

Impact of HepG2 Cells Glutathione Depletion on Neutral Sphingomyelinases mRNA Levels and Activity



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Cultures
2.3. BSO Treatment and Total Glutathione Detection Assay
2.4. Reactive Oxygen Species Assay
2.5. Isolation of Total RNA, Quantitative Reverse Transcription, and Real-Time PCR
2.6. Preparing Cell Lysates and Determination of Protein Content
2.7. Amplex Red Sphingomyelinase Assay
2.8. Ceramide Detection Assay
2.9. Cell Proliferation
2.10. Statistical Analysis
3. Results
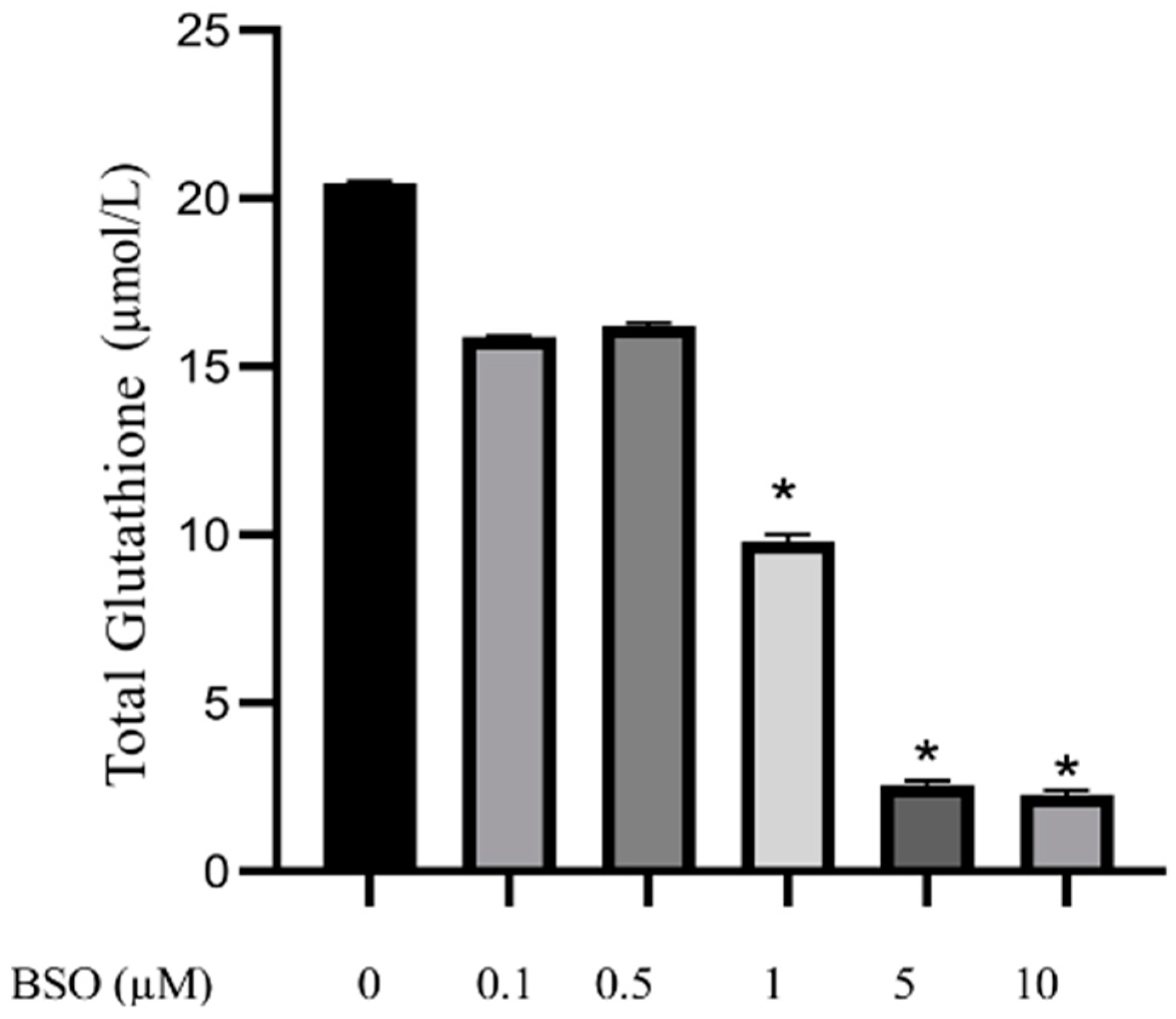
3.1. Effect of Different Concentrations of BSO on Total Glutathione Content in HepG2
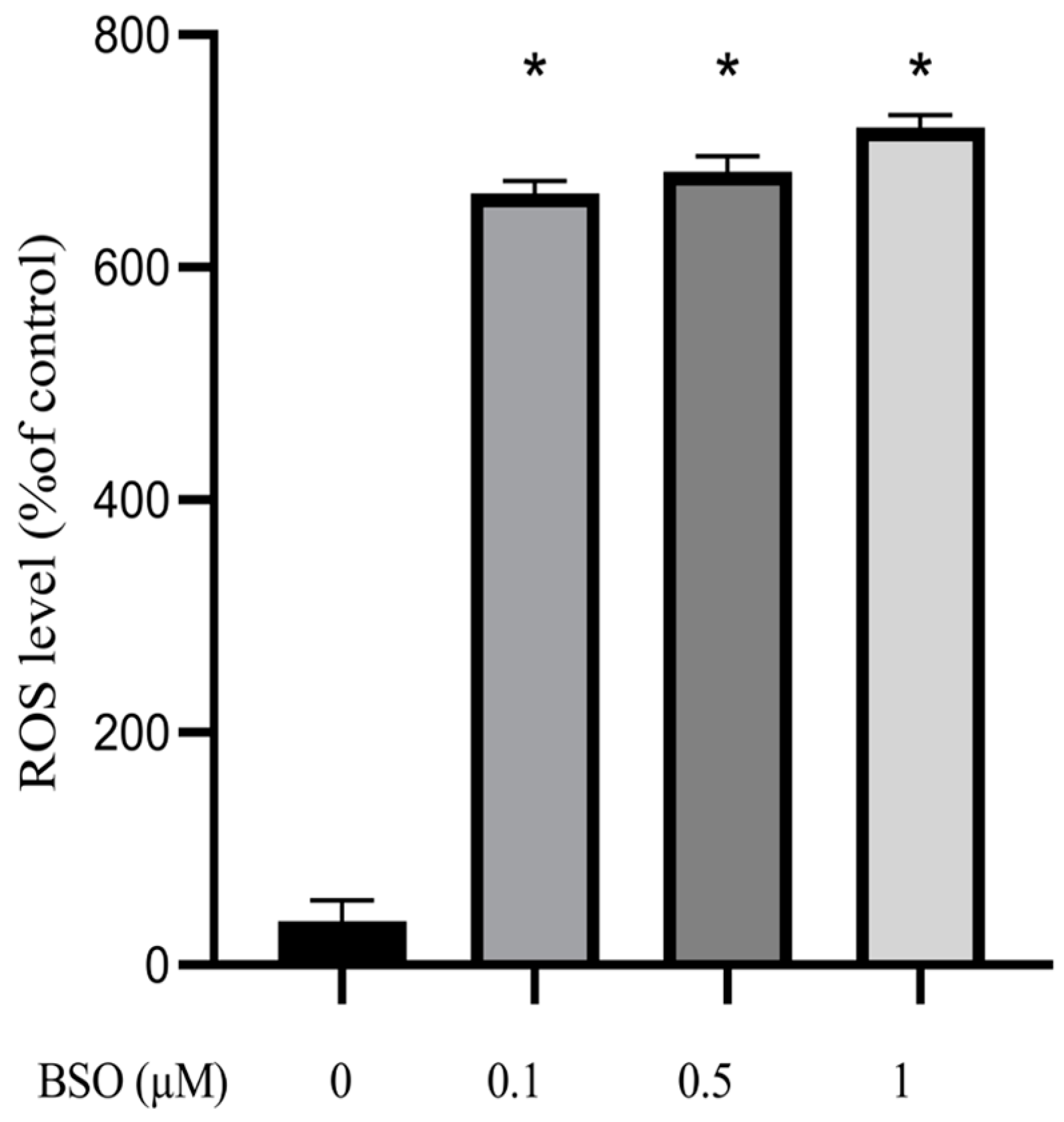
3.2. Effect of BSO on ROS Production in HepG2
3.3. Effect of BSO on nSMases mRNA Levels in HepG2
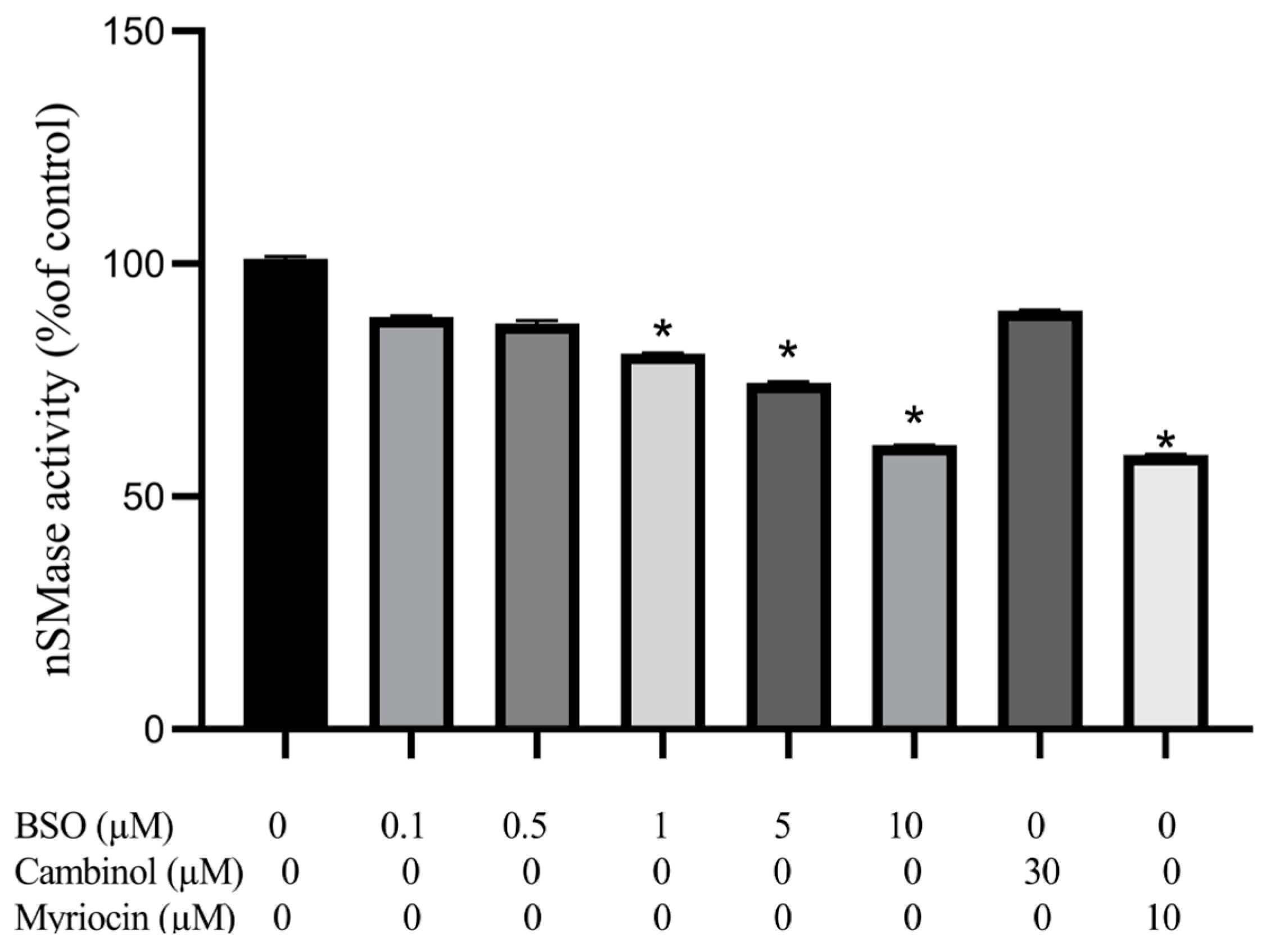
3.4. Effect of BSO, Cambinol, and Myriocin on nSMase Activity
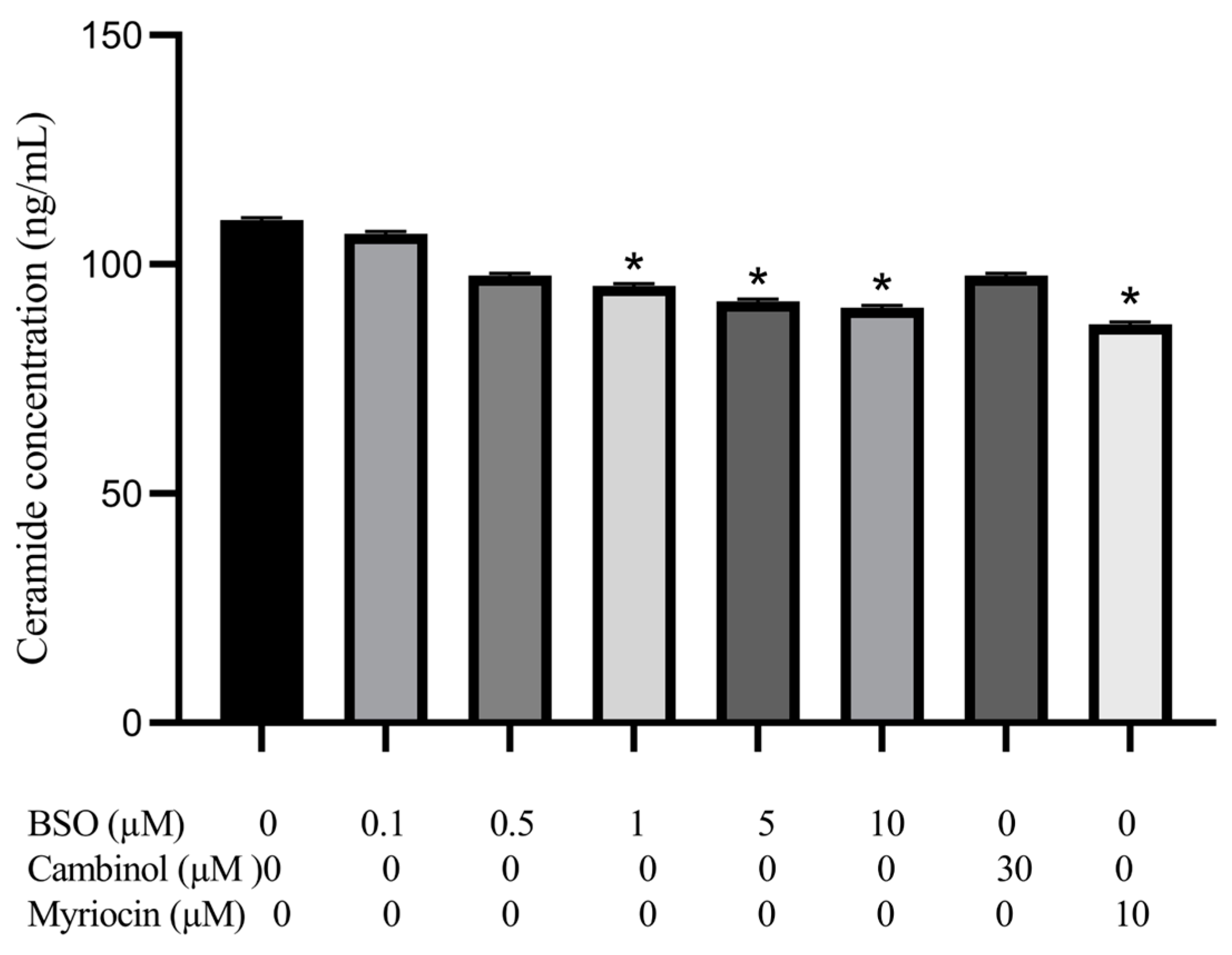
3.5. Effect of BSO, Cambinol and Myriocin on HepG2 Ceramide
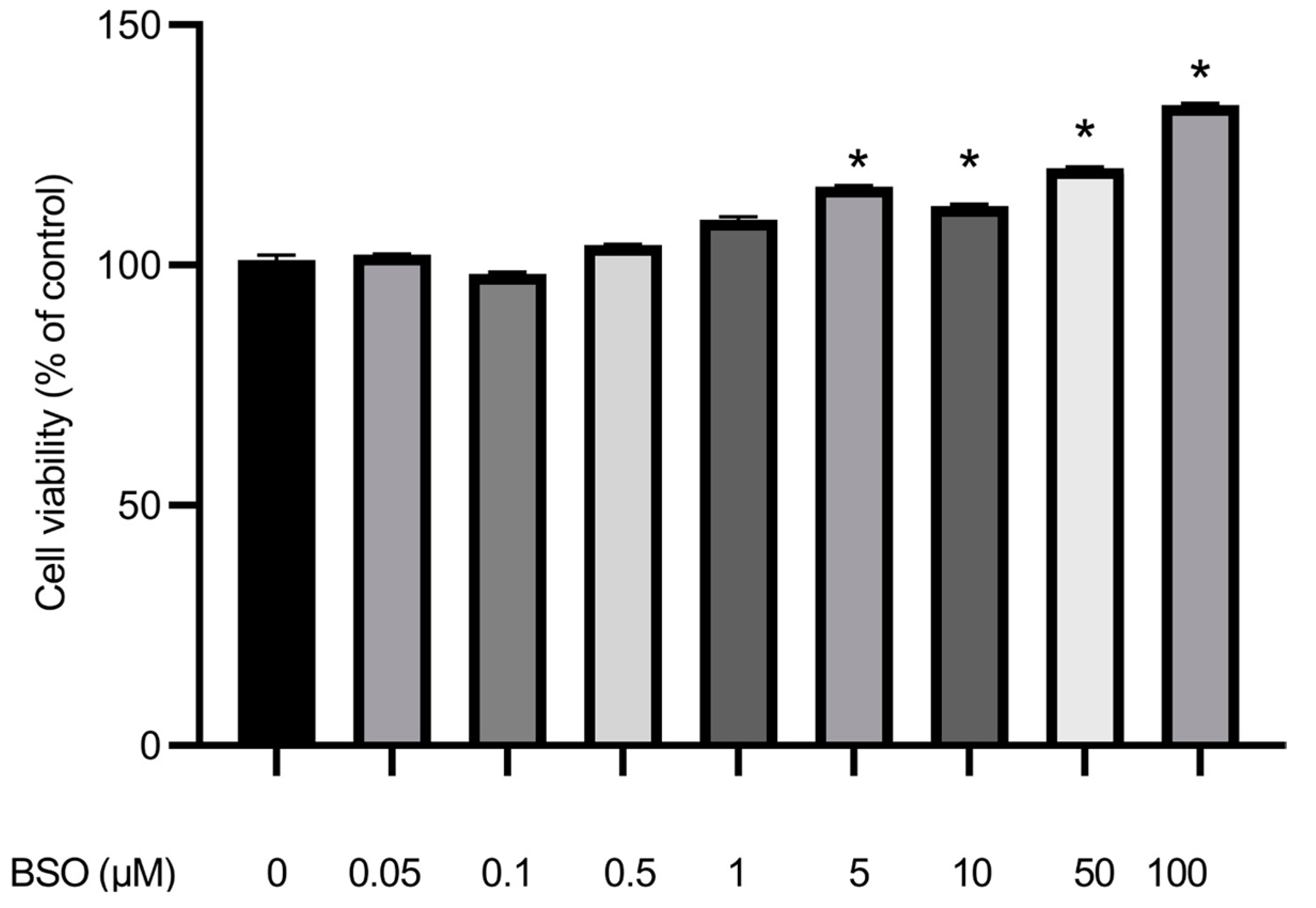
3.6. Effect of BSO on HepG2 Cell Proliferation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Rashed, W.M.; Kandeil, M.A.M.; Mahmoud, M.O.; Ezzat, S. Hepatocellular carcinoma (HCC) in Egypt: A comprehensive overview. J. Egypt. Natl. Canc. Inst. 2020, 32, 5. [Google Scholar] [CrossRef]
- Kulik, L.; El-Serag, H.B. Epidemiology and management of hepatocellular carcinoma. Gastroenterology 2019, 156, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Yegin, E.G.; Oymaci, E.; Karatay, E.; Coker, A. Progress in surgical and nonsurgical approaches for hepatocellular carcinoma treatment. Hepatobiliary Pancreat. Dis. Int. 2016, 15, 234–256. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yin, T.; Xu, Y.; Lu, X.J. Therapeutics for advanced hepatocellular carcinoma: Recent ad vances, current dilemma, and future directions. J. Cell. Physiol. 2019, 234, 12122–12132. [Google Scholar] [CrossRef]
- Meringer, H.; Shibolet, O.; Deutsch, L. Hepatocellular carcinoma in the post-hepatitis C virus era: Should we change the paradigm? World J. Gastroenterol. 2019, 25, 3929–3940. [Google Scholar] [CrossRef] [PubMed]
- Tallima, H.; Azzazy, H.M.E.; El Ridi, R. Cell surface sphingomyelin: Key role in cancer initiation, progression, and immune evasion. Lipids Health Dis. 2021, 20, 150. [Google Scholar] [CrossRef]
- Li, Z.; Guan, M.; Lin, Y.; Cui, X.; Zhang, Y.; Zhao, Z.; Zhu, J. Aberrant lipid metabolism in hepatocellular carcinoma revealed by liver lipidomics. Int. J. Mol. Sci. 2017, 18, 2550. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Huang, C.; Li, N.; Zou, L.; Chia, S.E.; Chen, S.; Yu, K.; Ling, Q.; Cheng, Q.; et al. Comparison of hepatic and serum lipid signatures in hepatocellular carcinoma patients leads to the discovery of diagnostic and prognostic biomarkers. Oncotarget 2017, 9, 5032–5043. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhou, L.; Zuo, H.; Le, W.; Hu, J.; Zhang, T.; Li, M.; Yuan, Y. Overriding sorafenib resistance via blocking lipid metabolism and Ras by sphingomyelin synthase 1 inhibition in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2021, 87, 217–228. [Google Scholar] [CrossRef]
- Revill, K.; Wang, T.; Lachenmayer, A.; Kojima, K.; Harrington, A.; Li, J.; Hoshida, Y.; Llovet, J.M.; Powers, S. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology 2013, 145, 1424–1435.e25. [Google Scholar] [CrossRef]
- Karakashian, A.A.; Giltiay, N.V.; Smith, G.M.; Nikolova-Karakashian, M.N. Expression of neutral sphingomyelinase-2 (NSMase-2) in primary rat hepatocytes modulates IL-beta-induced JNK activation. FASEB J. 2004, 18, 968–970. [Google Scholar] [CrossRef]
- Zhong, L.; Kong, J.N.; Dinkins, M.B.; Leanhart, S.; Zhu, Z.; Spassieva, S.D.; Qin, H.; Lin, H.P.; Elsherbini, A.; Wang, R.; et al. Increased liver tumor formation in neutral sphingomyelinase-2-deficient mice. J. Lipid Res. 2018, 59, 795–804. [Google Scholar] [CrossRef]
- Wu, B.X.; Clarke, C.J.; Hannun, Y.A. Mammalian neutral sphingomyelinases: Regulation and roles in cell signaling responses. Neuromolecular Med. 2010, 12, 320–330. [Google Scholar] [CrossRef]
- Airola, M.V.; Hannun, Y.A. Sphingolipid metabolism and neutral sphingomyelinases. Handb. Exp. Pharmacol. 2013, 215, 57–76. [Google Scholar]
- Insausti-Urkia, N.; Solsona-Vilarrasa, E.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Sphingomyelinases and liver diseases. Biomolecules 2020, 10, 1497. [Google Scholar] [CrossRef]
- Lin, M.; Liao, W.; Dong, M.; Zhu, R.; Xiao, J.; Sun, T.; Chen, Z.; Wu, B.; Jin, J. Exosomal neutral sphingomyelinase 1 suppresses hepatocellular carcinoma via decreasing the ratio of sphingomyelin/ceramide. FEBS J. 2018, 285, 3835–3848. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Yang, X.B.; Sang, X.T. Construction of a lipid metabolism-related and immune-associated prognostic signature for hepatocellular carcinoma. Cancer Med. 2020, 9, 7646–7662. [Google Scholar] [CrossRef] [PubMed]
- Fensome, A.C.; Rodrigues-Lima, F.; Josephs, M.; Paterson, H.F.; Katan, M. A neutral magnesium-dependent sphingomyelinase isoform associated with intracellular membranes and reversibly inhibited by reactive oxygen species. J. Biol. Chem. 2000, 275, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Josephs, M.; Katan, M.; Rodrigues-Lima, F. Irreversible inactivation of magnesium-dependent neutral sphingomyelinase 1 (NSM1) by peroxynitrite, a nitric oxide-derived oxidant. FEBS Lett. 2002, 531, 329–334. [Google Scholar] [CrossRef]
- Rutkute, K.; Asmis, R.H.; Nikolova-Karakashian, M.N. Regulation of neutral sphingomyelinase-2 by GSH: A new insight to the role of oxidative stress in aging-associated inflammation. J. Lipid Res. 2007, 48, 2443–2452. [Google Scholar] [CrossRef]
- Deevska, G.; Sunkara, M.; Karakashian, C.; Peppers, B.; Morris, A.J.; Nikolova-Karakashian, M.N. Effect of procysteine on aging-associated changes in hepatic GSH and SMase: Evidence for transcriptional regulation of smpd3. J. Lipid Res. 2014, 55, 2041–2052. [Google Scholar] [CrossRef]
- Liu, B.; Hannun, Y.A. Inhibition of the neutral magnesium-dependent sphingomyelinase by glutathione. J. Biol. Chem. 1997, 272, 16281–16287. [Google Scholar] [CrossRef]
- Levy, M.; Castillo, S.S.; Goldkorn, T. nSMase2 activation and trafficking are modulated by oxidative stress to induce apoptosis. Biochem. Biophys. Res. Commun. 2006, 344, 900–905. [Google Scholar] [CrossRef]
- Dotson, P.P., 2nd; Karakashian, A.A.; Nikolova-Karakashian, M.N. Neutral sphingomyelinase-2 is a redox sensitive enzyme: Role of catalytic cysteine residues in regulation of enzymatic activity through changes in oligomeric state. Biochem. J. 2015, 465, 371–382. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxid. Med. Cell. Longev. 2019, 2019, 3150145. [Google Scholar] [CrossRef]
- Kennedy, L.; Sandhu, J.K.; Harper, M.E.; Cuperlovic-Culf, M. Role of glutathione in cancer: From mechanisms to therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef]
- Liebmann, J.E.; Hahn, S.M.; Cook, J.A.; Lipschultz, C.; Mitchell, J.B.; Kaufman, D.C. Glutathione depletion by L-buthionine sulfoximine antagonizes taxol cytotoxicity. Cancer Res. 1993, 53, 2066–2070. [Google Scholar]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2′,7′-Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: Forty years of application and controversy. Free Radic. Res. 2010, 44, 587–604. [Google Scholar] [CrossRef]
- Rajneesh Pathak, J.; Chatterjee, A.; Singh, S.P.; Sinha, R.P. Detection of reactive oxygen species (ROS) in Cyanobacteria Using the oxidant-sensing probe 2’,7’-dichlorodihydrofluo-rescein diacetate (DCFH-DA). Bio Protoc. 2017, 7, e2545. [Google Scholar]
- Han, Y.H.; Kee, J.Y.; Kim, D.S.; Mun, J.G.; Jeong, M.Y.; Park, S.H.; Choi, B.M.; Park, S.J.; Kim, H.J.; Um, J.Y.; et al. Arctigenin inhibits lung metastasis of colorectal cancer by regulating cell viability and metastatic phenotypes. Molecules 2016, 21, 1135. [Google Scholar] [CrossRef]
- Figuera-Losada, M.; Stathis, M.; Dorskind, M.; Thomas, A.; RatnamBandaru, V.; Yoo, S.; Westwood, N.; Rogers, G.; McArthur, J.; Haughey, N.; et al. Cambinol, a novel inhibitor of neutral sphingomyelinase 2 shows neuroprotective properties. PLoS ONE 2015, 10, e0124481. [Google Scholar] [CrossRef]
- Park, T.S.; Panek, R.L.; Mueller, S.B.; Hanselman, J.C.; Rosebury, W.S.; Robertson, A.W.; Kindt, E.K.; Homan, R.; Karathanasis, S.K.; Rekhter, M.D. Inhibition of sphingomyelin synthesis reduces atherogenesis in apolipoprotein E-knockout mice. Circulation 2004, 110, 3465–3471. [Google Scholar] [CrossRef]
- Kruger, N.J. The Bradford method for protein quantitation. Methods Mol. Biol. 1994, 32, 9–15. [Google Scholar]
- Tallima, H.; Al-Halbosiy, M.F.; El Ridi, R. Enzymatic activity and immunolocalization of Schistosoma mansoni and Schistosoma haematobium neutral sphingomyelinase. Mol. Biochem. Parasitol. 2011, 178, 23–28. [Google Scholar]
- İzgördü, H.; Vejselova Sezer, C.; Çömlekçi, E.; Kutlu, H.M. Characteristics of apoptosis induction in human breast cancer cells treated with a ceramidase inhibitor. Cytotechnology 2020, 72, 907–919. [Google Scholar] [CrossRef]
- İzgördü, H.; Sezer, C.V.; Bayçelebi, K.; Baloğlu, M.; Kutlu, H.M. Cytotoxic impact of n-oleoylethanolamine on bone cancer cells. Anticancer Agents Med. Chem. 2022, 22, 1119. [Google Scholar] [CrossRef]
- Griffith, O.W. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J. Biol. Chem. 1982, 257, 13704–137012. [Google Scholar] [CrossRef]
- Donohue, T.M., Jr.; Curry-McCoy, T.V.; Todero, S.L.; White, R.L.; Kharbanda, K.K.; Nanji, A.A.; Osna, N.A. L-Buthionine (S,R) sulfoximine depletes hepatic glutathione but protects against ethanol-induced liver injury. Alcohol. Clin. Exp. Res. 2007, 31, 1053–1060. [Google Scholar] [CrossRef]
- Skácel, J.; Slusher, B.S.; Tsukamoto, T. Small molecule inhibitors targeting biosynthesis of ceramide, the central hub of the sphingolipid network. J. Med. Chem. 2021, 64, 279–297. [Google Scholar] [CrossRef]
- Krautbauer, S.; Meier, E.M.; Rein-Fischboeck, L.; Pohl, R.; Weiss, T.S.; Sigruener, A.; Aslanidis, C.; Liebisch, G.; Buechler, C. Ceramide and polyunsaturated phospholipids are strongly reduced in human hepatocellular carcinoma. Biochim. Biophys. Acta 2016, 1861, 1767–1774. [Google Scholar] [CrossRef]
- Tomiuk, S.; Hofmann, K.; Nix, M.; Zumbansen, M.; Stoffel, W. Cloned mammalian neutral sphingomyelinase: Functions in sphingolipid signaling? Proc. Natl. Acad. Sci. USA 1998, 95, 3638–3643. [Google Scholar] [CrossRef]
- Tepper, A.D.; Ruurs, P.; Borst, J.; van Blitterswijk, W.J. Effect of overexpression of a neutral sphingomyelinase on CD95-induced ceramide production and apoptosis. Biochem. Biophys. Res. Commun. 2001, 280, 634–639. [Google Scholar] [CrossRef]
- Akhtar, M.J.; Ahamed, M.; Alhadlaq, H.; Alrokayan, S. Pt-coated Au nanoparticle toxicity is preferentially triggered via mitochondrial nitric oxide/reactive oxygen species in human liver cancer (hepg2) cells. ACS Omega 2021, 6, 15431–15441. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.S.; Kong, Z.L.; Shen, Z.F.; Jin, Y.Z.; Zhang, W.K.; Chen, G.F. Regulation of hypoxia inducible factor-1α expression by the alteration of redox status in HepG2 cells. J. Exp. Clin. Cancer Res. 2011, 30, 61. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, M.; Cerella, C.; Amici, C.; Pesce, C.; Coppola, S.; Fanelli, C.; De Nicola, M.; Cristofanon, S.; Clavarino, G.; Bergamaschi, A.; et al. Glutathione depletion up-regulates Bcl-2 in BSO-resistant cells. FASEB J. 2004, 18, 1609–1611. [Google Scholar] [CrossRef] [PubMed]
- Lavrentiadou, S.N.; Chan, C.; Kawcak, T.; Ravid, T.; Tsaba, A.; van der Vliet, A.; Rasooly, R.; Goldkorn, T. Ceramide-mediated apoptosis in lung epithelial cells is regulated by glutathione. Am. J. Respir. Cell Mol. Biol. 2001, 25, 676–684. [Google Scholar] [CrossRef]
- You, B.R.; Shin, H.R.; Han, B.R.; Kim, S.H.; Park, W.H. Auranofin induces apoptosis and necrosis in HeLa cells via oxidative stress and glutathione depletion. Mol. Med. Rep. 2015, 11, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Park, W.H. Auranofin induces mesothelioma cell death through oxidative stress and GSH depletion. Oncol. Rep. 2016, 35, 546–551. [Google Scholar] [CrossRef]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Leonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Klaunig, J.E. Oxidative stress and cancer. Curr. Pharm. Des. 2018, 24, 4771–4778. [Google Scholar] [CrossRef]
- Florean, C.; Song, S.; Dicato, M.; Diederich, M. Redox biology of regulated cell death in cancer: A focus on necroptosis and ferroptosis. Free Radic. Biol. Med. 2019, 134, 177–189. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence 5′ to 3′ |
|---|---|
| nSMase1-F | TTTGGTGTCCGCATTGACTA |
| nSMase1-R | TAGAGCTGGGGTTCTGCTGT |
| nSMase2-F | GGAAGGCCGAGGTGGAA |
| nSMase2-R | CCCCCGAAGACACCATCA |
| nSMase3-F | CACCCAGGATGAGAATGGAAA |
| nSMase3-R | GTCCGTCCTCACCCACGAT |
| GAPDH-F | AGCCACATCGCTCAGACAC |
| GAPDH-R | GCCCAATACGACCAAATCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamal, M.; Tallima, H.; Azzazy, H.M.E.; Abdelnaser, A. Impact of HepG2 Cells Glutathione Depletion on Neutral Sphingomyelinases mRNA Levels and Activity. Curr. Issues Mol. Biol. 2023, 45, 5005-5017. https://doi.org/10.3390/cimb45060318
Gamal M, Tallima H, Azzazy HME, Abdelnaser A. Impact of HepG2 Cells Glutathione Depletion on Neutral Sphingomyelinases mRNA Levels and Activity. Current Issues in Molecular Biology. 2023; 45(6):5005-5017. https://doi.org/10.3390/cimb45060318
Chicago/Turabian StyleGamal, Marie, Hatem Tallima, Hassan M. E. Azzazy, and Anwar Abdelnaser. 2023. "Impact of HepG2 Cells Glutathione Depletion on Neutral Sphingomyelinases mRNA Levels and Activity" Current Issues in Molecular Biology 45, no. 6: 5005-5017. https://doi.org/10.3390/cimb45060318
APA StyleGamal, M., Tallima, H., Azzazy, H. M. E., & Abdelnaser, A. (2023). Impact of HepG2 Cells Glutathione Depletion on Neutral Sphingomyelinases mRNA Levels and Activity. Current Issues in Molecular Biology, 45(6), 5005-5017. https://doi.org/10.3390/cimb45060318