_Kim.png)
Integrative Network Pharmacology, Molecular Docking, and Dynamics Simulations Reveal the Mechanisms of Cinnamomum tamala in Diabetic Nephropathy Treatment: An In Silico Study

 ,
, 
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Active Compounds and Correlated Target Database Establishment
2.2. Network Construction of Active Compounds–Potential Targets
2.3. Collection of Potential DN-Associated Targets
2.4. Screening Compound–Disease Overlapping Targets
2.5. Network Construction of Compound–Disease Common Targets
2.6. GO and KEGG Pathway Enrichment Analyses
2.7. Construction of Compound–Target–Pathway Network
2.8. Molecular Docking
2.9. MD Simulation
2.10. Free Energy Calculation (MM-GBSA)
2.11. PCA
3. Results
3.1. Active Compounds and Correlated Target Network
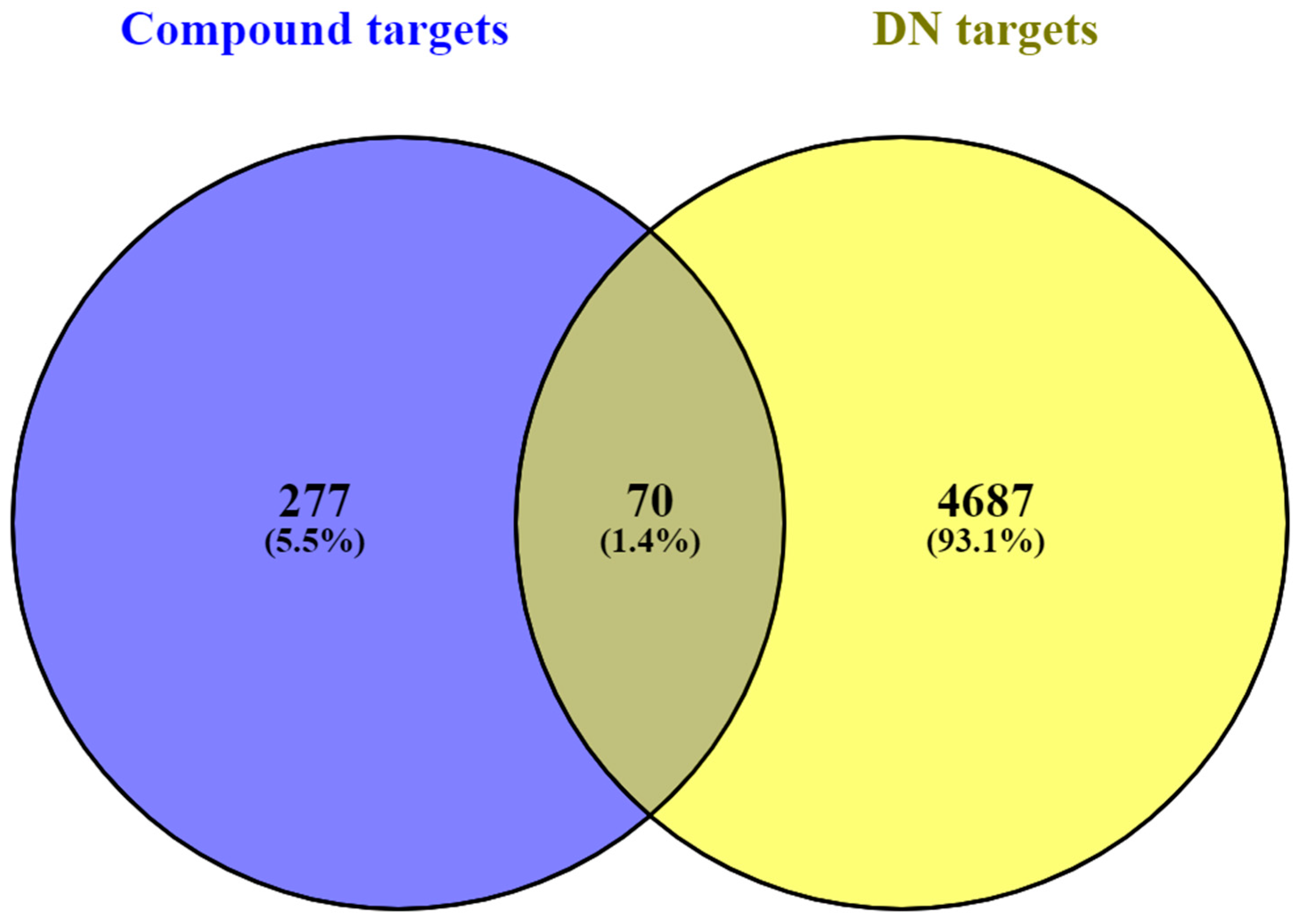
3.2. Screening of Common Targets
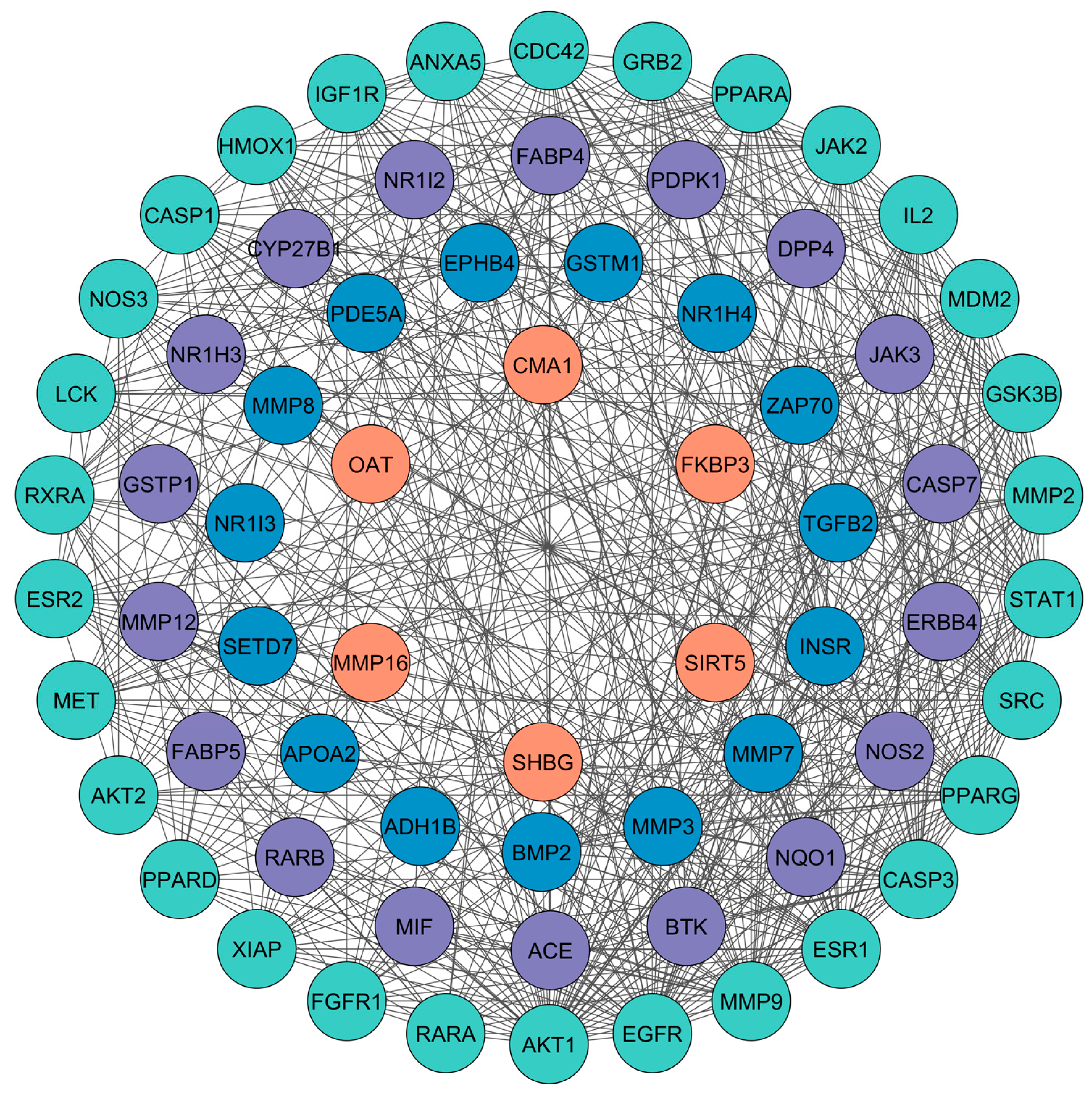
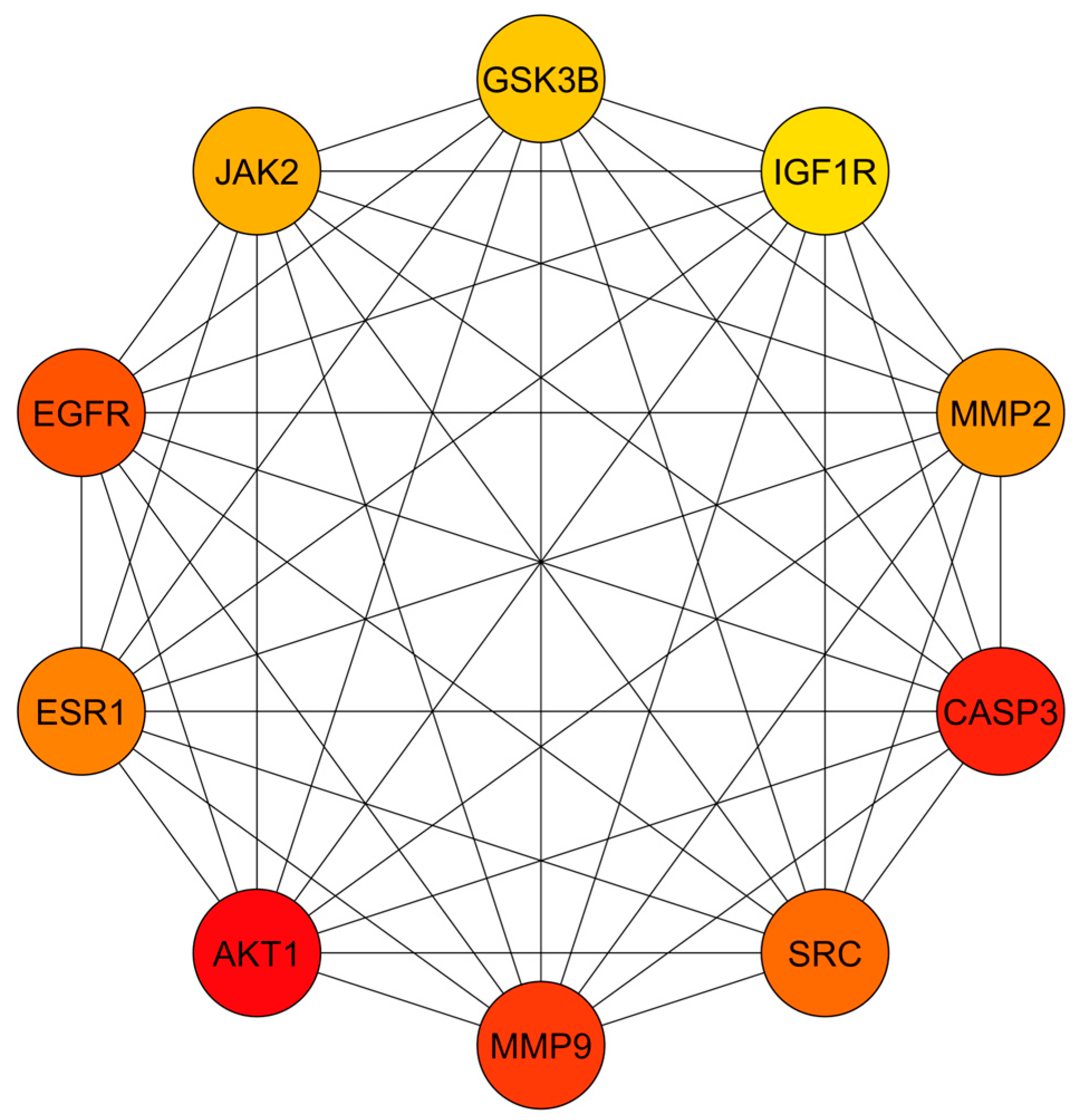
3.3. PPI Network Analysis
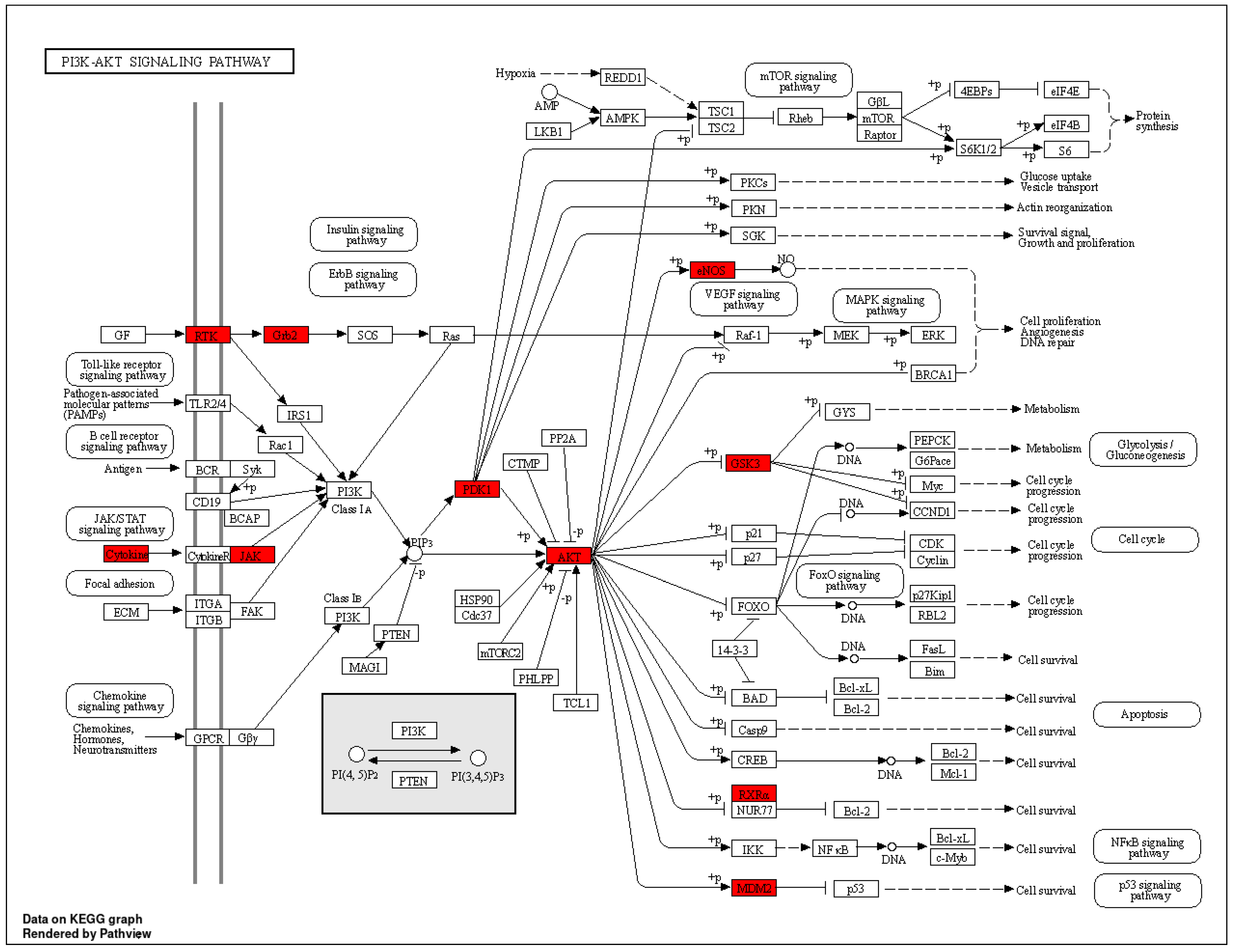
3.4. GO and KEGG Enrichment Analyses
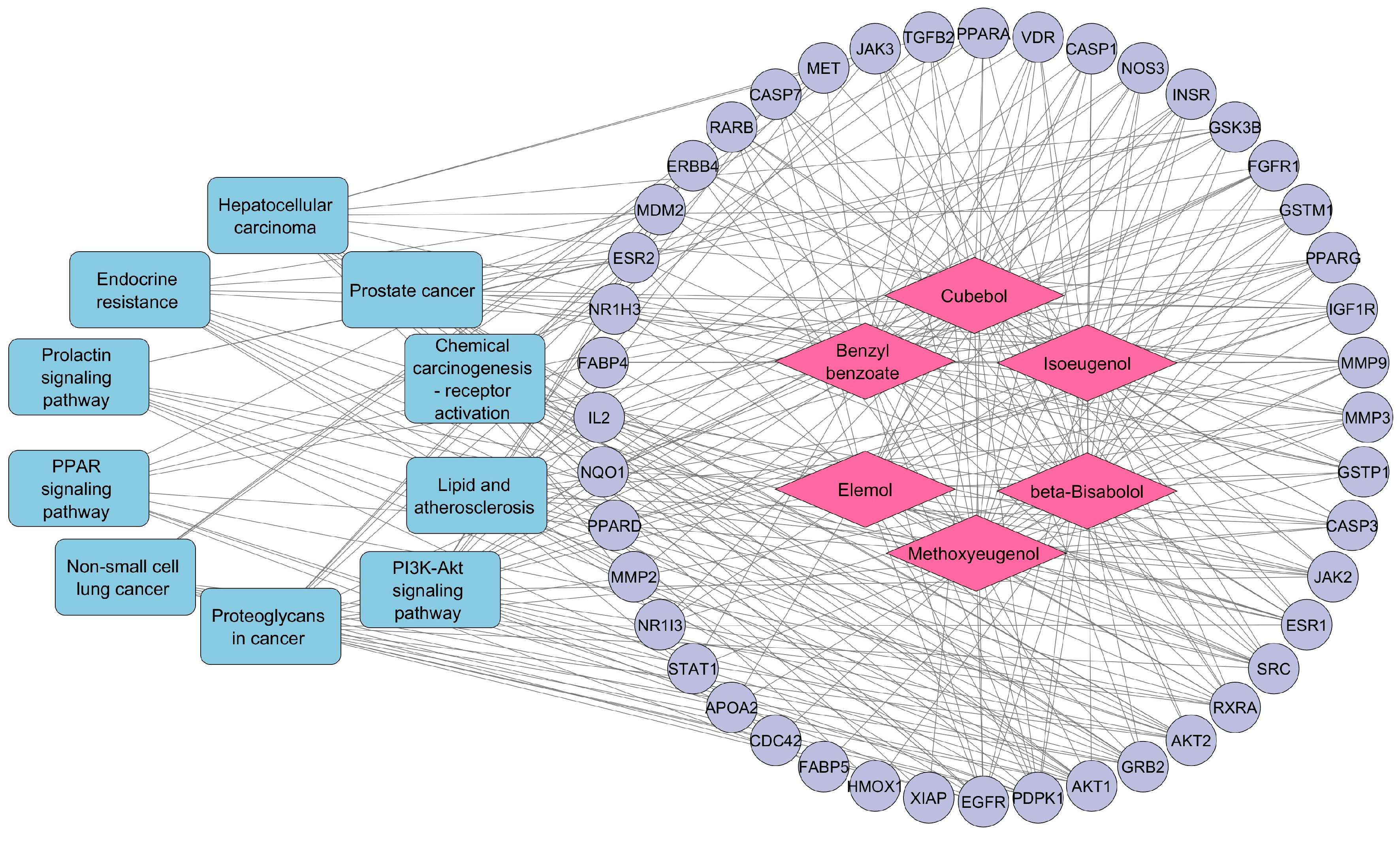
3.5. Compound–Target–Pathway Network Construction
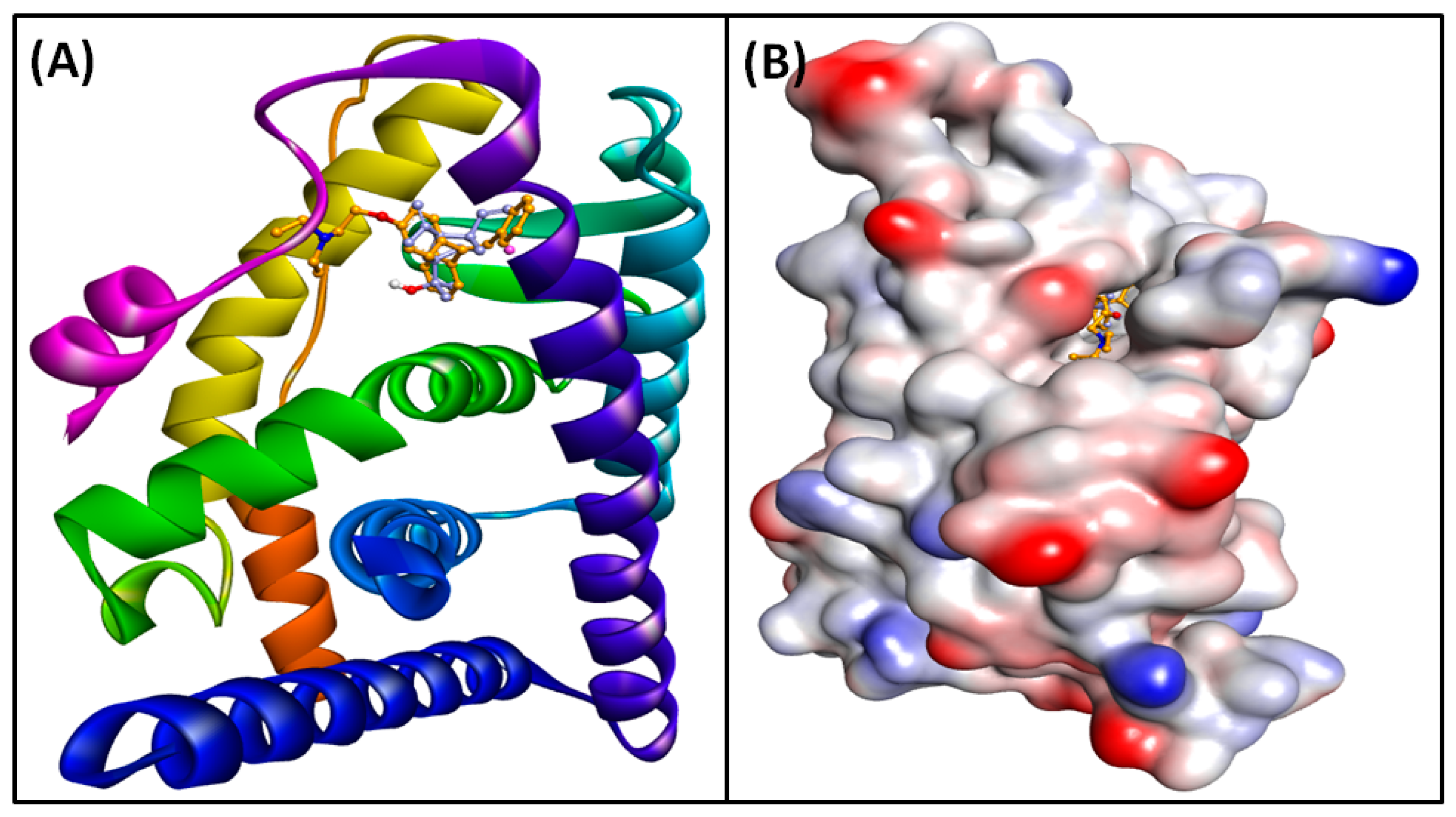
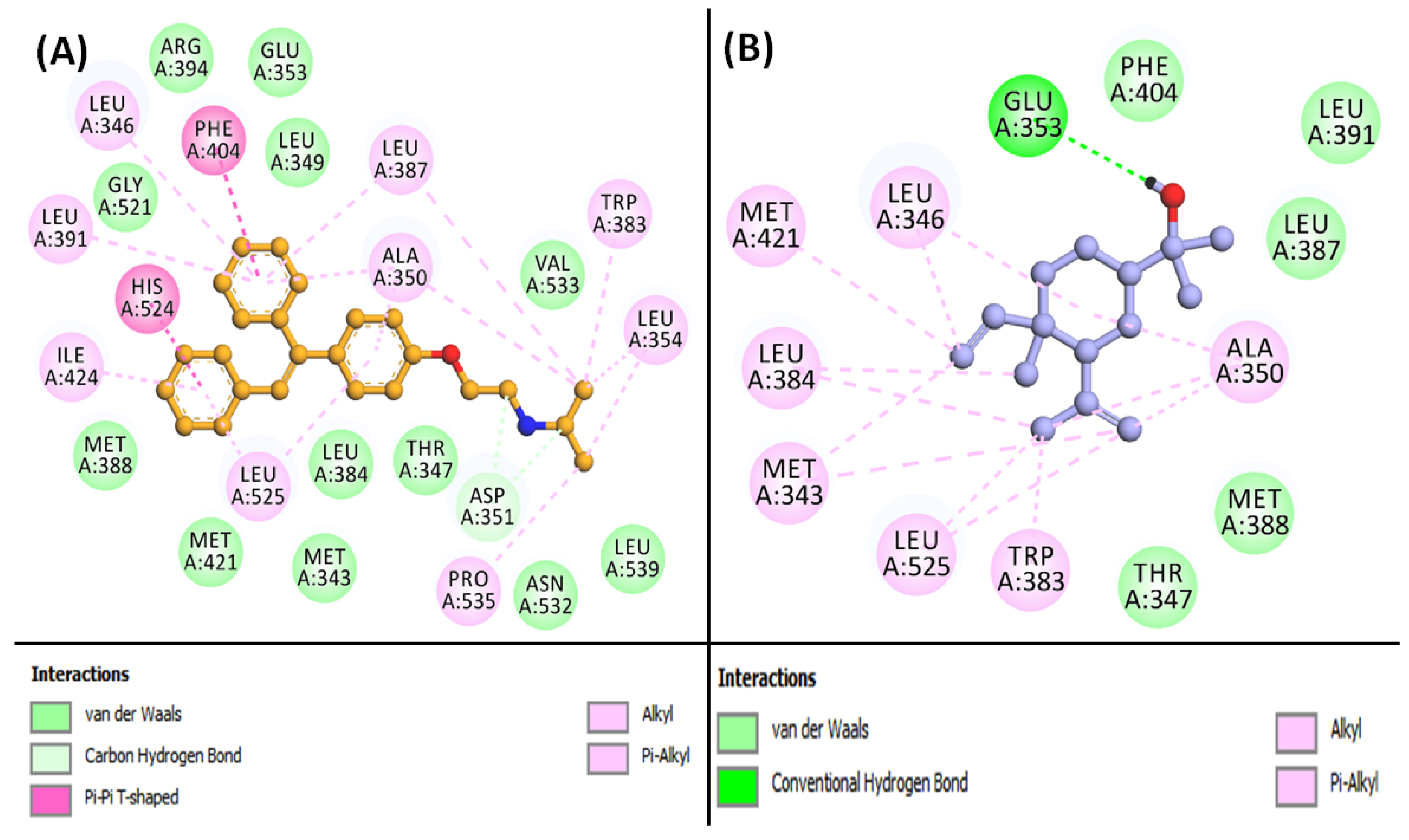
3.6. Molecular Docking of Compounds and Targets
3.7. Analysis of MD Simulation
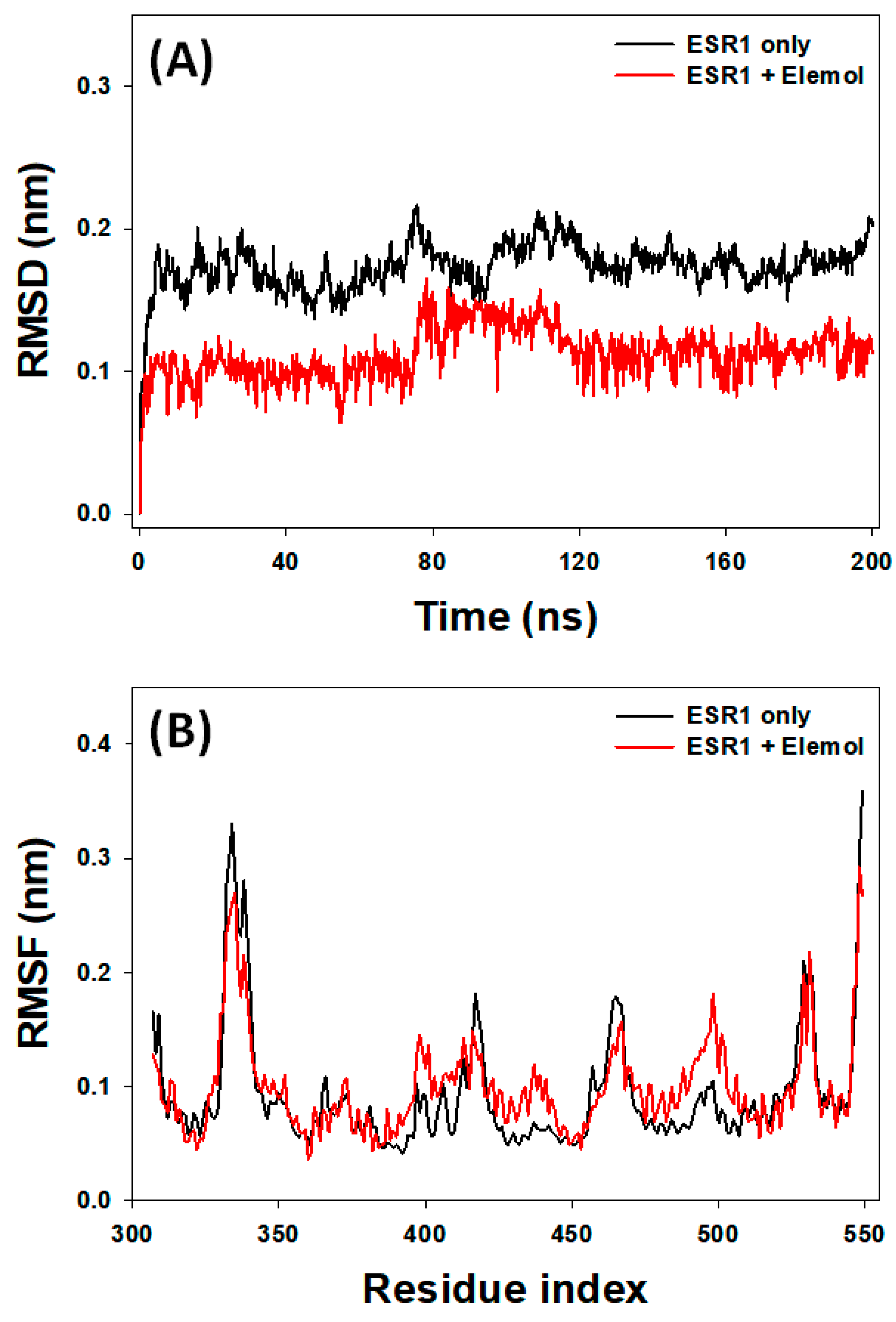
3.7.1. RMSD Analysis
3.7.2. RMSF Analysis
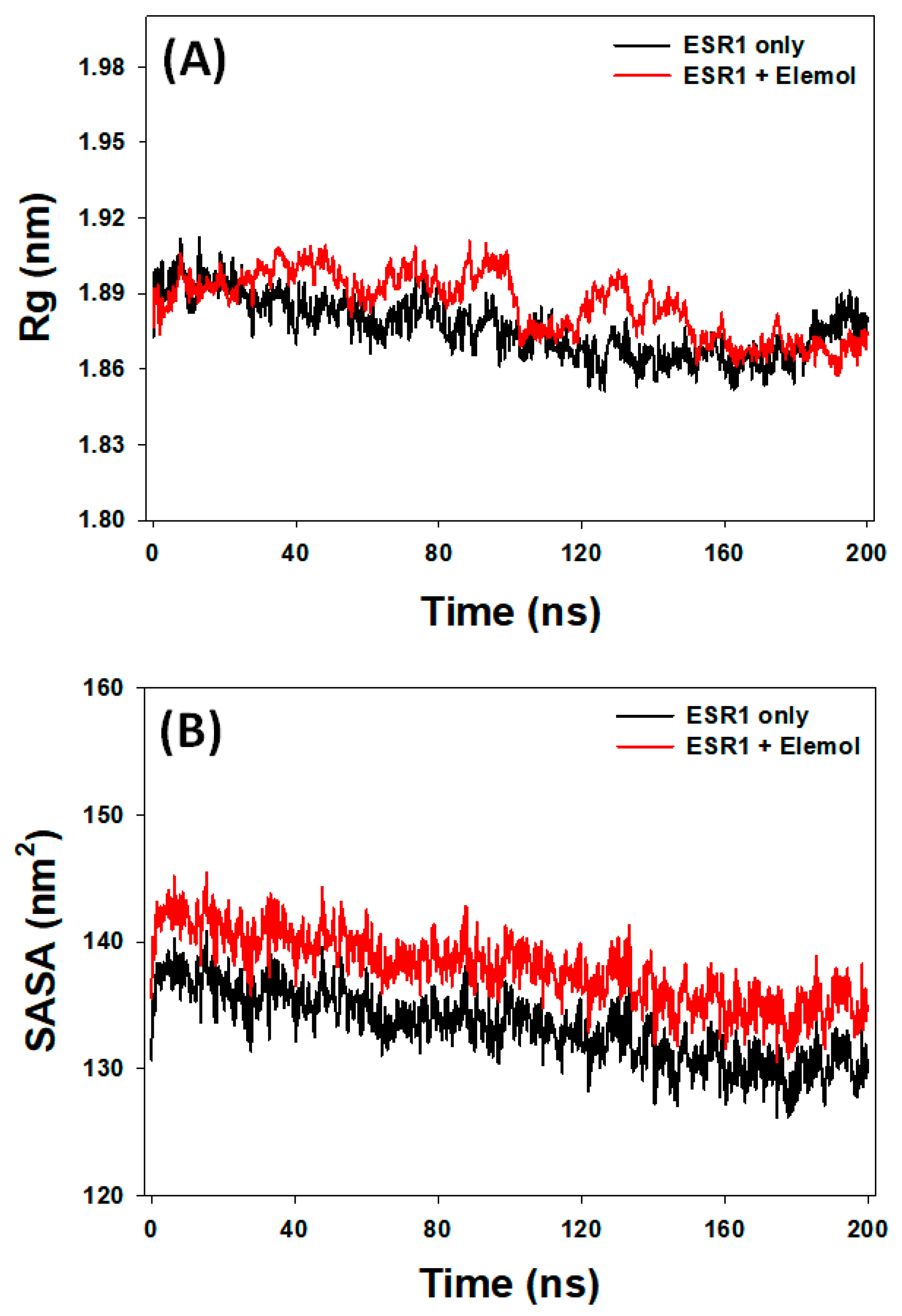
3.7.3. Rg Analysis
3.7.4. SASA Analysis
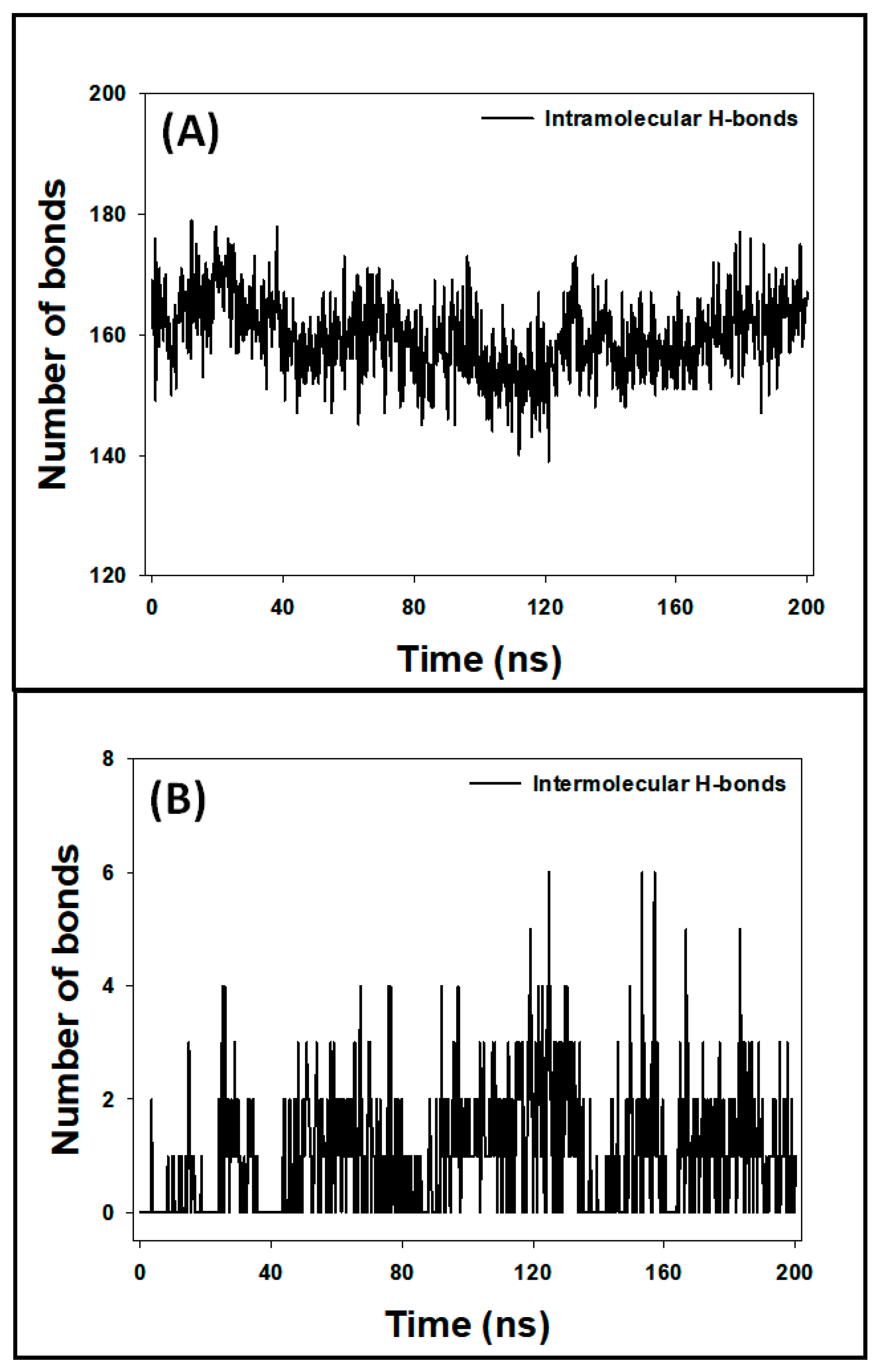
3.7.5. Analysis of Hydrogen Bonds
3.8. Analysis of Free Energy Calculations (MM/GBSA)
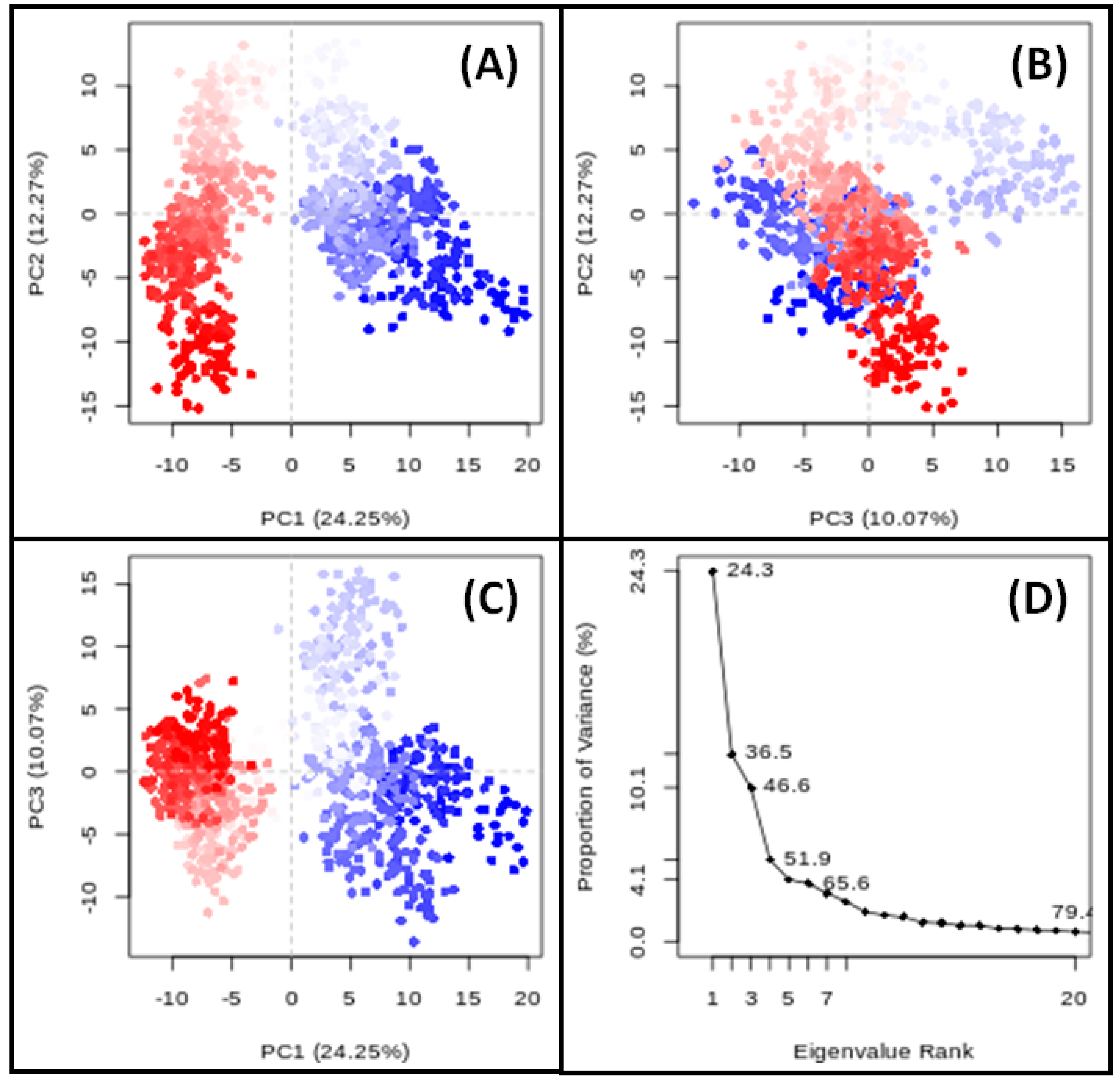
3.9. PCA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Viswanathan, V.; Mirshad, R. The Burden of Diabetic Nephropathy in India: Need for Prevention. Diabet. Nephrop. 2023, 3, 25–28. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Opazo-Ríos, L.; Guerrero-Hue, M.; García-Caballero, C.; Vázquez-Carballo, C.; Mas, S.; Sanz, A.B.; Herencia, C.; Mezzano, S.; et al. Pathogenic Pathways and Therapeutic Approaches Targeting Inflammation in Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 3798. [Google Scholar] [CrossRef]
- Liyanage, T.; Toyama, T.; Hockham, C.; Ninomiya, T.; Perkovic, V.; Woodward, M.; Fukagawa, M.; Matsushita, K.; Praditpornsilpa, K.; Hooi, L.S.; et al. Prevalence of Chronic Kidney Disease in Asia: A Systematic Review and Analysis. BMJ Glob. Health 2022, 7, e007525. [Google Scholar] [CrossRef]
- Unnikrishnan, I.R.; Rema, M.; Pradeepa, R.; Deepa, M.; Shanthirani, C.S.; Deepa, R.; Mohan, V. Prevalence and Risk Factors of Diabetic Nephropathy in an Urban South Indian Population: The Chennai Urban Rural Epidemiology Study (CURES 45). Diabetes Care 2007, 30, 2019–2024. [Google Scholar] [CrossRef]
- Huang, L.; Khardori, R. Pathogenesis of Diabetic Nephropathy. In Managing Diabetic Nephropathies in Clinical Practice; Springer: Berlin/Heidelberg, Germany, 2017; pp. 23–45. [Google Scholar] [CrossRef]
- Nordheim, E.; Jenssen, T.G. Chronic Kidney Disease in Patients with Diabetes Mellitus. Endocr. Connect. 2021, 10, R151–R159. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Reeves, W.B.; Awad, A.S. Pathophysiology of Diabetic Kidney Disease: Impact of SGLT2 Inhibitors. Nat. Rev. Nephrol. 2021, 17, 319–334. [Google Scholar] [CrossRef]
- Jiang, J.; Zhao, C.; Han, T.; Shan, H.; Cui, G.; Li, S.; Xie, Z.; Wang, J. Advanced Glycation End Products, Bone Health, and Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2022, 130, 671–677. [Google Scholar] [CrossRef]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the Fire: Emerging Role of the Hexosamine Biosynthetic Pathway in Cancer. BMC Biol. 2019, 17, 52. [Google Scholar] [CrossRef]
- Navale, A.M.; Paranjape, A.N. Glucose Transporters: Physiological and Pathological Roles. Biophys. Rev. 2016, 8, 5–9. [Google Scholar] [CrossRef]
- Bates, D.O.; Beazley-Long, N.; Benest, A.V.; Ye, X.; Ved, N.; Hulse, R.P.; Barratt, S.; Machado, M.J.; Donaldson, L.F.; Harper, S.J.; et al. Physiological Role of Vascular Endothelial Growth Factors as Homeostatic Regulators. Compr. Physiol. 2018, 8, 955–979. [Google Scholar] [CrossRef]
- Calle, P.; Hotter, G. Macrophage Phenotype and Renal Fibrosis in Obstructive Nephropathy. Nephron Exp. Nephrol. 2008, 110, e31–e36. [Google Scholar]
- Hughes, C.E.; Nibbs, R.J.B. A Guide to Chemokines and Their Receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef] [PubMed]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key Chemokines Direct Migration of Immune Cells in Solid Tumors. Cancer Gene Ther. 2022, 29, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.L.; Harrison, R.E. Microbial Phagocytic Receptors and Their Potential Involvement in Cytokine Induction in Macrophages. Front. Immunol. 2021, 12, 662063. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Mimura, I.; Tanaka, T.; Nangaku, M. Treatment of Diabetic Kidney Disease: Current and Future. Diabetes Metab. J. 2021, 45, 11–26. [Google Scholar] [CrossRef]
- Modak, M.; Dixit, P.; Londhe, J.; Ghaskadbi, S.; Devasagayam, T.P.A. Indian Herbs and Herbal Drugs Used for the Treatment of Diabetes. J. Clin. Biochem. Nutr. 2007, 40, 163–173. [Google Scholar] [CrossRef]
- Lysiuk, R.; Mansuetusmboya, J. Herbal Drugs for the Treatment of Diabetic Nephropathy: Current Status and Prospects for the Application. J. Kidney Treat. Diagn. 2020, 3, 3–4. [Google Scholar]
- Sharma, V.; Rao, L.J.M. An Overview on Chemical Composition, Bioactivity and Processing of Leaves of Cinnamomum Tamala. Crit. Rev. Food Sci. Nutr. 2014, 54, 433–448. [Google Scholar] [CrossRef]
- Mohanty, D.; Padhee, S.; Priyadarshini, A.; Champati, B.B.; Das, P.K.; Jena, S.; Sahoo, A.; Chandra Panda, P.; Nayak, S.; Ray, A. Elucidating the Anti-Cancer Potential of Cinnamomum Tamala Essential Oil against Non-Small Cell Lung Cancer: A Multifaceted Approach Involving GC-MS Profiling, Network Pharmacology, and Molecular Dynamics Simulations. Heliyon 2024, 10, e28026. [Google Scholar] [CrossRef]
- Sudan, R.; Bhagat, M.; Gupta, S.; Chitrarakha; Devi, T. Comparative Analysis of Cytotoxic and Antioxidant Potential of Edible Cinnamomum Verum (Bark) and Cinnamomum Tamala (Indian Bay Leaf). Free. Radic. Antioxid. 2013, 3, S70–S73. [Google Scholar] [CrossRef]
- Sahu, N.; Madan, S.; Walia, R.; Tyagi, R.; Fantoukh, O.I.; Hawwal, M.F.; Akhtar, A.; Almarabi, I.; Alam, P.; Saxena, S. Multi-Target Mechanism of Solanum Xanthocarpum for Treatment of Psoriasis Based on Network Pharmacology and Molecular Docking. Saudi Pharm. J. 2023, 31, 101788. [Google Scholar] [CrossRef] [PubMed]
- Afolabi, R.; Chinedu, S.; Ajamma, Y.; Adam, Y.; Koenig, R.; Adebiyi, E. Computational Identification of Plasmodium Falciparum RNA Pseudouridylate Synthase as a Viable Drug Target, Its Physicochemical Properties, 3D Structure Prediction and Prediction of Potential Inhibitors. Infect. Genet. Evol. 2022, 97, 105194. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Liu, S.; Zeng, F.; Rao, Z.; Yan, C.; Xing, Q.; Chen, Y. Exploring the Protective Effect of Sangggua Drink against Type 2 Diabetes Mellitus in Db/Db Mice Using a Network Pharmacological Approach and Experimental Validation. Heliyon 2023, 9, e18026. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, K.; Yang, J.; Zhang, A.; Dong, X.; Zhou, Z.; Li, T.; Fan, R. Mechanism of Ferroptosis in Hypertensive Nephropathy. Transl. Androl. Urol. 2022, 11, 617–626. [Google Scholar] [CrossRef]
- Zhao, W.M.; Wang, Z.J.; Shi, R.; Zhu, Y.; Li, X.L.; Wang, D.G. Analysis of the Potential Biological Mechanisms of Diosmin against Renal Fibrosis Based on Network Pharmacology and Molecular Docking Approach. BMC Complement. Med. Ther. 2023, 23, 157. [Google Scholar] [CrossRef]
- Beg, M.R.; Laeeq, A.; Sathaye, S. Pharmacological Target and the Biological Mechanism of Gallic Acid for Anticataract Effect: A Network Analysis. Phytomedicine Plus 2022, 2, 100258. [Google Scholar] [CrossRef]
- Liu, H.; Mohammed, S.A.D.; Lu, F.; Chen, P.; Wang, Y.; Liu, S. Network Pharmacology and Molecular Docking-Based Mechanism Study to Reveal Antihypertensive Effect of Gedan Jiangya Decoction. BioMed Res. Int. 2022, 2022. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, H.; Wang, D.; Mi, J.; Chen, G.; Li, F.; Wang, Y.; Zhang, Y. Network Pharmacology Analysis on the Mechanism of Huangqi Sijunzi Decoction in Treating Cancer-Related Fatigue. J. Healthc. Eng. 2021, 2021, 9780677. [Google Scholar] [CrossRef]
- Yang, X.; Tao, Y.; Xu, R.; Luo, W.; Lin, T.; Zhou, F.; Tang, L.; He, L.; He, Y. Analysis of Active Components and Molecular Mechanism of Action of Rubia Cordifolia L. in the Treatment of Nasopharyngeal Carcinoma Based on Network Pharmacology and Experimental Verification. Heliyon 2023, 9, e17078. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, A.; Yang, H.; Zhang, C. Identification of IL-8 in CSF as a Potential Biomarker in Sepsis-Associated Encephalopathy. Cytokine 2023, 172, 156390. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Y.; Ma, Z. Multi-Target Mechanism of Tripteryguim Wilfordii Hook for Treatment of Ankylosing Spondylitis Based on Network Pharmacology and Molecular Docking. Ann. Med. 2021, 53, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Liu, Y.; Tan, J.; Sun, Y.; Guan, W.; Jiang, P.; Yang, B.; Kuang, H. Integrated Serum Metabolomics and Network Pharmacology Approach to Reveal the Potential Mechanisms of Withanolides from the Leaves of Datura Metel L. on Psoriasis. J. Pharm. Biomed. Anal. 2020, 186, 113277. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.C.; Ji, H.F. A Search for Medications to Treat COVID-19 via in Silico Molecular Docking Models of the SARS-CoV-2 Spike Glycoprotein and 3CL Protease. Travel Med. Infect. Dis. 2020, 35, 101646. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Xu, F. Impact of the Subtle Differences in MMP-12 Structure on Glide-Based Molecular Docking for Pose Prediction of Inhibitors. J. Mol. Struct. 2014, 1076, 153–159. [Google Scholar] [CrossRef]
- Oyinloye, B.E.; Adekiya, T.A.; Aruleba, R.T.; Ojo, O.A.; Ajiboye, B.O. Structure-Based Docking Studies of GLUT4 Towards Exploring Selected Phytochemicals from Solanum Xanthocarpum as a Therapeutic Target for the Treatment of Cancer. Curr. Drug Discov. Technol. 2018, 16, 406–416. [Google Scholar] [CrossRef]
- Sahu, N.; Tyagi, R.; Kumar, N.; Mujeeb, M.; Akhtar, A.; Alam, P.; Madan, S. Forecasting the Pharmacological Mechanisms of Plumbago Zeylanica and Solanum Xanthocarpum in Diabetic Retinopathy Treatment: A Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation Study. Biology 2024, 13, 732. [Google Scholar] [CrossRef]
- Dong, Y.; Zhao, Q.; Wang, Y. Network Pharmacology-Based Investigation of Potential Targets of Astragalus Membranaceous-Angelica Sinensis Compound Acting on Diabetic Nephropathy. Sci. Rep. 2021, 11, 19496. [Google Scholar] [CrossRef]
- Saputro, A.H.; Amelia, T.; Mahardhika, A.B.; Widyawaruyanti, A.; Wahyuni, T.S.; Permanasari, A.A.; Artarini, A.A.; Tjahjono, D.H.; Damayanti, S. Alpha-Mangostin, Piperine and Beta-Sitosterol as Hepatitis C Antivirus (HCV): In Silico and in Vitro Studies. Heliyon 2023, 9, e20141. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Brooks, C.L. A Modified TIP3P Water Potential for Simulation with Ewald Summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Friesner, R.A.; Beachy, M.D. Quantum Mechanical Calculations on Biological Systems. Curr. Opin. Struct. Biol. 1998, 8, 257–262. [Google Scholar] [CrossRef]
- Menon, A.; Pascazio, L.; Nurkowski, D.; Farazi, F.; Mosbach, S.; Akroyd, J.; Kraft, M. OntoPESScan: An Ontology for Potential Energy Surface Scans. ACS Omega 2023, 8, 2462–2475. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Applequist, J.; Carl, J.R.; Fung, K.K. An Atom Dipole Interaction Model for Molecular Polarizability. Application to Polyatomic Molecules and Determination of Atom Polarizabilities. J. Am. Chem. Soc. 1972, 94, 2952–2960. [Google Scholar] [CrossRef]
- Sangster, M.J.L.; Atwood, R.M. Interionic Potentials for Alkali Halides. II. Completely Crystal Independent Specification of Born-Mayer Potentials. J. Phys. C Solid State Phys. 1978, 11, 1541–1555. [Google Scholar] [CrossRef]
- Tong, J.; Peng, B.; Kontogeorgis, G.M.; Liang, X. Behavior of the Aqueous Sodium Chloride Solutions from Molecular Simulations and Theories. J. Mol. Liq. 2023, 371, 121086. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hockney, R.W.; Goel, S.P.; Eastwood, J.W. Quiet High-Resolution Computer Models of a Plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Jairajpuri, D.S.; Hussain, A.; Nasreen, K.; Mohammad, T.; Anjum, F.; Tabish Rehman, M.; Mustafa Hasan, G.; Alajmi, M.F.; Imtaiyaz Hassan, M. Identification of Natural Compounds as Potent Inhibitors of SARS-CoV-2 Main Protease Using Combined Docking and Molecular Dynamics Simulations. Saudi J. Biol. Sci. 2021, 28, 2423–2431. [Google Scholar] [CrossRef]
- Ichiye, T.; Karplus, M. Collective Motions in Proteins: A Covariance Analysis of Atomic Fluctuations in Molecular Dynamics and Normal Mode Simulations. Proteins Struct. Funct. Bioinform. 1991, 11, 205–217. [Google Scholar] [CrossRef]
- Shamsi, A.; Mohammad, T.; Khan, M.S.; Shahwan, M.; Husain, F.M.; Rehman, M.T.; Hassan, M.I.; Ahmad, F.; Islam, A. Unraveling Binding Mechanism of Alzheimer’s Drug Rivastigmine Tartrate with Human Transferrin: Molecular Docking and Multi-Spectroscopic Approach towards Neurodegenerative Diseases. Biomolecules 2019, 9, 495. [Google Scholar] [CrossRef]
- Choudhary, S.; Kesavan, A.K.; Juneja, V.; Thakur, S. Molecular Modeling, Simulation and Docking of Rv1250 Protein from Mycobacterium Tuberculosis. Front. Bioinform. 2023, 3, 1125479. [Google Scholar] [CrossRef]
- Belapure, J.; Sorokina, M.; Kastritis, P.L. IRAA: A Statistical Tool for Investigating a Protein–Protein Interaction Interface from Multiple Structures. Protein Sci. 2023, 32, e4523. [Google Scholar] [CrossRef]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. ING Estimates for the Year 2000 and Projections for 2030. World Health 2004, 27, 1047–1053. [Google Scholar]
- Zharkikh, E.; Dremin, V.; Zherebtsov, E.; Dunaev, A.; Meglinski, I. Biophotonics Methods for Functional Monitoring of Complications of Diabetes Mellitus. J. Biophotonics 2020, 13, e202000203. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, Z.; Gong, R. Long Noncoding RNA: An Emerging Player in Diabetes and Diabetic Kidney Disease. Clin. Sci. 2019, 133, 1321–1339. [Google Scholar] [CrossRef] [PubMed]
- Byard, R.W. A Review of the Potential Forensic Significance of Traditional Herbal Medicines. J. Forensic Sci. 2010, 55, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Nedungadi, P.; Salethoor, S.N.; Puthiyedath, R.; Nair, V.K.; Kessler, C.; Raman, R. Ayurveda Research: Emerging Trends and Mapping to Sustainable Development Goals. J. Ayurveda Integr. Med. 2023, 14, 100809. [Google Scholar] [CrossRef] [PubMed]
- Ullah, N.; Azam Khan, M.; Khan, T.; Ahmad, W. Protective Effect of Cinnamomum Tamala Extract on Gentamicin-Induced Nephrotic Damage in Rabbits. Trop. J. Pharm. Res. 2013, 12, 215–219. [Google Scholar] [CrossRef]
- Fakhruddin, S.; Alanazi, W.; Jackson, K.E. Diabetes-Induced Reactive Oxygen Species: Mechanism of Their Generation and Role in Renal Injury. J. Diabetes Res. 2017, 2017. [Google Scholar] [CrossRef]
- Kim, D.; Li, H.Y.; Lee, J.H.; Oh, Y.S.; Jun, H.S. Lysophosphatidic Acid Increases Mesangial Cell Proliferation in Models of Diabetic Nephropathy via Rac1/MAPK/KLF5 Signaling. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef]
- Xia, M.; Liu, D.; Liu, H.; Zhao, J.; Tang, C.; Chen, G.; Liu, Y.; Liu, H. Based on Network Pharmacology Tools to Investigate the Mechanism of Tripterygium Wilfordii Against IgA Nephropathy. Front. Med. 2021, 8, 794962. [Google Scholar] [CrossRef]
- Luan, Z.L.; Zhang, C.; Ming, W.H.; Huang, Y.Z.; Guan, Y.F.; Zhang, X.Y. Nuclear Receptors in Renal Health and Disease. eBioMedicine 2022, 76, 103855. [Google Scholar] [CrossRef]
- Makino, Y.; Haneda, M. Diabetic Nephropathy and Transcription Factors. Diabetol. Int. 2016, 7, 1–3. [Google Scholar] [CrossRef]
- Althubiti, M. Tyrosine Kinase Targeting: A Potential Therapeutic Strategy for Diabetes. Saudi J. Med. Med. Sci. 2022, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Jia, K.; Wang, H.; Gao, F.; Zhao, S.; Li, F.; Hao, J. METTL14-Regulated PI3K/Akt Signaling Pathway via PTEN Affects HDAC5-Mediated Epithelial–Mesenchymal Transition of Renal Tubular Cells in Diabetic Kidney Disease. Cell Death Dis. 2021, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Uzu, T.; Isshiki, K.; Koya, D. Peroxisome Proliferator-Activated Receptors in Diabetic Nephropathy. PPAR Res. 2008, 2008, 879523. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Gao, P. Diabetes Mellitus and Risk of Hepatocellular Carcinoma. BioMed Res. Int. 2017, 2017, 5202684. [Google Scholar] [CrossRef] [PubMed]
- Jayashankar, C.A.; Manohar, A.; Joshi, A.; Dwarakanathan, V.; Pinnelli, V.B.K.; Sarathi, V.; Gada, L.M. Association of Serum Prolactin with Type 2 Diabetes Mellitus: A Comparative Cross-Sectional Study From South India. Cureus 2022, 14, e23721. [Google Scholar] [CrossRef]
- Febres-Aldana, C.A.; Poppiti, R.; Varlotto, J.M.; Voland, R.; Zaleski, M.; Sharzehi, S.; Rassaei, N. Diabetes Mellitus Type 2 Is Associated with Increased Tumor Expression of Programmed Death-Ligand 1 (PD-L1) in Surgically Resected Non-Small Cell Lung Cancer—A Matched Case-Control Study. Cancer Treat. Res. Commun. 2020, 23, 100170. [Google Scholar] [CrossRef]
- Lu, Q.; Wang, W.; Zhang, M.; Ma, Z.; Qiu, X.; Shen, M.; Yin, X. ROS Induces Epithelial-mesenchymal Transition via the TGF-β1/PI3K/Akt/MTOR Pathway in Diabetic Nephropathy. Exp. Ther. Med. 2018, 17, 835–846. [Google Scholar] [CrossRef]
- Huang, W.J.; Fu, Q.; Xiao, Y.H.; Gong, Q.; Wu, W.J.; Shen, Z.L.; Zhang, H.; Jia, X.; Huang, X.M.; Zhang, Y.X.; et al. Effect of Qufengtongluo Decoction on PI3K/Akt Signaling Pathway in the Kidney of Type 2 Diabetes Mellitus Rat (GK Rat) with Diabetic Nephropathy. Evid. Based Complement. Altern. Med. 2018, 2018, 8421979. [Google Scholar] [CrossRef]
- Rane, M.J.; Song, Y.; Jin, S.; Barati, M.T.; Wu, R.; Kausar, H.; Tan, Y.; Wang, Y.; Zhou, G.; Klein, J.B.; et al. Interplay between Akt and P38 MAPK Pathways in the Regulation of Renal Tubular Cell Apoptosis Associated with Diabetic Nephropathy. Am. J. Physiol. Ren. Physiol. 2010, 298, F49–F61. [Google Scholar] [CrossRef]
- Hou, D.; Shang, S.; Lv, J.; Wang, S. Tripterygium Glycoside Ameliorates Kidney Injury in Diabetic Rats by Regulating the PI3K/Akt Signaling Pathway. Food Sci. Technol. 2022, 42, e124721. [Google Scholar] [CrossRef]
- Marrero, M.B.; Banes-Berceli, A.K.; Stern, D.M.; Eaton, D.C. Role of the JAK/STAT Signaling Pathway in Diabetic Nephropathy. Am. J. Physiol. Ren. Physiol. 2006, 290, 762–768. [Google Scholar] [CrossRef] [PubMed]
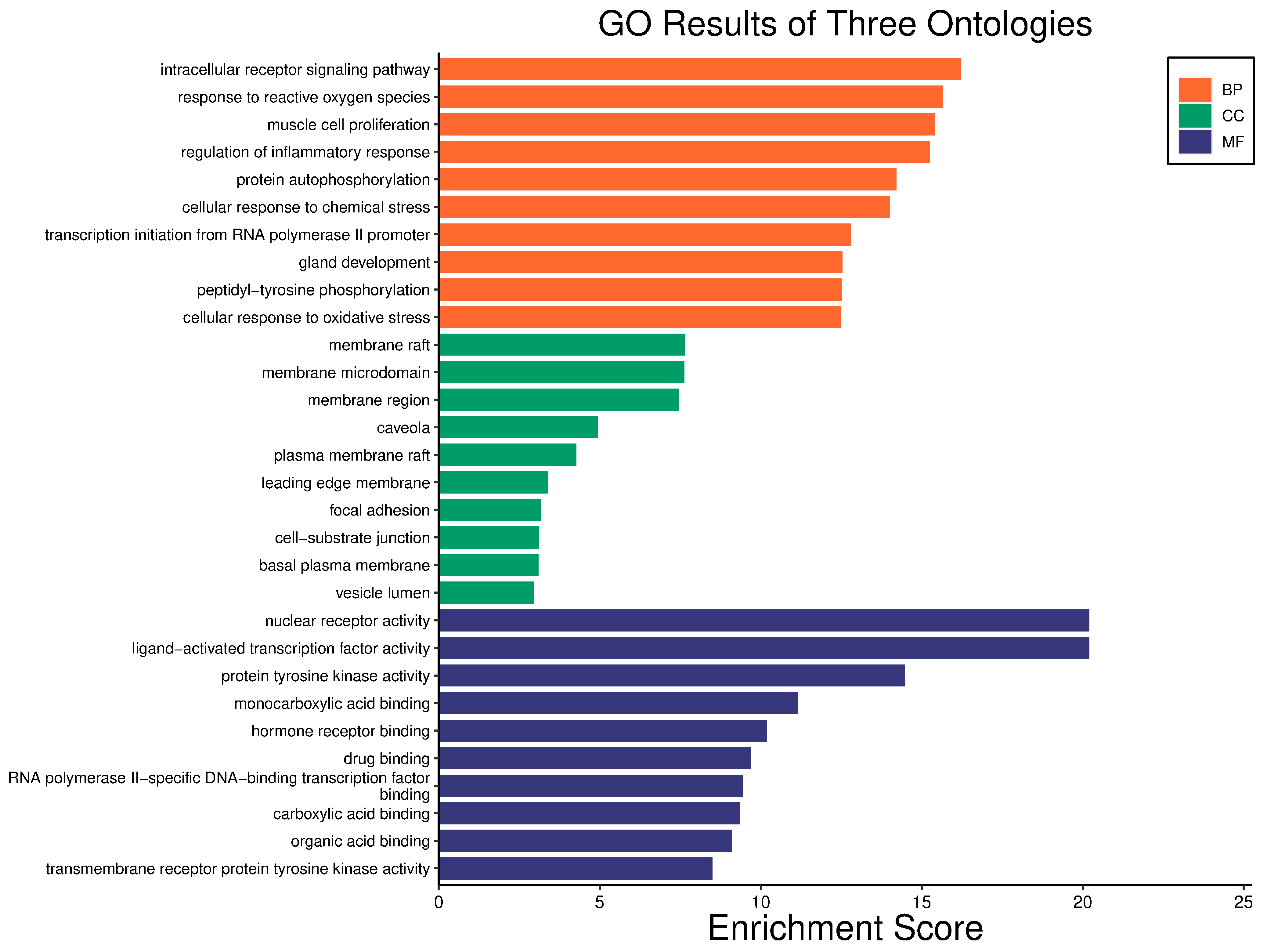



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COMPOUND | CID | SMILES | BS | DL | Molecular Weight (g/mol) | Num. Heavy Atoms | TPSA(Å2) | GI Absorption |
|---|---|---|---|---|---|---|---|---|
| Cubebol | 11276107 | CC1CCC(C2C13C2C(CC3)(C)O)C(C)C | 0.55 | 0.72 | 222.37 | 16 | 20.23 | High |
| Methoxyeugenol | 226486 | COC1=CC(=CC(=C1O)OC)CC=C | 0.55 | 0.75 | 194.23 | 14 | 38.69 | High |
| Benzyl benzoate | 2345 | C1=CC=C(C=C1)COC(=O)C2=CC=CC=C2 | 0.55 | 0.73 | 212.24 | 16 | 26.30 | High |
| Isoeugenol | 853433 | CC=CC1=CC(=C(C=C1)O)OC | 0.55 | 0.73 | 164.20 | 12 | 29.46 | High |
| Elemol | 92138 | CC(=C)C1CC(CCC1(C)C=C)C(C)(C)O | 0.55 | 0.72 | 222.37 | 16 | 20.23 | High |
| beta-Bisabolol | 12300146 | CC1=CCC(CC1)(C(C)CCC=C(C)C)O | 0.55 | 0.71 | 222.37 | 16 | 20.23 | High |
| Pathway ID | Description | p-Value | Gene IDs | Gene Count |
|---|---|---|---|---|
| hsa05205 | Proteoglycans in cancer | 1.05552 × 10−13 | SRC/FGFR1/PDPK1/EGFR/ESR1/MET/IGF1R/MMP9/TGFB2/CASP3/MDM2/ERBB4/AKT2/MMP2/GRB2/AKT1/CDC42 | 17 |
| hsa05215 | Prostate cancer | 5.88521 × 10−12 | MMP3/FGFR1/PDPK1/EGFR/IGF1R/GSTP1/MMP9/GSK3B/MDM2/AKT2/GRB2/AKT1 | 12 |
| hsa05417 | Lipid and atherosclerosis | 4.51366 × 10−11 | MMP3/SRC/PDPK1/RXRA/PPARG/MMP9/CASP3/CASP1/CASP7/NOS3/GSK3B/JAK2/AKT2/AKT1/CDC42 | 15 |
| hsa01522 | Endocrine resistance | 1.43526 × 10−10 | SRC/EGFR/ESR1/IGF1R/MMP9/ESR2/MDM2/AKT2/MMP2/GRB2/AKT1 | 11 |
| hsa05207 | Chemical carcinogenesis—receptor activation | 4.57818 × 10−10 | SRC/EGFR/RXRA/PPARA/ESR1/VDR/GSTM1/ESR2/JAK2/NR1I3/AKT2/GRB2/XIAP/AKT1 | 14 |
| hsa04151 | PI3K-Akt signaling pathway | 6.77448 × 10−10 | FGFR1/PDPK1/EGFR/RXRA/MET/IGF1R/IL2/INSR/NOS3/GSK3B/MDM2/ERBB4/JAK2/JAK3/AKT2/GRB2/AKT1 | 17 |
| hsa04917 | Prolactin signaling pathway | 2.27113 × 10−9 | SRC/ESR1/STAT1/ESR2/GSK3B/JAK2/AKT2/GRB2/AKT1 | 9 |
| hsa05223 | Non-small cell lung cancer | 2.93718 × 10−9 | PDPK1/EGFR/RXRA/MET/JAK3/AKT2/RARB/GRB2/AKT1 | 9 |
| hsa05225 | Hepatocellular carcinoma | 3.85957 × 10−9 | EGFR/MET/IGF1R/GSTP1/NQO1/TGFB2/GSTM1/GSK3B/AKT2/HMOX1/GRB2/AKT1 | 12 |
| hsa03320 | PPAR signaling pathway | 4.25749 × 10−9 | PDPK1/RXRA/PPARA/PPARG/FABP4/NR1H3/APOA2/FABP5/PPARD | 9 |
|
Benzyl Benzoate (2345) |
Beta-Bisabolol (12300146) |
Cubebol (11276107) |
Elemol (92138) |
Isoeugenol (853433) |
Methoxyeugenol (226486) | |
|---|---|---|---|---|---|---|
| AKT1 | −6.404 (−0.400) | −6.753 (−0.466) | −7.449 (−0.466) | −6.217 (−0.389) | −6.113 (−0.509) | −6.141 (−0.439) |
| CASP3 | −1.893 (−0.118) | −2.449 (−0.153) | −3.448 (−0.216) | −2.100 (−0.131) | −3.969 (−0.331) | −5.118 (−0.366) |
| EGFR | 0.099 (0.006) | −4.415 (−0.276) | −3.851 (−0.241) | −4.041 (−0.253) | −5.323 (−0.444) | −5.268 (−0.376) |
| ESR1 | −8.199 (−0.512) | −7.686 (−0.480) | −8.413 (−0.526) | −8.674 (−0.542) | −6.709 (−0.559) | −6.964 (−0.497) |
| GSK3B | −2.490 (−0.156) | −2.483 (−0.155) | −2.589 (−0.162) | −2.198 (−0.137) | −2.887 (−0.241) | −3.233 (−0.231) |
| IGF1R | −7.167 (−0.448) | −4.553 (−0.285) | −4.930 (−0.308) | −4.005 (−0.250) | −5.430 (−0.453) | −6.158 (−0.440) |
| JAK2 | −6.092 (−0.381) | −3.758 (−0.235) | −5.009 (−0.313) | −4.302 (−0.269) | −6.800 (−0.567) | −6.222 (−0.444) |
| MMP2 | _ | _ | _ | _ | −1.235 (−0.103) | −3.227 (−0.231) |
| MMP9 | −5.624 (−0.352) | −5.236 (−0.327) | −4.694 (−0.293) | −3.901 (−0.244) | −5.243 (−0.437) | −4.793 (−0.342) |
| SRC | −6.497 (−0.406) | −5.730 (−0.358) | −2.170 (−0.136) | −5.927 (−0.370) | −5.251 (−0.438) | −6.038 (−0.431) |
| System | ∆G or ∆GBind | ∆GCoulomb | ∆GCovalent | ∆GH-bond | ∆GSA or ∆GSol_Lipo | ∆GSolv or ∆GSolGB | ∆GPacking | ∆GvdW |
|---|---|---|---|---|---|---|---|---|
| ESR1–Elemol | −49.85 | −31.73 | 2.89 | −3.03 | −8.09 | 30.21 | −1.71 | −38.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, R.; Sahu, N.; Tyagi, R.; Alam, P.; Akhtar, A.; Walia, R.; Chandra, A.; Madan, S. Integrative Network Pharmacology, Molecular Docking, and Dynamics Simulations Reveal the Mechanisms of Cinnamomum tamala in Diabetic Nephropathy Treatment: An In Silico Study. Curr. Issues Mol. Biol. 2024, 46, 11868-11889. https://doi.org/10.3390/cimb46110705
Singh R, Sahu N, Tyagi R, Alam P, Akhtar A, Walia R, Chandra A, Madan S. Integrative Network Pharmacology, Molecular Docking, and Dynamics Simulations Reveal the Mechanisms of Cinnamomum tamala in Diabetic Nephropathy Treatment: An In Silico Study. Current Issues in Molecular Biology. 2024; 46(11):11868-11889. https://doi.org/10.3390/cimb46110705
Chicago/Turabian StyleSingh, Rashmi, Nilanchala Sahu, Rama Tyagi, Perwez Alam, Ali Akhtar, Ramanpreet Walia, Amrish Chandra, and Swati Madan. 2024. "Integrative Network Pharmacology, Molecular Docking, and Dynamics Simulations Reveal the Mechanisms of Cinnamomum tamala in Diabetic Nephropathy Treatment: An In Silico Study" Current Issues in Molecular Biology 46, no. 11: 11868-11889. https://doi.org/10.3390/cimb46110705
APA StyleSingh, R., Sahu, N., Tyagi, R., Alam, P., Akhtar, A., Walia, R., Chandra, A., & Madan, S. (2024). Integrative Network Pharmacology, Molecular Docking, and Dynamics Simulations Reveal the Mechanisms of Cinnamomum tamala in Diabetic Nephropathy Treatment: An In Silico Study. Current Issues in Molecular Biology, 46(11), 11868-11889. https://doi.org/10.3390/cimb46110705