.; Zhang, L. Targeting F13 from Monkeypox Virus and Variola Virus by Tecovirimat: Molecular Simulation Analysis. J. Infect. 2022, 85, e99–e101. [Google Scholar] [CrossRef]
- Yefet, R.; Friedel, N.; Tamir, H.; Polonsky, K.; Mor, M.; Cherry-Mimran, L.; Taleb, E.; Hagin, D.; Sprecher, E.; Israely, T.; et al. Monkeypox Infection Elicits Strong Antibody and B Cell Response against A35R and H3L Antigens. iScience 2023, 26, 105957. [Google Scholar] [CrossRef] [PubMed]
- Benedyk, T.H.; Connor, V.; Caroe, E.R.; Shamin, M.; Svergun, D.I.; Deane, J.E.; Jeffries, C.M.; Crump, C.M.; Graham, S.C. Herpes Simplex Virus 1 Protein PUL21 Alters Ceramide Metabolism by Activating the Interorganelle Transport Protein CERT. J. Biol. Chem. 2022, 298, 102589. [Google Scholar] [CrossRef] [PubMed]
- Collantes, T.M.A.; Clark, C.M.; Musarrat, F.; Jambunathan, N.; Jois, S.; Kousoulas, K.G. Predicted Structure and Functions of the Prototypic Alphaherpesvirus Herpes Simplex Virus Type-1 UL37 Tegument Protein. Viruses 2022, 14, 2189. [Google Scholar] [CrossRef] [PubMed]
- Fieulaine, S.; Tubiana, T.; Bressanelli, S. De Novo Modelling of HEV Replication Polyprotein: Five-Domain Breakdown and Involvement of Flexibility in Functional Regulation. Virology 2023, 578, 128–140. [Google Scholar] [CrossRef]
- Liu, H.; Peck, X.Y.; Choong, Y.K.; Ng, W.S.; Engl, W.; Raghuvamsi, P.V.; Zhao, Z.W.; Anand, G.S.; Zhou, Y.; Sivaraman, J.; et al. Identification of Putative Binding Interface of PI(3,5)P2 Lipid on Rice Black-Streaked Dwarf Virus (RBSDV) P10 Protein. Virology 2022, 570, 81–95. [Google Scholar] [CrossRef]
- Chen, L.; Chen, L.; Chen, H.; Zhang, H.; Dong, P.; Sun, L.; Huang, X.; Lin, P.; Wu, L.; Jing, D.; et al. Structural Insights into the CP312R Protein of the African Swine Fever Virus. Biochem. Biophys. Res. Commun. 2022, 624, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kwak, J.S.; Jung, W.; Kim, M.S.; Kim, K.H. Compensatory Mutations in the Matrix Protein of Viral Hemorrhagic Septicemia Virus (VHSV) Genotype IVa in Response to Artificial Mutation of Two Amino Acids (D62A E181A). Virus Res. 2023, 326, 199067. [Google Scholar] [CrossRef] [PubMed]
- Veit, M.; Gadalla, M.R.; Zhang, M. Using Alphafold2 to Predict the Structure of the Gp5/M Dimer of Porcine Respiratory and Reproductive Syndrome Virus. Int. J. Mol. Sci. 2022, 23, 13209. [Google Scholar] [CrossRef]
- Hötzel, I. Domain Organization of Lentiviral and Betaretroviral Surface Envelope Glycoproteins Modeled with AlphaFold. J. Virol. 2022, 96, e01348-21. [Google Scholar] [CrossRef]
- Weaver, G.C.; Arya, R.; Schneider, C.L.; Hudson, A.W.; Stern, L.J. Structural Models for Roseolovirus U20 And U21: Non-Classical MHC-I Like Proteins From HHV-6A, HHV-6B, and HHV-7. Front. Immunol. 2022, 13, 864898. [Google Scholar] [CrossRef]
- Al-Shayeb, B.; Skopintsev, P.; Soczek, K.M.; Stahl, E.C.; Li, Z.; Groover, E.; Smock, D.; Eggers, A.R.; Pausch, P.; Cress, B.F.; et al. Diverse Virus-Encoded CRISPR-Cas Systems Include Streamlined Genome Editors. Cell 2022, 185, 4574–4586.e16. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, J.; Dunne, M.; Loessner, M.J. A Perfect Fit: Bacteriophage Receptor-Binding Proteins for Diagnostic and Therapeutic Applications. Curr. Opin. Microbiol. 2023, 71, 102240. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Cambillau, C. Structure and Topology Prediction of Phage Adhesion Devices Using AlphaFold2: The Case of Two Oenococcus Oeni Phages. Microorganisms 2021, 9, 2151. [Google Scholar] [CrossRef]
- Evseev, P.; Lukianova, A.; Tarakanov, R.; Tokmakova, A.; Popova, A.; Kulikov, E.; Shneider, M.; Ignatov, A.; Miroshnikov, K. Prophage-Derived Regions in Curtobacterium Genomes: Good Things, Small Packages. Int. J. Mol. Sci. 2023, 24, 1586. [Google Scholar] [CrossRef]
- Hawkins, N.C.; Kizziah, J.L.; Hatoum-Aslan, A.; Dokland, T. Structure and Host Specificity of Staphylococcus Epidermidis Bacteriophage Andhra. Sci. Adv. 2022, 8, eade0459. [Google Scholar] [CrossRef]
- Nieweglowska, E.S.; Brilot, A.F.; Méndez-Moran, M.; Kokontis, C.; Baek, M.; Li, J.; Cheng, Y.; Baker, D.; Bondy-Denomy, J.; Agard, D.A. The ΦPA3 Phage Nucleus Is Enclosed by a Self-Assembling 2D Crystalline Lattice. Nat. Commun. 2023, 14, 927. [Google Scholar] [CrossRef] [PubMed]
- Šiborová, M.; Füzik, T.; Procházková, M.; Nováček, J.; Benešík, M.; Nilsson, A.S.; Plevka, P. Tail Proteins of Phage SU10 Reorganize into the Nozzle for Genome Delivery. Nat. Commun. 2022, 13, 5622. [Google Scholar] [CrossRef]
- Conners, R.; McLaren, M.; Łapińska, U.; Sanders, K.; Stone, M.R.L.; Blaskovich, M.A.T.; Pagliara, S.; Daum, B.; Rakonjac, J.; Gold, V.A.M. CryoEM Structure of the Outer Membrane Secretin Channel PIV from the F1 Filamentous Bacteriophage. Nat. Commun. 2021, 12, 6316. [Google Scholar] [CrossRef]
- Eskenazi, A.; Lood, C.; Wubbolts, J.; Hites, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Kvachadze, L.; van Noort, V.; Wagemans, J.; et al. Combination of Pre-Adapted Bacteriophage Therapy and Antibiotics for Treatment of Fracture-Related Infection Due to Pandrug-Resistant Klebsiella Pneumoniae. Nat. Commun. 2022, 13, 302. [Google Scholar] [CrossRef]
- McGinnis, R.J.; Brambley, C.A.; Stamey, B.; Green, W.C.; Gragg, K.N.; Cafferty, E.R.; Terwilliger, T.C.; Hammel, M.; Hollis, T.J.; Miller, J.M.; et al. A Monomeric Mycobacteriophage Immunity Repressor Utilizes Two Domains to Recognize an Asymmetric DNA Sequence. Nat. Commun. 2022, 13, 4105. [Google Scholar] [CrossRef]
- Zhang, T.; Tamman, H.; Coppieters ’t Wallant, K.; Kurata, T.; LeRoux, M.; Srikant, S.; Brodiazhenko, T.; Cepauskas, A.; Talavera, A.; Martens, C.; et al. Direct Activation of a Bacterial Innate Immune System by a Viral Capsid Protein. Nature 2022, 612, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.; Gutnik, D.; Shneider, M.; Miroshnikov, K. Use of an Integrated Approach Involving AlphaFold Predictions for the Evolutionary Taxonomy of Duplodnaviria Viruses. Biomolecules 2023, 13, 110. [Google Scholar] [CrossRef]
- Liu, Y.; Demina, T.A.; Roux, S.; Aiewsakun, P.; Kazlauskas, D.; Simmonds, P.; Prangishvili, D.; Oksanen, H.M.; Krupovic, M. Diversity, Taxonomy, and Evolution of Archaeal Viruses of the Class Caudoviricetes. PLOS Biol. 2021, 19, e3001442. [Google Scholar] [CrossRef] [PubMed]
- Podgorski, J.M.; Freeman, K.; Gosselin, S.; Huet, A.; Conway, J.F.; Bird, M.; Grecco, J.; Patel, S.; Jacobs-Sera, D.; Hatfull, G.; et al. A Structural Dendrogram of the Actinobacteriophage Major Capsid Proteins Provides Important Structural Insights into the Evolution of Capsid Stability. Structure 2023, 31, 282–294.e5. [Google Scholar] [CrossRef]
- Evseev, P.; Shneider, M.; Miroshnikov, K. Evolution of Phage Tail Sheath Protein. Viruses 2022, 14, 1148. [Google Scholar] [CrossRef]
- Hötzel, I. Deep-Time Structural Evolution of Retroviral and Filoviral Surface Envelope Proteins. J. Virol. 2022, 96, e00063-22. [Google Scholar] [CrossRef]
- Callaway, E. “The Entire Protein Universe”: AI Predicts Shape of Nearly Every Known Protein. Nature 2022, 608, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Perrakis, A.; Sixma, T.K. AI Revolutions in Biology. EMBO Rep. 2021, 22, e54046. [Google Scholar] [CrossRef]
- Akdel, M.; Pires, D.E.V.; Pardo, E.P.; Jänes, J.; Zalevsky, A.O.; Mészáros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A Structural Biology Community Assessment of AlphaFold2 Applications. Nat. Struct. Mol. Biol. 2022, 29, 1056–1067. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, I.; Pei, J.; Baek, M.; Krishnakumar, A.; Anishchenko, I.; Ovchinnikov, S.; Zhang, J.; Ness, T.J.; Banjade, S.; Bagde, S.R.; et al. Computed Structures of Core Eukaryotic Protein Complexes. Science 2021, 374, eabm4805. [Google Scholar] [CrossRef]
- Gomes, P.S.F.C.; Gomes, D.E.B.; Bernardi, R.C. Protein Structure Prediction in the Era of AI: Challenges and Limitations When Applying to in Silico Force Spectroscopy. Front. Bioinform. 2022, 2. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Kleywegt, G. A Paradigm Shift in Structural Biology. Nat. Methods 2022, 19, 20–23. [Google Scholar] [CrossRef]
- Drake, Z.C.; Seffernick, J.T.; Lindert, S. Protein Complex Prediction Using Rosetta, AlphaFold, and Mass Spectrometry Covalent Labeling. Nat. Commun. 2022, 13, 7846. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Lin, P.; Chen, J.; Cao, H.; Huang, S.Y. Model Building of Protein Complexes from Intermediate-Resolution Cryo-EM Maps with Deep Learning-Guided Automatic Assembly. Nat. Commun. 2022, 13, 4066. [Google Scholar] [CrossRef]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved Prediction of Protein-Protein Interactions Using AlphaFold2. Nat. Commun. 2022, 13, 1265. [Google Scholar] [CrossRef] [PubMed]
- Bryant, P.; Pozzati, G.; Zhu, W.; Shenoy, A.; Kundrotas, P.; Elofsson, A. Predicting the Structure of Large Protein Complexes Using AlphaFold and Monte Carlo Tree Search. Nat. Commun. 2022, 13, 6028. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef]
- Laurents, D.V. AlphaFold 2 and NMR Spectroscopy: Partners to Understand Protein Structure, Dynamics and Function. Front. Mol. Biosci. 2022, 9, 906437. [Google Scholar] [CrossRef] [PubMed]
- Edich, M.; Briggs, D.C.; Kippes, O.; Gao, Y.; Thorn, A. The Impact of AlphaFold on Experimental Structure Solution. Faraday Discuss. 2022, 240, 184–195. [Google Scholar] [CrossRef]
- Wong, F.; Krishnan, A.; Zheng, E.J.; Stärk, H.; Manson, A.L.; Earl, A.M.; Jaakkola, T.; Collins, J.J. Benchmarking AlphaFold -enabled Molecular Docking Predictions for Antibiotic Discovery. Mol. Syst. Biol. 2022, 18, e11081. [Google Scholar] [CrossRef]
- Hekkelman, M.L.; de Vries, I.; Joosten, R.P.; Perrakis, A. AlphaFill: Enriching AlphaFold Models with Ligands and Cofactors. Nat. Methods 2022, 20, 205–213. [Google Scholar] [CrossRef]
- Bagdonas, H.; Fogarty, C.; Fadda, E.; Agirre, J. The Case for Post-Predictional Modifications in the AlphaFold Protein Structure Database. Nat. Struct. Mol. Biol. 2021, 28, 869–870. [Google Scholar] [CrossRef] [PubMed]
- Van Breugel, M.; Rosa e Silva, I.; Andreeva, A. Structural Validation and Assessment of AlphaFold2 Predictions for Centrosomal and Centriolar Proteins and Their Complexes. Commun. Biol. 2022, 5, 312. [Google Scholar] [CrossRef]
- Lane, T.J. Protein Structure Prediction Has Reached the Single-Structure Frontier. Nat. Methods 2023, 20, 170–173. [Google Scholar] [CrossRef]
- Bertoline, L.M.F.; Lima, A.N.; Krieger, J.E.; Teixeira, S.K. Before and after AlphaFold2: An Overview of Protein Structure Prediction. Front. Bioinform. 2023, 3, 1120370. [Google Scholar] [CrossRef] [PubMed]
- Buel, G.; Walters, K. Can AlphaFold2 Predict the Impact of Missense Mutations on Structure? Nat. Struct. Mol. Biol. 2022, 29, 1–2. [Google Scholar] [CrossRef]
- Pak, M.A.; Markhieva, K.A.; Novikova, M.S.; Petrov, D.S.; Vorobyev, I.S.; Maksimova, E.S.; Kondrashov, F.A.; Ivankov, D.N. Using AlphaFold to Predict the Impact of Single Mutations on Protein Stability and Function. PLOS ONE 2023, 18, e0282689. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Král, P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yang, R.; Wang, W.; Jiang, S.; Yang, C.; Liu, N.; Dai, H.; Lee, I.; Meng, X.; Yuan, Z. Probing the Formation, Structure and Free Energy Relationships of M Protein Dimers of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 573–582. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. High-accuracy protein structures by combining machine-learning with physics-based refinement. Proteins 2020, 88, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Hiranuma, N.; Park, H.; Baek, M.; Anishchenko, I.; Dauparas, J.; Baker, D. Improved Protein Structure Refinement Guided by Deep Learning Based Accuracy Estimation. Nat. Commun. 2021, 12, 1340. [Google Scholar] [CrossRef]
- Li, Z.; Hirst, J.D. Computed Optical Spectra of SARS-CoV-2 Proteins. Chem. Phys. Lett. 2020, 758, 137935. [Google Scholar] [CrossRef]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The TrRosetta Server for Fast and Accurate Protein Structure Prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef]
- Rizk, J.G.; Lippi, G.; Henry, B.M.; Forthal, D.N.; Rizk, Y. Prevention and Treatment of Monkeypox. Drugs 2022, 82, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Delaune, D.; Iseni, F. Drug Development against Smallpox: Present and Future. Antimicrob. Agents Chemother. 2020, 64, e01683-19. [Google Scholar] [CrossRef]
- Peng, Q.; Xie, Y.; Kuai, L.; Wang, H.; Qi, J.; Gao, G.F.; Shi, Y. Structure of Monkeypox Virus DNA Polymerase Holoenzyme. Science 2023, 379, 100–105. [Google Scholar] [CrossRef]
- Sehrawat, S.; Kumar, D.; Rouse, B.T. Herpesviruses: Harmonious Pathogens but Relevant Cofactors in Other Diseases? Front. Cell. Infect. Microbiol. 2018, 8, 177. [Google Scholar] [CrossRef] [PubMed]
- Current ICTV Taxonomy Release | ICTV. Available online: https://ictv.global/taxonomy (accessed on 9 November 2022).
- Nahas, K.L.; Connor, V.; Scherer, K.M.; Kaminski, C.F.; Harkiolaki, M.; Crump, C.M.; Graham, S.C. Near-Native State Imaging by Cryo-Soft-X-Ray Tomography Reveals Remodelling of Multiple Cellular Organelles during HSV-1 Infection. PLOS Pathog. 2022, 18, e1010629. [Google Scholar] [CrossRef]
- Bigalke, J.M.; Heldwein, E.E. Nuclear Exodus: Herpesviruses Lead the Way. Annu. Rev. Virol. 2016, 3, 387–409. [Google Scholar] [CrossRef]
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in Aquatic Ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus Statement: Virus Taxonomy in the Age of Metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W.; Smith, M.C.M.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary Relationships among Diverse Bacteriophages and Prophages: All the World’s a Phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S.T. Pros and Cons of Phage Therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef]
- Fischetti, V.A. Bacteriophage Endolysins: A Novel Anti-Infective to Control Gram-Positive Pathogens. Int J. Med. Microbiol. 2010, 300, 357–362. [Google Scholar] [CrossRef]
- Ouyang, R.; Costa, A.R.; Cassidy, C.K.; Otwinowska, A.; Williams, V.C.J.; Latka, A.; Stansfeld, P.J.; Drulis-Kawa, Z.; Briers, Y.; Pelt, D.M.; et al. High-Resolution Reconstruction of a Jumbo-Bacteriophage Infecting Capsulated Bacteria Using Hyperbranched Tail Fibers. Nat. Commun. 2022, 13, 7241. [Google Scholar] [CrossRef]
- Krupovic, M.; Koonin, E.V. Multiple Origins of Viral Capsid Proteins from Cellular Ancestors. Proc. Natl. Acad. Sci. USA 2017, 114, E2401–E2410. [Google Scholar] [CrossRef]
- Salemme, F.R.; Miller, M.D.; Jordan, S.R. Structural Convergence during Protein Evolution. Proc. Natl. Acad. Sci. USA 1977, 74, 2820–2824. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Using Dali for Protein Structure Comparison. Methods Mol. Biol. 2020, 2112, 29–42. [Google Scholar] [CrossRef]
- Bisio, H.; Legendre, M.; Giry, C.; Philippe, N.; Alempic, J.-M.; Jeudy, S.; Abergel, C. Evolution of Giant Pandoravirus Revealed by CRISPR/Cas9. Nat. Commun. 2023, 14, 428. [Google Scholar] [CrossRef] [PubMed]
- Fokine, A.; Leiman, P.G.; Shneider, M.M.; Ahvazi, B.; Boeshans, K.M.; Steven, A.C.; Black, L.W.; Mesyanzhinov, V.V.; Rossmann, M.G. Structural and Functional Similarities between the Capsid Proteins of Bacteriophages T4 and HK97 Point to a Common Ancestry. Proc. Natl. Acad. Sci. USA 2005, 102, 7163–7168. [Google Scholar] [CrossRef]
- Fang, Q.; Tang, W.-C.; Fokine, A.; Mahalingam, M.; Shao, Q.; Rossmann, M.G.; Rao, V.B. Structures of a Large Prolate Virus Capsid in Unexpanded and Expanded States Generate Insights into the Icosahedral Virus Assembly. Proc. Natl. Acad. Sci. USA 2022, 119, e2203272119. [Google Scholar] [CrossRef]
- Steven, A.C.; Greenstone, H.L.; Booy, F.P.; Black, L.W.; Ross, P.D. Conformational Changes of a Viral Capsid Protein. Thermodynamic Rationale for Proteolytic Regulation of Bacteriophage T4 Capsid Expansion, Co-Operativity, and Super-Stabilization by Soc Binding. J. Mol. Biol. 1992, 228, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Bowman, B.R.; Baker, M.L.; Rixon, F.J.; Chiu, W.; Quiocho, F.A. Structure of the Herpesvirus Major Capsid Protein. EMBO J. 2003, 22, 757–765. [Google Scholar] [CrossRef]
- Hark Gan, H.; Perlow, R.A.; Roy, S.; Ko, J.; Wu, M.; Huang, J.; Yan, S.; Nicoletta, A.; Vafai, J.; Sun, D.; et al. Analysis of Protein Sequence/Structure Similarity Relationships. Biophys. J. 2002, 83, 2781–2791. [Google Scholar] [CrossRef]
- An, H.; Froehlich, J.; Lebrilla, C. Determination of Glycosylation Sites and Site-Specific Heterogeneity in Glycoproteins. Curr. Opin. Chem. Biol. 2009, 13, 421–426. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).