Exploring the Genetic Basis of Calonectria spp. Resistance in Eucalypts


Abstract
1. Introduction
1.1. Background and Significance of Research on Eucalypt Leaf Blight
1.2. Eucalyptus pellita
1.3. Variety Improvement Is the Solution to the Impacts of Disease
2. Materials and Methods
2.1. Genetic Material
2.2. Transcriptome Sequencing and Analysis
3. Results
3.1. Quality Control of Sequence Data (QC)
3.1.1. Sequencing Data Quality
3.1.2. Mapping Sequencing Information to E. grandis Reference Genome and Its Regional Distribution
3.2. Quantitative Analysis of the Genes Sequenced
3.2.1. Detection of Expression Distribution of Genes Sequenced in Different Eucalypt Genotypes
3.2.2. The Correlation of Gene Expression of Eucalypt Genotypes Sequenced
3.2.3. Principal Component Analysis of Gene Expression
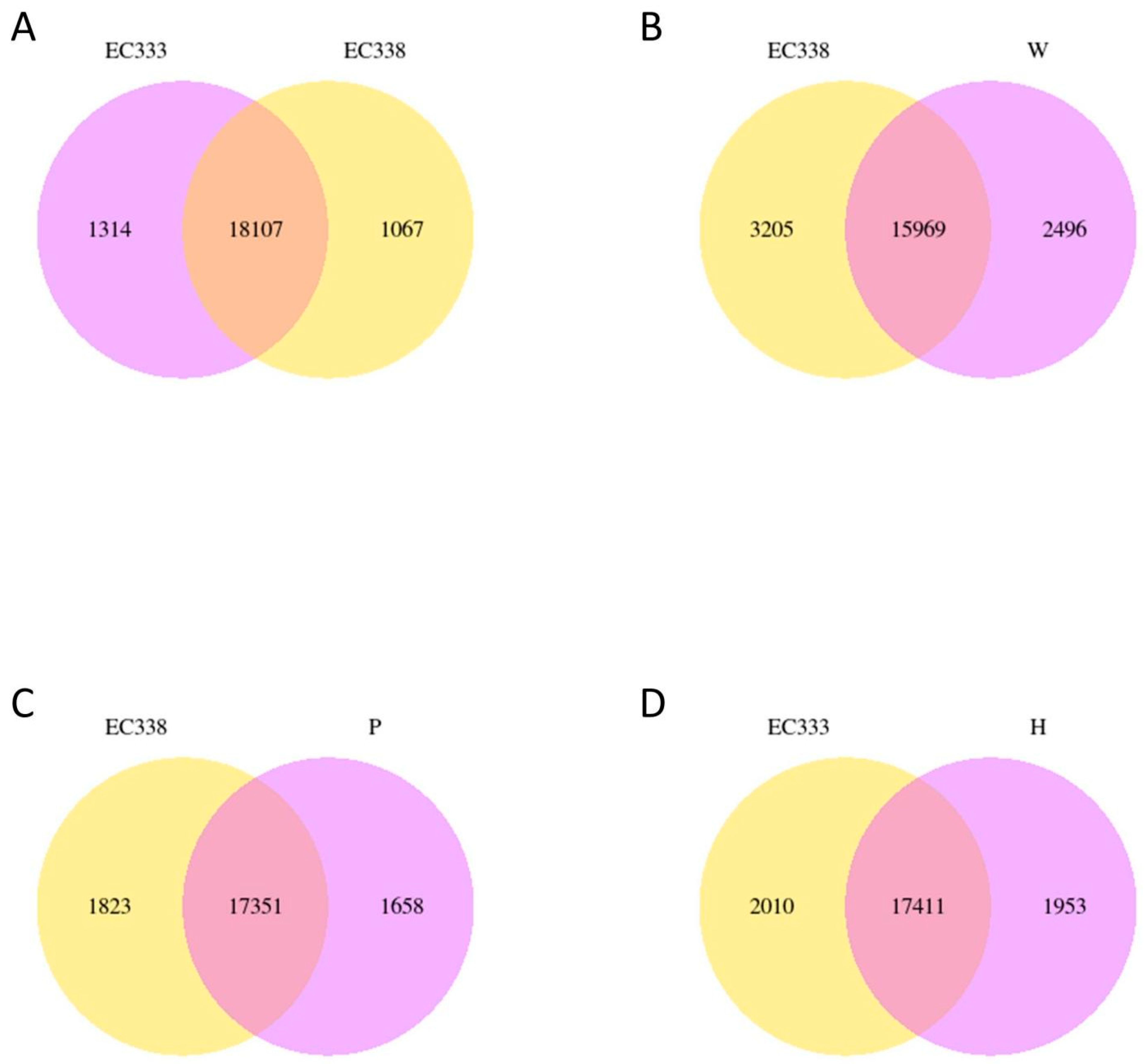
3.3. Gene Co-Expression Venn Diagram and Differential Gene Venn Diagram
3.4. Gene Expression Analysis (Differential Gene Screening and Clustering)
3.5. Enrichment Analysis of Differential Genes in Comparison Groups
3.6. Alternative Splicing Event Analysis
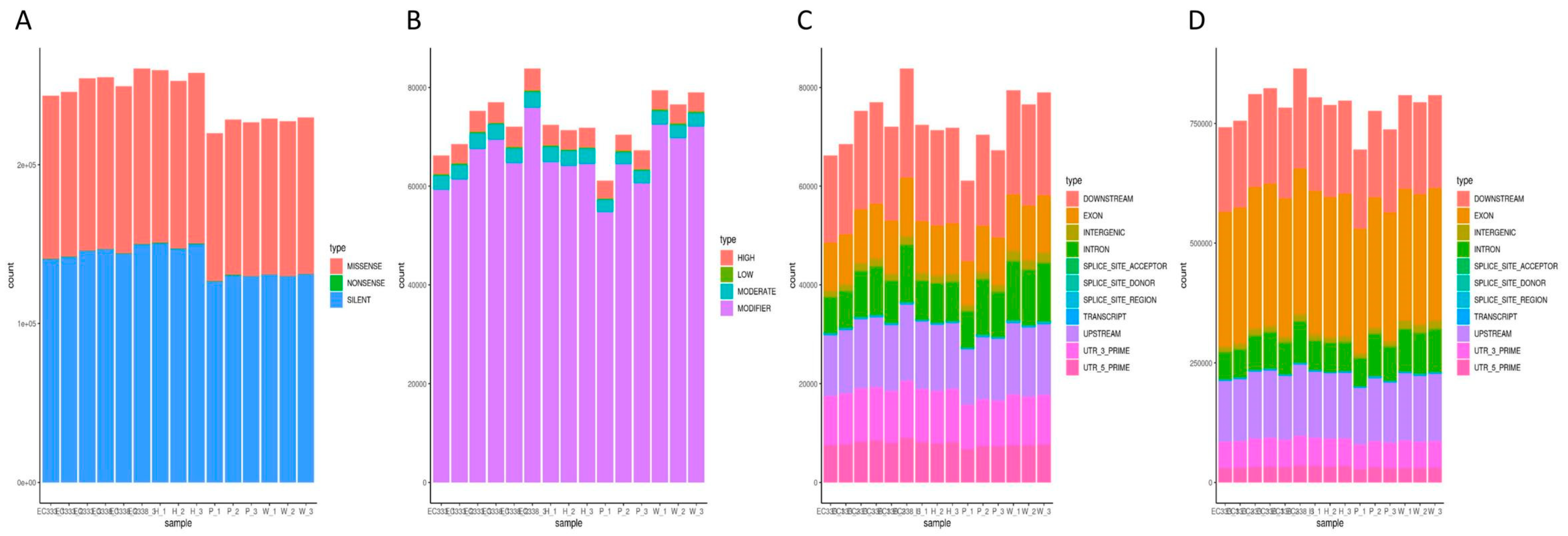
3.7. SNP Variation Loci Analysis
3.8. Weighted Gene Co-Expression Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, G.Q.; Chen, S.F.; Wu, Z.H.; Zhou, X.D.; Xie, Y.J. Preliminary analyses on diversity and pathogenicity of Calonectria spp. on eucalyptus in China. Chin. J. Trop. Crops 2014, 35, 1183–1191. (In Chinese) [Google Scholar]
- Chen, S.F.; Lombard, L.; Roux, J.; Xie, Y.J.; Wingfield, M.J.; Zhou, X.D. Novel species of Calonectria associated with Eucalyptus leaf blight in southeast China. Persoonia 2011, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Guo, W.S.; Chen, H.M.; Wu, J.Q.; Chen, Q.Z.; Meng, X.M. Loss estimation of eucalyptus growth caused by of eucalyptus dieback. For. Pest Dis. 2011, 30, 6–10. (In Chinese) [Google Scholar]
- Liu, Q.L.; Chen, S.F. Two novel species of Calonectria isolated from soil in a natural forest in China. Mycokeys 2017, 26, 25–60. [Google Scholar] [CrossRef]
- Wang, Q.C.; Chen, S.F. Calonectria pentaseptata causes severe leaf disease of cultivated eucalyptus on the Leizhou peninsula of southern China. Plant Dis. 2020, 104, 493–509. [Google Scholar] [CrossRef]
- Li, G.Q.; Liu, F.F.; Li, J.Q.; Liu, Q.L.; Chen, S.F. Botryosphaeriaceae from eucalyptus plantations and adjacent plants in China. Persoonia 2018, 40, 63–95. [Google Scholar] [CrossRef]
- Wang, C.B.; Lan, J.; Wang, J.Z.; He, W.L.; Lu, W.H.; Lin, Y.; Luo, J.Z. Population structure and genetic diversity in Eucalyptus pellita based on SNP markers. Front. Plant Sci. 2023, 14, 1278427. [Google Scholar] [CrossRef] [PubMed]
- Harwood, C.E.; Nikles, D.G.; Pomroy, P.C. Genetic improvement of Eucalyptus pellita in north Queensland, Australia. In Proceedings of the IUFRO Conference on Siliviculture and Improvement of Eucalypt. BAAPA, Salvador, Brazil, 24–29 August 1997; Volume 1, pp. 219–226. [Google Scholar]
- Poubel, D.S.; Garcia, R.A.; Latorraca, J.V.F.; Carvalho, A.M. Estrutura Anatômica e Propriedades Físicas da Madeira de Eucalyptus pellita F. Muell (Anatomical structure and physical properties of Eucalyptus pellita F. Muell Wood). Floresta E Ambiente 2011, 18, 117–126. [Google Scholar] [CrossRef]
- Leksono, B.; Kurinobu, S.; Ide, Y. Optimum age for selection based on a time trend of genetic parameters related to diameter growth in seedling seed orchards of Eucalyptus pellita, in Indonesia. J. For. Res. 2006, 11, 359–364. [Google Scholar] [CrossRef]
- Qin, L.; Zhao, Y.K.; Huang, R.F.; Lu, J.X.; Luo, J.Z. Genetic variation of growth strains and growth of 8-year-old Eucalyptus pellita. J. Cent. South Univ. For. Technol. 2008, 28, 58–63. (In Chinese) [Google Scholar]
- Nirsatmanto, A.; Leksono, B.; Kurinobu, S.; Shiraishi, S. Realized genetic gain observed in second-generation seedling seed orchards of Acacia mangium, in South Kalimantan, Indonesia. J. For. Res. 2004, 9, 265–269. [Google Scholar] [CrossRef]
- Leksono, B.; Kurinobu, S. Trend of within family-plot selection practised in three seedling seed orchards of Eucalyptus pellita in Indonesia. J. Trop. For. Sci. 2005, 18, 121–126. [Google Scholar]
- Brawner, J.T.; Bush, D.J.; Macdonell, P.F.; Warburton, P.M.; Clegg, P.A. Genetic parameters of red mahogany breeding populations grown in the tropics. Aust. For. 2010, 73, 177–183. [Google Scholar] [CrossRef]
- Liu, X.H.; Luo, J.Z.; Lu, W.H.; Lin, Y.; Wang, C.B.; Qi, J. Genetic characteristics of 2 consecutive Eucalyptus pellita generations in growth and typhoon resistance. Mol. Plant Breed. 2017, 15, 5103–5111. (In Chinese) [Google Scholar]
- Shang, L.G.; Gao, Z.Y.; Qian, Q. Progress in understanding the genetic basis of heterosis in crops. Chin. Bull. Bot. 2017, 52, 10–18. (In Chinese) [Google Scholar]
- Velasco, T.; Toro, M.A.; Fuentes, M.C. Heterosis and genotype-environment interaction: An experimental study with Tribolium castaneum. J. Anim. Breed. Genet. 1987, 104, 273–282. [Google Scholar] [CrossRef]
- Maphumulo, S.; Derera, J.; Sibiya, J.; Mathew, I. Combining ability, gene action and heterosis analyses of maize lines that were developed for maize streak virus resistance and acidic soil tolerance. Euphytica 2021, 217, 23. [Google Scholar] [CrossRef]
- Ji, J.L.; Huang, J.X.; Yang, L.M.; Fang, Z.Y.; Zhang, Y.Y.; Zhuang, M.; Lv, H.H.; Wang, Y.; Liu, Y.M.; Li, Z.S.; et al. Advances in Research and Application of Male Sterility in Brassica oleracea. Horticulturae 2020, 6, 101. [Google Scholar] [CrossRef]
- Alisoltani, A.; Fallahi, H.; Shiran, B.; Alisoltani, A.; Ebrahimie, E. RNA-Seq SSRs and small RNA-Seq SSRs: New approaches in cancer biomarker discovery. Gene 2015, 560, 34–43. [Google Scholar] [CrossRef]
- Feng, M.F.; Zhao, J.H.; Li, S.C.; Wei, N.; Kuang, B.W.; Yang, X.P. Molecular genetic mechanisms of heterosis in sugarcane cultivars using a comparative transcriptome analysis of hybrids and ancestral parents. Agronomy 2023, 13, 348. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, F.X.; Zhang, X.G.; Wei, Q.Y.; Dong, J.L.; Bo, C.; Cheng, B.J.; Ma, Q. Comparative transcriptome analysis reveals important roles of nonadditive genes in maize hybrid An’nong 591 under heat stress. BMC Plant Biol. 2019, 19, 273. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.J.; Li, Y.M.; Li, X.S.; Liu, Z.Q.; Zhang, G.Y.; Zhao, Y.L. De novo transcriptome analysis reveals possible heterosis for growth, immunity and anti-oxidation of Macrobrachium nipponense hybrid offspring and parent populations. Aquac. Res. 2022, 53, 1–12. [Google Scholar] [CrossRef]
- Howlader, J.; Robin, A.H.K.; Natarajan, S.; Biswas, M.K.; Sumi, K.R.; Song, C.Y.; Park, J.I.; Nou, I.S. Transcriptome Analysis by RNA-Seq Reveals Genes Related to Plant Height in Two Sets of Parent-hybrid Combinations in Easter lily (Lilium longiflorum). Sci. Rep. 2020, 10, 9082. [Google Scholar] [CrossRef]
- Fang, Z.D. Research Methods for Plant Diseases; China Agriculture Press: Beijing, China, 1998. (In Chinese) [Google Scholar]
- Liang, X.Y.; Lin, Y.; Lu, W.H. Resistance of 14 eucalypt genotypes to Calonectria leaf blight. Eucalypt Sci. Technol. 2023, 40, 1–10. (In Chinese) [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.D.; Cao, Y.; Pau, G.; Lawrence, M.; Wu, T.D.; Seshagiri, S.; Gentleman, R. Prediction and Quantification of Splice Events from RNA-Seq Data. PloS ONE 2016, 11, e0156132. [Google Scholar] [CrossRef]
- He, Z.J.; Zhao, X.; Lu, Z.Y.; Wang, H.F.; Liu, P.F.; Zeng, F.Q.; Zhang, Y.J. Comparative transcriptome and gene co-expression network analysis reveal genes and signaling pathways adaptively responsive to varied adverse stresses in the insect fungal pathogen, Beauveria bassiana. J. Invertebr. Pathol. 2018, 151, 169–181. [Google Scholar] [CrossRef]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomicfeatures. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. NAR 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A.; Hellsten, U.; Hayes, R.D.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D.; et al. The genome of Eucalyptus grandis. Nature 2014, 510, 356–362. [Google Scholar] [CrossRef]
- Xiong, Z.Q.; Fan, Y.Z.; Song, X.; Xia, Y.J.; Zhang, H.; Ai, L.Z. Short communication: Genome-wide identification of new reference genes for reverse-transcription quantitative PCR in Streptococcus thermophilus based on RNA-sequencing analysis. J. Dairy Sci. 2020, 103, 10001–10005. [Google Scholar] [CrossRef]
- Guo, T.T.; Yang, N.; Tong, H.; Pan, Q.C.; Yang, X.H.; Tang, J.H.; Wang, J.K.; Li, J.S.; Yan, J.B. Genetic basis of grain yield heterosis in an “immortalized F2” maize population. Theor. Appl. Genet. 2014, 127, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Chen, Y.; Yao, W.; Zhang, C.J.; Xie, W.B.; Hua, J.P.; Xing, Y.Z.; Xiao, J.H.; Zhang, Q.F. Genetic composition of yield heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 2012, 109, 15847–15852. [Google Scholar] [CrossRef]
- Zheng, X.T.; Chen, J.F.; Nie, R.X.; Miao, H.H.; Chen, Z.W.; He, J.H.; Xie, Y.K.; Zhang, H. Differential expression of ASIP transcripts reveals genetic mechanism underpinning black-tail independence from body plumage in yellow-bodied chickens. Anim. Genet. 2024, 55, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.L.; Li, X.; Ai, J.; Wang, Z.X.; Sun, D.; Liu, X.Y.; Geng, J.Q.; Li, S.Z.; Zhu, B.H.; Zhao, Z.T.; et al. Inheritance analysis of major lignan contents in Schisandra chinensis hybrid progeny. Ind. Crops Prod. 2024, 215, 118684. [Google Scholar] [CrossRef]
- Abtahi, M.; Mirlohi, A.; Ataii, E.; Sabzalian, M.R.; Jafari, P. Flax seed and flower color inheritance is more complicated than once thought. Crop Sci. 2023, 63, 2713–2726. [Google Scholar] [CrossRef]
- Keadtidumrongkul, P.; Suttangkakul, A.; Pinmanee, P.; Pattana, K.; Kittiwongwattana, C.; Apisitwanich, S.; Vuttipongchaikij, S. Growth modulation effects of CBM2a under the control of AtEXP4 and CaMV35S promoters in Arabidopsis thaliana, Nicotiana tabacum and Eucalyptus camaldulensis. Transgenic Res. 2017, 26, 447–463. [Google Scholar] [CrossRef]
- Obembe, O.O.; Jacobsen, E.; Timmers, J.; Gilbert, H.; Blake, A.W.; Knox, J.P.; Visser, R.G.F.; Vincken, J.P. Promiscuous, non-catalytic, tandem carbohydrate-binding modules modulate the cell-wall structure and development of transgenic tobacco (Nicotiana tabacum) plants. J. Plant Res. 2007, 120, 605–617. [Google Scholar] [CrossRef]
- Peter, G.F. Breeding and Engineering Trees to Accumulate High Levels of Terpene Metabolites for Plant Defense and Renewable Chemicals. Front. Plant Sci. 2018, 9, 1672. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, V.R.; Krokene, P.; Christiansen, E.; Krekling, T. Anatomical and chemical defenses of conifer bark against bark beetles and other pests. New Phytol. 2005, 167, 353–375. [Google Scholar] [CrossRef]
- King, D.J.; Gleadow, R.M.; Woodrow, I.E. Regulation of oil accumulation in single glands of Eucalyptus Polybractea. New Phytol. 2006, 172, 440–451. [Google Scholar] [CrossRef]
- Naidoo, S.; Kulheim, C.; Zwart, L.; Mangwanda, R.; Oates, C.N.; Visser, E.A. Uncovering the defence responses of eucalyptus to pests and pathogens in the genomics age. Tree Physiol. 2014, 34, 931–943. [Google Scholar] [CrossRef]
- Trapp, S.; Croteau, R. Defensive resin biosynthesis in conifers. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 689–724. [Google Scholar] [CrossRef] [PubMed]
- Hodges, J.D.; Nebeker, T.E.; Deangelis, J.D.; Karr, B.L.; Blanche, C.A. Host resistance and mortality: A hypothesis based on southern pine beetle-microorganism-host interactions. Bull. Entomol. Soc. Am. 1985, 31, 31–35. [Google Scholar] [CrossRef]
- Strom, B.L.; Goyer, R.A.; Ingram, L.L.; Boyd, G.D.L.; Lott, L.H. Oleoresin characteristics of progeny of loblolly pines that escaped attack by the southern pine beetle. For. Ecol. Manag. 2002, 158, 169–178. [Google Scholar] [CrossRef]
- Klepzig, K.D.; Robison, D.J.; Fowler, G.; Minchin, P.R.; Hain, F.P.; Allen, H.L. Effects of mass inoculation on induced oleoresin response in intensively managed loblolly pine. Tree Physiol. 2005, 25, 681–688. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Li, Y.; Sun, L.; Chu, S.Y.; Xu, H.W.; Zhou, X.F. Integration of transcriptomic and proteomic analyses of Rhododendron chrysanthum Pall. in response to cold stress in the Changbai Mountains. Mol. Biol. Rep. 2023, 50, 3607–3616. [Google Scholar] [CrossRef]
- Volkov, A.; Liavonchanka, A.; Kamneva, O.; Fiedler, T.; Goebel, C.; Kreikemeyer, B.; Feussner, I. Myosin Cross-reactive Antigen of Streptococcus pyogenes M49 Encodes a Fatty Acid Double Bond Hydratase that plays a role in oleic acid detoxification and bacterial virulence. J. Biol. Chem. 2010, 285, 10353–10361. [Google Scholar] [CrossRef]
- Ma, S.S.; Sun, C.Z.; Su, W.N.; Zhao, W.J.; Zhang, S.; Su, S.Y.; Xie, B.Y.; Kong, L.J.; Zheng, J.S. Transcriptomic and physiological analysis of atractylodes chinensis in response to drought stress reveals the putative genes related to sesquiterpenoid biosynthesis. BMC Plant Biol. 2024, 24, 91. [Google Scholar] [CrossRef]
- Singh, P.P.; Joshi, R.; Kumar, R.; Kumar, A.; Sharma, U. Comparative phytochemical analysis of Ferula assa-foetida with Ferula jaeschkeana and commercial oleo-gum resins using GC-MS and UHPLC-PDA-QTOF-IMS. Food Res. Int. 2023, 164, 112434. [Google Scholar] [CrossRef]
- Xu, J.R.; Du, R.Y.; Wang, Y.; Chen, J.H. RNA-Sequencing reveals the involvement of sesquiterpene biosynthesis genes and transcription factors during an early response to mechanical wounding of Aquilaria sinensis. Genes 2023, 14, 464. [Google Scholar] [CrossRef] [PubMed]
- Gan, T.T.; Lin, Z.W.; Bao, L.J.; Hui, T.; Cui, X.P.; Huang, Y.Z.; Wang, H.X.; Su, C.; Jiao, F.; Zhang, M.J.; et al. Comparative proteomic analysis of tolerant and sensitive varieties reveals that phenylpropanoid biosynthesis contributes to salt tolerance in mulberry. Int. J. Mol. Sci. 2021, 22, 9402. [Google Scholar] [CrossRef]
- Chen, Q.L.; Zhang, R.Z.; Li, D.L.; Wang, F. Transcriptomic and coexpression network analyses revealed pine chalcone synthase genes associated with pine wood nematode infection. Int. J. Mol. Sci. 2021, 22, 11195. [Google Scholar] [CrossRef]
- Li, J.B.; Ai, M.T.; Hou, J.; Zhu, P.Q.; Cui, X.M.; Yang, Q. Plant-pathogen interaction with root rot of panax notoginseng as a model: Insight into pathogen pathogenesis, plant defence response and biological control. Mol. Plant Pathol. 2024, 25, e13427. [Google Scholar] [CrossRef]
- Ngou, B.P.M.; Ahn, H.K.; Ding, P.T.; Jones, J.D.G. Mutual potentiation of plant immunity by cell-surface and intracellular receptors. Nature 2021, 592, 110–115. [Google Scholar] [CrossRef]
- Yuan, M.H.; Jiang, Z.Y.; Bi, G.Z.; Nomura, K.Y.; Liu, M.H.; Wang, Y.P.; Cai, B.Y.; Zhou, J.M.; He, S.Y.; Xin, X.F. Pattern-recognition receptors are required for NLR-mediated plant immunity. Nature 2021, 592, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Gorshkov, V.; Tsers, I. Plant susceptible responses: The underestimated side of plant-pathogen interactions. Biol. Rev. Camb. Philos. Soc. 2022, 97, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Seybold, H.; Demetrowitsch, T.J.; Hassani, M.A.; Szymczak, S.; Reim, E.; Haueisen, J.; Lübbers, L.; Rühlemann, M.; Franke, A.; Schwarz, K.; et al. A fungal pathogen induces systemic susceptibility and systemic shifts in wheat metabolome and microbiome composition. Nat. Commun. 2020, 11, 1910. [Google Scholar] [CrossRef] [PubMed]
- Bonora, F.S.; Nahrung, H.F.; Hayes, R.A.; Scharaschkin, T.; Pegg, G.; Lee, D.J. Changes in leaf chemistry and anatomy of Corymbia citriodora subsp. variegata (Myrtaceae) in response to native and exotic pathogens. Australas. Plant Pathol. 2020, 49, 641–653. [Google Scholar] [CrossRef]
- Wang, R.; Li, G.; Li, Y. Effects of inducers on relevant resistant substance in colored cotton. Cotton Sci. 2005, 17, 107–111. (In Chinese) [Google Scholar]
- Yin, Y.Q.; Qi, F.; Gao, L.; Rao, S.; Yang, Z.; Fang, W. iTRAQ-based quantitativeproteomic analysis of dark-germinated soybeans in responseto salt stress. RSC Adv. 2018, 8, 17905–17913. [Google Scholar] [CrossRef]
- Yang, Q.; Li, J.B.; Sun, J.L.; Cui, X.M. Comparative transcriptomic and proteomic analyses to determine the lignin synthesis pathway involved in the fungal stress response in Panax notoginseng. Physiol. Mol. Plant Pathol. 2022, 119, 101814. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrids | Susceptibility | Female | Species | Male | Species |
|---|---|---|---|---|---|
| EC338 | Resistant | W1767 | E. wetarensis | P9060 | E. pellita |
| EC333 | Susceptible | H1522 | E. urophylla × E. pellita | Unknown | E. urophylla |
| Comparisons | GO | KEGG |
|---|---|---|
| EC338 vs. EC333 | Carbohydrate binding, pattern binding, polysaccharide binding (−log10(padj) > 6) | Phenylpropanoid biosynthesis, flavonoid biosynthesis, plant–pathogen interaction, sesquiterpenoid and triterpenoid biosynthesis (−log10(padj) > 2.5) |
| EC338 vs. P9060 | Terpene synthase activity, carbon–oxygen lyase acting on phosphates, carbon−oxygen lyase activity (−log10(padj) > 10) | Phenylpropanoid biosynthesis, sesquiterpenoid and triterpenoid biosynthesis (−log10(padj) > 5) |
| EC338 vs. W1767 | DNA binding transcription factor activity, terpene synthase activity, carbon–oxygen lyase acting on phosphates, iron ion binding, transcription regulator activity (−log10(padj) > 6) | Phenylpropanoid biosynthesis, plant hormone signal transduction, sesquiterpenoid and triterpenoid biosynthesis, plant–pathogen interaction, biosynthesis of various plant secondary, flavonoid biosynthesis (−log10(padj) > 4) |
| EC333 vs. H1522 | Carbohydrate binding, terpene synthase activity, carbon–oxygen lyase acting on phosphates, carbon–oxygen lyase activity (−log10(padj) > 4) | Phenylpropanoid biosynthesis (−log10(padj) > 7.5) |
| Differential Genotypes | Genotypes | Name | Size | ES | NES | NOM p-Value | FDR q-Value | FWER p-Value | Rank at Max | Leading Edge |
|---|---|---|---|---|---|---|---|---|---|---|
| EC338 vs. EC333 | EC333 | Carbohydrate binding (GO:0030246) | 286 | −0.519 | −1.580 | 0 | 0.171 | 0.75 | 5450 | tags = 40%, list = 17%, signal = 48% |
| EC338 vs. P9060 | EC338 | Terpene synthase activity (GO:0010333) | 86 | 0.469 | 1.952 | 0 | 0.201 | 0.188 | 2191 | tags = 36%, list = 7%, signal = 39% |
| EC338 | Carbon–oxygen lyase acting on phosphates (GO:0016838) | 89 | 0.462 | 1.939 | 0 | 0.149 | 0.188 | 2191 | tags = 35%, list = 7%, signal = 37% | |
| EC338 | Carbon–oxygen lyase activity (GO:0016835) | 112 | 0.429 | 2.024 | 0 | 0.098 | 0.047 | 3561 | tags = 35%, list = 11%, signal = 39% |
| Differential Genotypes | Genotypes | Name | Size | ES | NES | NOM p-Value | FDR q-Value | FWER p-Value | Rank at Max | Leading Edge |
|---|---|---|---|---|---|---|---|---|---|---|
| EC338 vs. EC333 | EC338 | Sesquiterpenoid and triterpenoid biosynthesis (EGR00909) | 65 | 0.610 | 1.714 | 0 | 0.066 | 0.093 | 3071 | tags = 35%, list = 10%, signal = 39% |
| EC338 vs. W1767 | EC338 | Phenylpropanoid biosynthesis (EGR00940) | 224 | 0.369 | 1.900 | 0 | 0.048 | 0.048 | 3594 | tags = 27%, list = 11%, signal = 30% |
| W1767 | Plant–pathogen interaction (EGR04626) | 281 | −0.455 | −1.501 | 0 | 0.178 | 0.599 | 4752 | tags = 37%, list = 15%, signal = 43% | |
| EC338 | Flavonoid biosynthesis (EGR00941) | 89 | 0.521 | 1.455 | 0 | 0.103 | 0.760 | 4985 | tags = 42%, list = 15%, signal = 49% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Z.; Lu, W.; Lin, Y.; Luo, J.; Liu, G.; Huang, A. Exploring the Genetic Basis of Calonectria spp. Resistance in Eucalypts. Curr. Issues Mol. Biol. 2024, 46, 10854-10879. https://doi.org/10.3390/cimb46100645
Su Z, Lu W, Lin Y, Luo J, Liu G, Huang A. Exploring the Genetic Basis of Calonectria spp. Resistance in Eucalypts. Current Issues in Molecular Biology. 2024; 46(10):10854-10879. https://doi.org/10.3390/cimb46100645
Chicago/Turabian StyleSu, Zhiyi, Wanhong Lu, Yan Lin, Jianzhong Luo, Guo Liu, and Anying Huang. 2024. "Exploring the Genetic Basis of Calonectria spp. Resistance in Eucalypts" Current Issues in Molecular Biology 46, no. 10: 10854-10879. https://doi.org/10.3390/cimb46100645
APA StyleSu, Z., Lu, W., Lin, Y., Luo, J., Liu, G., & Huang, A. (2024). Exploring the Genetic Basis of Calonectria spp. Resistance in Eucalypts. Current Issues in Molecular Biology, 46(10), 10854-10879. https://doi.org/10.3390/cimb46100645