
Spotlight on the Mechanism of Action of Semaglutide


Abstract
:1. Introduction
2. The Role of Semaglutide in Insulin Resistance
3. Mechanisms in the Role of Semaglutide in Obesity
3.1. Adipose Tissue Function
3.2. Fat Browning and Markers of Adipose Tissue Differentiation
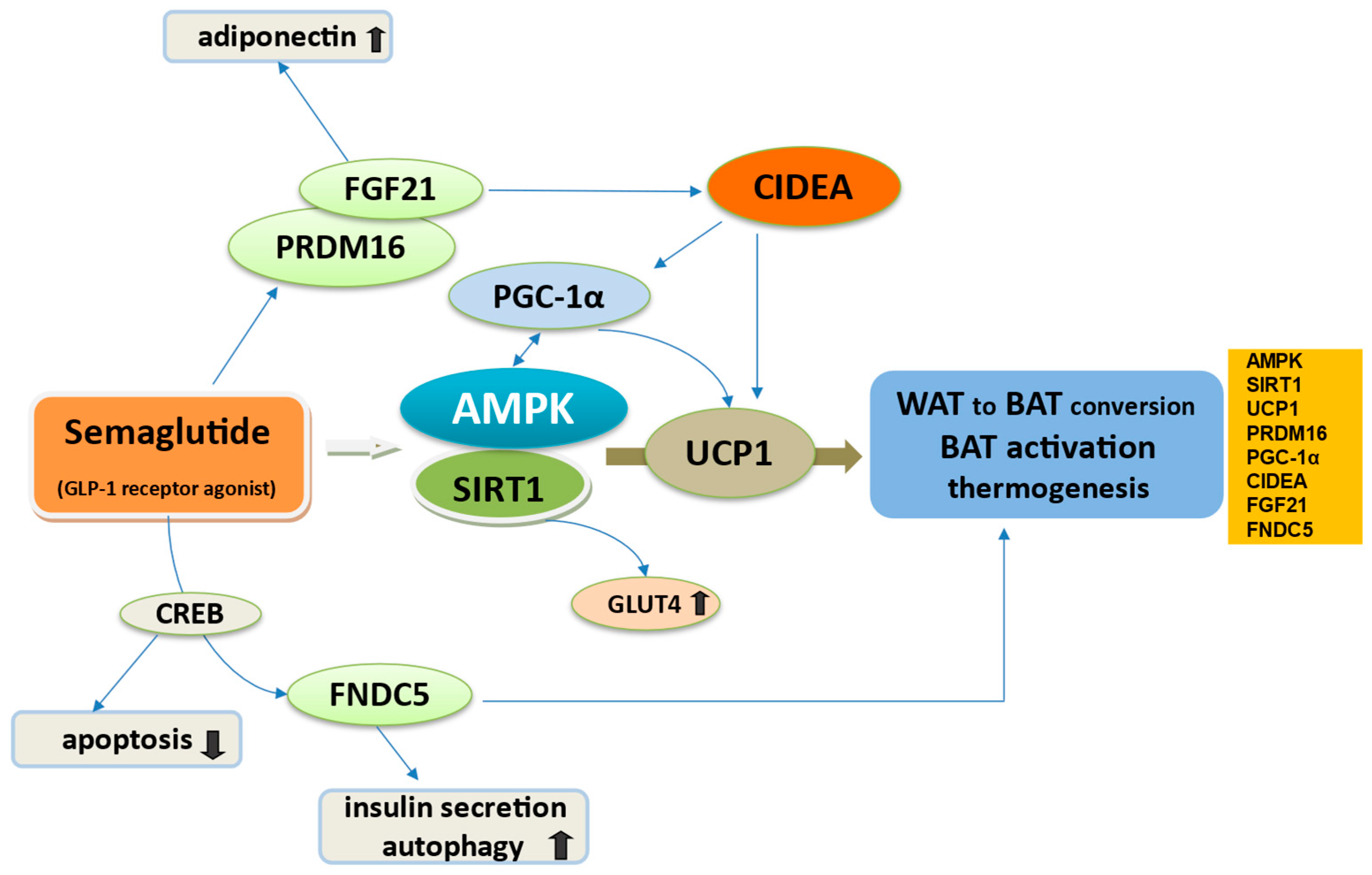
3.3. Semaglutide Regulates WAT to BAT Conversion
3.4. Action of Semaglutide in Dysfunctional Adiposity
4. The Role of Semaglutide in Appetite Regulation
4.1. Semaglutide and Direct GLP-1 Regulation of Appetite in the Brain
4.2. The Input of Semaglutide in Leptin Signaling
5. Semaglutide and Oxidative Stress
5.1. Drivers of Mitochondrial and Endoplasmic Reticulum Oxidative Stress
5.2. Semaglutide and Oxidative Stress Regulation
6. Semaglutide and Body Composition
6.1. Semaglutide Weight Loss Effects and Impact on Muscle Mass
6.2. Skeletal Muscle Malfunction in Obesity and Diabetes
6.3. Molecular Role of Semaglutide in Skeletal Muscle Homeostasis
6.3.1. Semaglutide Improves Obesity-Induced Muscle Atrophy via SIRT1
6.3.2. Semaglutide Improves Obesity-Induced Muscle Atrophy via Inhibition of UPS Degradation
7. Semaglutide and Anti-Aging
7.1. The Crossroad of GLP-1 and Cellular Senescence
7.2. Is Semaglutide the Amplifier of Life?
8. Semaglutide and Infections
9. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Bu, T.; Sun, Z.; Pan, Y.; Deng, X.; Yuan, G. Glucagon-like Peptide-1: New Regulator in Lipid Metabolism. Diabetes Metab. J. 2024, 48, 354–372. [Google Scholar] [CrossRef]
- Chobot, A.; Górowska-Kowolik, K.; Sokołowska, M.; Jarosz-Chobot, P. Obesity and diabetes-Not only a simple link between two epidemics. Diabetes Metab. Res. Rev. 2018, 34, e3042. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Gastaldelli, A.; Yki-Järvinen, H.; Scherer, P.E. Why does obesity cause diabetes? Cell Metab. 2022, 34, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Masenga, S.K.; Kabwe, L.S.; Chakulya, M.; Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 7898. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Aroda, V.; Collins, B.S.; Gabbay, R.A.; Green, J.; Maruthur, N.M.; Rosas, S.E.; Del Prato, S.; Mathieu, C.; Mingrone, G.; et al. Management of hyperglycemia in type 2 diabetes, 2022. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2022, 45, 2753–2786. [Google Scholar] [CrossRef]
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. Pharmacologic approaches to glycemic treatment: Standards of care in diabetes—2023. Diabetes Care 2023, 46, S140–S157. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Brown-Frandsen, K.; Colhoun, H.M.; Deanfield, J.; Emerson, S.S.; Esbjerg, S.; Hardt-Lindberg, S.; Hovingh, G.K.; Kahn, S.E.; Kushner, R.F.; et al. SELECT Trial Investigators. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N. Engl. J. Med. 2023, 389, 2221–2232. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.H.; Lingvay, I.; Deanfield, J.; Kahn, S.E.; Barros, E.; Burguera, B.; Colhoun, H.M.; Cercato, C.; Dicker, D.; Horn, D.B.; et al. Long-term weight loss effects of semaglutide in obesity without diabetes in the SELECT trial. Nat. Med. 2024, 30, 2049–2057. [Google Scholar] [CrossRef]
- Schaffer, R.; Heymsfield, S.B.; Garvey, T.W.; FDA Approves Once-Weekly Semaglutide for Weight Loss. Healio Website. 2021. Available online: https://www.healio.com/news/endocrinology/20210604/fda-approves-onceweekly-semaglutide-for-weight-loss (accessed on 24 September 2024).
- Chao, A.M.; Tronieri, J.S.; Amaro, A.; Wadden, T.A. Semaglutide for the treatment of obesity. Trends Cardiovasc. Med. 2023, 33, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Wilding, J.P.H.; Batterham, R.L.; Calanna, S.; Davies, M.; Van Gaal, L.F.; Lingvay, I.; McGowan, B.M.; Rosenstock, J.; Tran, M.T.D.; Wadden, T.A.; et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N. Engl. J. Med. 2021, 384, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Rubino, D.M.; Greenway, F.L.; Khalid, U.; O’Neil, P.M.; Rosenstock, J.; Sørrig, R.; Wadden, T.A.; Wizert, A.; Garvey, W.T.; STEP 8 Investigators; et al. Effect of Weekly Subcutaneous Semaglutide vs Daily Liraglutide on Body Weight in Adults with Overweight or Obesity without Diabetes: The STEP 8 Randomized Clinical Trial. JAMA 2022, 327, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Scirica, B.M.; Lincoff, A.M.; Lingvay, I.; Bogdanski, P.; Buscemi, S.; Colhoun, H.; Craciun, A.E.; Ezhov, M.; Hardt-Lindberg, S.; Kleist Jeppesen, O.; et al. The Effect of Semaglutide on Mortality and COVID-19-Related Deaths: An Analysis From the SELECT Trial. J. Am. Coll. Cardiol. 2024, 84, 1632–1642. [Google Scholar] [CrossRef]
- Cornell, S. A review of GLP-1 receptor agonists in type 2 diabetes: A focus on the mechanism of action of once-weekly agents. J. Clin. Pharm. Ther. 2020, 45, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Blundell, J.; Finlayson, G.; Axelsen, M.; Flint, A.; Gibbons, C.; Kvist, T.; Hjerpsted, J.B. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes. Metab. 2017, 19, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. GLP-1 physiology informs the pharmacotherapy of obesity. Mol. Metab. 2021, 57, 101351. [Google Scholar] [CrossRef]
- Biddinger, J.E.; Lazarenko, R.M.; Scott, M.M.; Simerly, R. Leptin suppresses development of GLP-1 inputs to the paraventricular nucleus of the hypothalamus. eLife 2020, 9, e59857. [Google Scholar] [CrossRef] [PubMed]
- Angelini, G.; Russo, S.; Mingrone, G. Incretin hormones, obesity and gut microbiota. Peptides 2024, 178, 171216. [Google Scholar] [CrossRef]
- Lebrun, L.J.; Lenaerts, K.; Kiers, D.; Pais de Barros, J.P.; Le Guern, N.; Plesnik, J.; Thomas, C.; Bourgeois, T.; Dejong, C.H.; Kox, M.; et al. Enteroendocrine L Cells Sense LPS after Gut Barrier Injury to Enhance GLP-1 Secretion. Cell Rep. 2017, 21, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, M.; Wen, Z.; Lu, Z.; Cui, L.; Fu, C.; Xue, H.; Liu, Y.; Zhang, Y. GLP-1 Receptor Agonists: Beyond Their Pancreatic Effects. Front. Endocrinol. 2021, 12, 721135. [Google Scholar] [CrossRef]
- Tamayo-Trujillo, R.; Ruiz-Pozo, V.A.; Cadena-Ullauri, S.; Guevara-Ramírez, P.; Paz-Cruz, E.; Zambrano-Villacres, R.; Simancas-Racines, D.; Zambrano, A.K. Molecular mechanisms of semaglutide and liraglutide as a therapeutic option for obesity. Front. Nutr. 2024, 11, 1398059. [Google Scholar] [CrossRef] [PubMed]
- Bednarz, K.; Kowalczyk, K.; Cwynar, M.; Czapla, D.; Czarkowski, W.; Kmita, D.; Nowak, A.; Madej, P. The Role of Glp-1 Receptor Agonists in Insulin Resistance with Concomitant Obesity Treatment in Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2022, 23, 4334. [Google Scholar] [CrossRef] [PubMed]
- Scheel, A.K.; Espelage, L.; Chadt, A. Many Ways to Rome: Exercise, Cold Exposure and Diet-Do They All Affect BAT Activation and WAT Browning in the Same Manner? Int. J. Mol. Sci. 2022, 23, 4759. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, B.; Sanchez-Delgado, G.; John, L.M.; Ryan, D.H.; Raun, K.; Ravussin, E. Beyond appetite regulation: Targeting energy expenditure, fat oxidation, and lean mass preservation for sustainable weight loss. Obesity 2022, 30, 841–857. [Google Scholar] [CrossRef]
- Chang, Y.F.; Zhang, D.; Hu, W.M.; Liu, D.X.; Li, L. Semaglutide-mediated protection against Aβ correlated with enhancement of autophagy and inhibition of apotosis. J. Clin. Neurosci. 2020, 81, 234–239. [Google Scholar] [CrossRef]
- Liu, S.; Yao, S.; Yang, H.; Liu, S.; Wang, Y. Autophagy: Regulator of cell death. Cell Death Dis. 2023, 14, 648. [Google Scholar] [CrossRef]
- Kitada, M.; Koya, D. Autophagy in metabolic disease and ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661. [Google Scholar] [CrossRef]
- Katsiki, N.; Ferrannini, E. Anti-inflammatory properties of antidiabetic drugs: A “promised land” in the COVID-19 era? J. Diabetes Complicat. 2020, 34, 107723. [Google Scholar] [CrossRef]
- Guarente, L.; Sinclair, D.A.; Kroemer, G. Human trials exploring anti-aging medicines. Cell Metab. 2024, 36, 354–376. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Tuttle, K.R.; Rossing, P.; Mahaffey, K.W.; Mann, J.F.E.; Bakris, G.; Baeres, F.M.M.; Idorn, T.; Bosch-Traberg, H.; Lausvig, N.L.; et al. FLOW Trial Committees and Investigators. Effects of Semaglutide on Chronic Kidney Disease in Patients with Type 2 Diabetes. N. Engl. J. Med. 2024, 391, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Blazek, O.; Bakris, G.L. Slowing the Progression of Diabetic Kidney Disease. Cells 2023, 12, 1975. [Google Scholar] [CrossRef]
- Marzook, A.; Tomas, A.; Jones, B. The Interplay of Glucagon-like Peptide-1 Receptor Trafficking and Signalling in Pancreatic Beta Cells. Front. Endocrinol. 2021, 12, 678055. [Google Scholar] [CrossRef]
- Kolb, H.; Stumvoll, M.; Kramer, W.; Kempf, K.; Martin, S. Insulin translates unfavourable lifestyle into obesity. BMC Med. 2018, 16, 232. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuniga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tang, Y.; Luo, Y.; Gao, Y.; He, L. Role and mechanism of specialized pro-resolving mediators in obesity-associated insulin resistance. Lipids Health Dis. 2024, 23, 234. [Google Scholar] [CrossRef]
- La Sala, L.; Mrakic-Sposta, S.; Tagliabue, E.; Prattichizzo, F.; Micheloni, S.; Sangalli, E.; Specchia, C.; Uccellatore, A.C.; Lupini, S.; Spinetti, G.; et al. Circulating microRNA-21 is an early predictor of ROS-mediated damage in subjects with high risk of developing diabetes and in drug-naive T2D. Cardiovasc. Diabetol. 2019, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Ye, J. Mechanisms of insulin resistance in obesity. Front. Med. 2013, 7, 14–24. [Google Scholar] [CrossRef]
- De Fano, M.; Bartolini, D.; Tortoioli, C.; Vermigli, C.; Malara, M.; Galli, F.; Murdolo, G. Adipose Tissue Plasticity in Response to Pathophysiological Cues: A Connecting Link between Obesity and Its Associated Comorbidities. Int. J. Mol. Sci. 2022, 23, 5511. [Google Scholar] [CrossRef]
- Elagizi, A.; Kachur, S.; Lavie, C.J.; Carbone, S.; Pandey, A.; Ortega, F.B.; Milani, R.V. An Overview and Update on Obesity and the Obesity Paradox in Cardiovascular Diseases. Prog. Cardiovasc. Dis. 2018, 61, 142–150. [Google Scholar] [CrossRef]
- Zheng, Z.; Zong, Y.; Ma, Y.; Tian, Y.; Pang, Y.; Zhang, C.; Gao, J. Glucagon-like peptide-1 receptor: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 234. [Google Scholar] [CrossRef]
- Sharma, D.K.; Verma, S.; Vaidya, S.; Kalia, K.; Tiwari, V. Recent updates on GLP-1 agonists: Current advancements and challenges. Biomed. Pharmacother. 2018, 108, 952–962. [Google Scholar] [CrossRef]
- Mariam, Z.; Niazi, S.K. Glucagon-like peptide agonists: A prospective review. Endocrinol. Diabetes Metab. 2024, 7, e462. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, J.; Heng, J.; Newsholme, P.; Carlessi, R. Pleiotropic Effects of GLP-1 and Analogs on Cell Signaling, Metabolism, and Function. Front. Endocrinol. 2018, 9, 672. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Zhang, B.; Wu, B.; Xiao, H.; Li, Z.; Li, R.; Xu, X.; Li, T. Signaling pathways in obesity: Mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 298. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Choi, S.E.; Ha, E.S.; Lee, H.B.; Kim, T.H.; Han, S.J.; Kim, H.J.; Kim, D.J.; Kang, Y.; Lee, K.W. GLP-1 improves palmitate-induced insulin resistance in human skeletal muscle via SIRT1 activity. Int. J. Mol. Med. 2019, 44, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Feng, P.; Zhang, X.; Li, D.; Wang, R.; Ji, C.; Li, G.; Hölscher, C. The diabetes drug semaglutide reduces infarct size, inflammation, and apoptosis, and normalizes neurogenesis in a rat model of stroke. Neuropharmacology 2019, 158, 107748. [Google Scholar] [CrossRef]
- Rodríguez Jiménez, B.; Rodríguez de Vera Gómez, P.; Belmonte Lomas, S.; Mesa Díaz, Á.M.; Caballero Mateos, I.; Galán, I.; Morales Portillo, C.; Martínez-Brocca, M.A. Transforming body composition with semaglutide in adults with obesity and type 2 diabetes mellitus. Front. Endocrinol. 2024, 15, 1386542. [Google Scholar] [CrossRef]
- Hropot, T.; Herman, R.; Janez, A.; Lezaic, L.; Jensterle, M. Brown Adipose Tissue: A New Potential Target for Glucagon-like Peptide 1 Receptor Agonists in the Treatment of Obesity. Int. J. Mol. Sci. 2023, 24, 8592. [Google Scholar] [CrossRef] [PubMed]
- Reguero, M.; Gómez de Cedrón, M.; Wagner, S.; Reglero, G.; Quintela, J.C.; Ramírez de Molina, A. Precision Nutrition to Activate Thermogenesis as a Complementary Approach to Target Obesity and Associated-Metabolic-Disorders. Cancers 2021, 13, 866. [Google Scholar] [CrossRef]
- Sakers, A.; De Siqueira, M.K.; Seale, P.; Villanueva, C.J. Adipose-tissue plasticity in health and disease. Cell 2022, 185, 419–446. [Google Scholar] [CrossRef]
- Gómez-Hernández, A.; de Las Heras, N.; Gálvez, B.G.; Fernández-Marcelo, T.; Fernández-Millán, E.; Escribano, Ó. New Mediators in the Crosstalk between Different Adipose Tissues. Int. J. Mol. Sci. 2024, 25, 4659. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Z.; Xiao, Y.; Li, X. Cross-talks between perivascular adipose tissue and neighbors: Multifaceted nature of nereids. Front. Pharmacol. 2024, 15, 1442086. [Google Scholar] [CrossRef]
- Cypess, A.M. Reassessing Human Adipose Tissue. N. Engl. J. Med. 2022, 386, 768–779. [Google Scholar] [CrossRef]
- Brownstein, A.J.; Veliova, M.; Acin-Perez, R.; Liesa, M.; Shirihai, O.S. ATP-consuming futile cycles as energy dissipating mechanisms to counteract obesity. Rev. Endocr. Metab. Disord. 2022, 23, 121–131. [Google Scholar] [CrossRef]
- Noriega, L.; Yang, C.Y.; Wang, C.H. Brown Fat and Nutrition: Implications for Nutritional Interventions. Nutrients 2023, 15, 4072. [Google Scholar] [CrossRef] [PubMed]
- Lockie, S.H.; Stefanidis, A.; Oldfield, B.J.; Perez-Tilve, D. Brown adipose tissue thermogenesis in the resistance to and reversal of obesity: A potential new mechanism contributing to the metabolic benefits of proglucagon-derived peptides. Adipocyte 2013, 2, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y. Thermogenic Brown Fat in Humans: Implications in Energy Homeostasis, Obesity and Metabolic Disorders. World J. Men’s Health 2023, 41, 489–507. [Google Scholar] [CrossRef]
- Czech, M.P. Mechanisms of insulin resistance related to white, beige, and brown adipocytes. Mol. Metab. 2020, 34, 27–42. [Google Scholar] [CrossRef]
- Radziszewska, M.; Ostrowska, L.; Smarkusz-Zarzecka, J. The Impact of Gastrointestinal Hormones on Human Adipose Tissue Function. Nutrients 2024, 16, 3245. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- Mo, Y.Y.; Han, Y.X.; Xu, S.N.; Jiang, H.L.; Wu, H.X.; Cai, J.M.; Li, L.; Bu, Y.H.; Xiao, F.; Liang, H.D.; et al. Adipose Tissue Plasticity: A Comprehensive Definition and Multidimensional Insight. Biomolecules 2024, 14, 1223. [Google Scholar] [CrossRef] [PubMed]
- Kojta, I.; Chaci’nska, M.; Błachnio-Zabielska, A. Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients 2020, 12, 1305. [Google Scholar] [CrossRef] [PubMed]
- De Fano, M.; Malara, M.; Vermigli, C.; Murdolo, G. Adipose Tissue: A Novel Target of the Incretin Axis? A Paradigm Shift in Obesity-Linked Insulin Resistance. Int. J. Mol. Sci. 2024, 25, 8650. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Farias, M.; Fos-Domenech, J.; Serra, D.; Herrero, L.; Sánchez-Infantes, D. White adipose tissue dysfunction in obesity and aging. Biochem. Pharmacol. 2021, 192, 114723. [Google Scholar] [CrossRef]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef]
- Funcke, J.B.; Scherer, P.E. Beyond adiponectin and leptin: Adipose tissue-derived mediators of inter-organ communication. J. Lipid Res. 2019, 60, 1648–1684. [Google Scholar] [CrossRef] [PubMed]
- Flori, L.; Piragine, E.; Spezzini, J.; Citi, V.; Calderone, V.; Martelli, A. Influence of Polyphenols on Adipose Tissue: Sirtuins as Pivotal Players in the Browning Process. Int. J. Mol. Sci. 2023, 24, 9276. [Google Scholar] [CrossRef]
- An, S.M.; Cho, S.H.; Yoon, J.C. Adipose Tissue and Metabolic Health. Diabetes Metab. J. 2023, 47, 595–611. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, J.; Dai, H.; Duan, Y.; An, Y.; Shi, L.; Lv, Y.; Li, H.; Wang, C.; Ma, Q.; et al. Brown and beige adipose tissue: A novel therapeutic strategy for obesity and type 2 diabetes mellitus. Adipocyte 2021, 10, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Challa, T.D.; Beaton, N.; Arnold, M.; Rudofsky, G.; Langhans, W.; Wolfrum, C. Regulation of adipocyte formation by GLP-1/GLP-1R signaling. J. Biol. Chem. 2012, 287, 6421–6430. [Google Scholar] [CrossRef]
- Singh, R.; Barrios, A.; Dirakvand, G.; Pervin, S. Human Brown Adipose Tissue and Metabolic Health: Potential for Therapeutic Avenues. Cells 2021, 10, 3030. [Google Scholar] [CrossRef]
- Malavazos, A.E.; Iacobellis, G.; Dozio, E.; Basilico, S.; Di Vincenzo, A.; Dubini, C.; Menicanti, L.; Vianello, E.; Meregalli, C.; Ruocco, C.; et al. Human epicardial adipose tissue expresses glucose-dependent insulinotropic polypeptide, glucagon, and glucagon-like peptide-1 receptors as potential targets of pleiotropic therapies. Eur. J. Prev. Cardiol. 2023, 30, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.F.; Martins, B.C.; Teixeira, A.V.S.; Ajackson, M.; Souza-Mello, V.; Daleprane, J.B. Brown Adipose Tissue, Batokines, and Bioactive Compounds in Foods: An Update. Mol. Nutr. Food Res. 2024, 68, e2300634. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tao, Z.; Zheng, L.; Yang, J.; Hu, X.; Scott, K.; de Kloet, A.; Krause, E.; Collins, J.F.; Cheng, Z. FoxO1 regulates adipose transdifferentiation and iron influx by mediating Tgfβ1 signaling pathway. Redox Biol. 2023, 63, 102727. [Google Scholar] [CrossRef] [PubMed]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef]
- Townsend, L.K.; Steinberg, G.R. AMPK and the Endocrine Control of Metabolism. Endocr. Rev. 2023, 44, 910–933. [Google Scholar] [CrossRef]
- Li, M.; Meng, N.; Guo, X.; Niu, X.; Zhao, Z.; Wang, W.; Xie, X.; Lv, P. Dl-3-n-Butylphthalide Promotes Remyelination and Suppresses Inflammation by Regulating AMPK/SIRT1 and STAT3/NF-κB Signaling in Chronic Cerebral Hypoperfusion. Front. Aging Neurosci. 2020, 12, 137. [Google Scholar] [CrossRef]
- Ahmad, B.; Serpell, C.J.; Fong, I.L.; Wong, E.H. Molecular Mechanisms of Adipogenesis: The Anti-adipogenic Role of AMP-Activated Protein Kinase. Front. Mol. Biosci. 2020, 7, 76. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, H.; Zhang, C.; He, J.; Wang, Y. Semaglutide alleviates early brain injury following subarachnoid hemorrhage by suppressing ferroptosis and neuroinflammation via SIRT1 pathway. Am. J. Transl. Res. 2024, 16, 1102–1117. [Google Scholar] [CrossRef]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Cero, C.; Lea, H.J.; Zhu, K.Y.; Shamsi, F.; Tseng, Y.H.; Cypess, A.M. β3-Adrenergic receptors regulate human brown/beige adipocyte lipolysis and thermogenesis. JCI Insight 2021, 6, e139160. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Cai, B.; Nie, Q. PGC-1α affects skeletal muscle and adipose tissue development by regulating mitochondrial biogenesis. Mol. Genet. Genom. 2022, 297, 621–633. [Google Scholar] [CrossRef]
- Bouviere, J.; Fortunato, R.S.; Dupuy, C.; Werneck-de-Castro, J.P.; Carvalho, D.P.; Louzada, R.A. Exercise-stimulated ROS sensitive signaling pathways in skeletal muscle. Antioxidants 2021, 10, 537. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a Nodal Regulator of Mitochondrial Biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef]
- Kleiner, S.; Douris, N.; Fox, E.C.; Mepani, R.J.; Verdeguer, F.; Wu, J.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E.; Spiegelman, B.M. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012, 26, 271–281. [Google Scholar]
- Hemat Jouy, S.; Mohan, S.; Scichilone, G.; Mostafa, A.; Mahmoud, A.M. Adipokines in the Crosstalk between Adipose Tissues and Other Organs: Implications in Cardiometabolic Diseases. Biomedicines 2024, 12, 2129. [Google Scholar] [CrossRef]
- Cuevas-Ramos, D.; Mehta, R.; Aguilar-Salinas, C.A. Fibroblast Growth Factor 21 and Browning of White Adipose Tissue. Front. Physiol. 2019, 10, 37. [Google Scholar] [CrossRef]
- Tan, H.; Yue, T.; Chen, Z.; Wu, W.; Xu, S.; Weng, J. Targeting FGF21 in cardiovascular and metabolic diseases: From mechanism to medicine. Int. J. Biol. Sci. 2023, 19, 66–88. [Google Scholar] [CrossRef]
- Feng, J.N.; Shao, W.; Jin, T. Short-term semaglutide treatment improves FGF21 responsiveness in primary hepatocytes isolated from high fat diet challenged mice. Physiol. Rep. 2023, 11, e15620. [Google Scholar] [CrossRef]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Tu, Y.; Liu, J.; Kong, D.; Guo, X.; Li, J.; Long, Z.; Peng, J.; Wang, Z.; Wu, H.; Liu, P.; et al. Irisin drives macrophage anti-inflammatory differentiation via JAK2-STAT6-dependent activation of PPAR gamma and Nrf2 signaling. Free Radic. Biol. Med. 2023, 201, 98–110. [Google Scholar] [CrossRef]
- Marrano, N.; Biondi, G.; Borrelli, A.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F.; Natalicchio, A. Irisin and Incretin Hormones: Similarities, Differences, and Implications in Type 2 Diabetes and Obesity. Biomolecules 2021, 11, 286. [Google Scholar] [CrossRef]
- Li, H.; Donelan, W.; Wang, F.; Zhang, P.; Yang, L.; Ding, Y.; Tang, D.; Li, S. GLP-1 Induces the Expression of FNDC5 Derivatives That Execute Lipolytic Actions. Front. Cell Dev. Biol. 2021, 9, 777026. [Google Scholar] [CrossRef]
- Martins, F.F.; Marinho, T.S.; Cardoso, L.E.M.; Barbosa-da-Silva, S.; Souza-Mello, V.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Semaglutide (GLP-1 receptor agonist) stimulates browning on subcutaneous fat adipocytes and mitigates inflammation and endoplasmic reticulum stress in visceral fat adipocytes of obese mice. Cell Biochem. Funct. 2022, 40, 903–913. [Google Scholar] [CrossRef]
- Vecchie, A.; Dallegri, F.; Carbone, F.; Bonaventura, A.; Liberale, L.; Portincasa, P.; Fruhbeck, G.; Montecucco, F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur. J. Intern. Med. 2018, 48, 6–17. [Google Scholar] [CrossRef]
- White, U. Adipose tissue expansion in obesity, health, and disease. Front. Cell Dev. Biol. 2023, 11, 1188844. [Google Scholar] [CrossRef] [PubMed]
- Bakinowska, E.; Krompiewski, M.; Boboryko, D.; Kiełbowski, K.; Pawlik, A. The Role of Inflammatory Mediators in the Pathogenesis of Obesity. Nutrients 2024, 16, 2822. [Google Scholar] [CrossRef]
- Kang, K.W.; Ok, M.; Lee, S.K. Leptin as a Key between Obesity and Cardiovascular Disease. J. Obes. Metab. Syndr. 2020, 29, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Feng, X.; Li, Q.; Wang, Y.; Li, Q.; Hua, M. Adiponectin, TNF-α and Inflammatory Cytokines and Risk of Type 2 Diabetes: A Systematic Review and Meta-Analysis. Cytokine 2016, 86, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Straub, L.G.; Scherer, P.E. Metabolic Messengers: Adiponectin. Nat. Metab. 2019, 1, 334–339. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Vatner, D.F.; Goedeke, L.; Hirabara, S.M.; Zhang, Y.; Perry, R.J.; Shulman, G.I. Mechanisms by Which Adiponectin Reverses High Fat Diet-Induced Insulin Resistance in Mice. Proc. Natl. Acad. Sci. USA 2020, 117, 32584–32593. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, M.; Okada-Iwabu, M.; Yamauchi, T.; Kadowaki, T. Adiponectin/AdipoR Research and Its Implications for Lifestyle-Related Diseases. Front. Cardiovasc. Med. 2019, 6, 116. [Google Scholar] [CrossRef]
- Cushing, E.M.; Chi, X.; Sylvers, K.L.; Shetty, S.K.; Potthoff, M.J.; Davies, B.S.J. Angiopoietin-like 4 directs uptake of dietary fat away from adipose during fasting. Mol. Metab. 2017, 6, 809–818. [Google Scholar] [CrossRef]
- Li, X.; Ren, Y.; Chang, K.; Wu, W.; Griffiths, H.R.; Lu, S.; Gao, D. Adipose tissue macrophages as potential targets for obesity and metabolic diseases. Front. Immunol. 2023, 14, 1153915. [Google Scholar] [CrossRef]
- Rouault, C.; Pellegrinelli, V.; Schilch, R.; Cotillard, A.; Poitou, C.; Tordjman, J.; Sell, H.; Clément, K.; Lacasa, D. Roles of chemokine ligand-2 (CXCL2) and neutrophils in influencing endothelial cell function and inflammation of human adipose tissue. Endocrinology 2013, 154, 1069–1079. [Google Scholar] [CrossRef]
- Rakotoarivelo, V.; Variya, B.; Langlois, M.F.; Ramanathan, S. Chemokines in human obesity. Cytokine 2020, 127, 154953. [Google Scholar] [CrossRef]
- Guo, L.Y.; Yang, F.; Peng, L.J.; Li, Y.B.; Wang, A.P. CXCL2, a new critical factor and therapeutic target for cardiovascular diseases. Clin. Exp. Hypertens. 2020, 42, 428–437. [Google Scholar] [CrossRef]
- Riuzzi, F.; Chiappalupi, S.; Arcuri, C.; Giambanco, I.; Sorci, G.; Donato, R. S100 proteins in obesity: Liaisons dangereuses. Cell. Mol. Life Sci. 2020, 77, 129–147. [Google Scholar] [CrossRef]
- Pan, X.; Yang, L.; Wang, S.; Liu, Y.; Yue, L.; Chen, S. Semaglutide ameliorates obesity-induced cardiac inflammation and oxidative stress mediated via reduction of neutrophil Cxcl2, S100a8, and S100a9 expression. Mol. Cell. Biochem. 2024, 479, 1133–1147. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Sowers, J.R. Hypertension in Diabetes: An Update of Basic Mechanisms and Clinical Disease. Hypertension 2021, 78, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Chen, S. Proteomic analysis reveals semaglutide impacts lipogenic protein expression in epididymal adipose tissue of obese mice. Front. Endocrinol. 2023, 14, 1095432. [Google Scholar] [CrossRef]
- Varin, E.M.; Mulvihill, E.E.; Baggio, L.L.; Koehler, J.A.; Cao, X.; Seeley, R.J.; Drucker, D.J. Distinct Neural Sites of GLP-1R Expression Mediate Physiological versus Pharmacological Control of Incretin Action. Cell Rep. 2019, 27, 3371–3384.e3. [Google Scholar] [CrossRef]
- Caussy, C.; Aubin, A.; Loomba, R. The Relationship Between Type 2 Diabetes, NAFLD, and Cardiovascular Risk. Curr. Diabetes Rep. 2021, 21, 15. [Google Scholar] [CrossRef]
- Ard, J.; Fitch, A.; Fruh, S.; Herman, L. Weight Loss and Maintenance Related to the Mechanism of Action of Glucagon-like Peptide 1 Receptor Agonists. Adv. Ther. 2021, 38, 2821–2839. [Google Scholar] [CrossRef]
- Vilariño-García, T.; Polonio-González, M.L.; Pérez-Pérez, A.; Ribalta, J.; Arrieta, F.; Aguilar, M.; Obaya, J.C.; Gimeno-Orna, J.A.; Iglesias, P.; Navarro, J.; et al. Role of Leptin in Obesity, Cardiovascular Disease, and Type 2 Diabetes. Int. J. Mol. Sci. 2024, 25, 2338. [Google Scholar] [CrossRef] [PubMed]
- López-Ojeda, W.; Hurley, R.A. Glucagon-like Peptide 1: An Introduction and Possible Implications for Neuropsychiatry. J. Neuropsychiatry Clin. Neurosci. 2024, 36, A4-86. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; He, Y.; Bai, J.; Zhang, C.; Zhang, F.; Yang, Y.; Luo, H.; Yu, M.; Liu, H.; Tu, L.; et al. Hypothalamic Grb10 enhances leptin signalling and promotes weight loss. Nat. Metab. 2023, 5, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Wang, Z.J.; Li, X.R.; Chai, S.F.; Li, W.R.; Li, S.; Hou, M.; Li, J.L.; Ye, Y.C.; Cai, H.Y.; Hölscher, C.; et al. Semaglutide ameliorates cognition and glucose metabolism dysfunction in the 3xTg mouse model of Alzheimer’s disease via the GLP-1R/SIRT1/GLUT4 pathway. Neuropharmacology 2023, 240, 109716. [Google Scholar] [CrossRef]
- Laurindo, L.F.; Barbalho, S.M.; Guiguer, E.L.; da Silva Soares de Souza, M.; de Souza, G.A.; Fidalgo, T.M.; Araújo, A.C.; de Souza Gonzaga, H.F.; de Bortoli Teixeira, D.; de Oliveira Silva Ullmann, T.; et al. GLP-1a: Going beyond Traditional Use. Int. J. Mol. Sci. 2022, 23, 739. [Google Scholar] [CrossRef]
- Zeng, N.; Cutts, E.J.; Lopez, C.B.; Kaur, S.; Duran, M.; Virkus, S.A.; Hardaway, J.A. Anatomical and Functional Characterization of Central Amygdala Glucagon-like Peptide 1 Receptor Expressing Neurons. Front. Behav. Neurosci. 2021, 15, 724030. [Google Scholar] [CrossRef]
- Aranäs, C.; Edvardsson, C.E.; Shevchouk, O.T.; Zhang, Q.; Witley, S.; Blid Sköldheden, S.; Zentveld, L.; Vallöf, D.; Tufvesson-Alm, M.; Jerlhag, E. Semaglutide reduces alcohol intake and relapse-like drinking in male and female rats. EBioMedicine 2023, 93, 104642. [Google Scholar] [CrossRef]
- Perakakis, N.; Farr, O.M.; Mantzoros, C.S. Leptin in Leanness and Obesity: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 77, 745–760. [Google Scholar] [CrossRef]
- Liu, H.; Du, T.; Li, C.; Yang, G. STAT3 phosphorylation in central leptin resistance. Nutr. Metab. 2021, 18, 39. [Google Scholar] [CrossRef]
- Pereira, S.; Cline, D.L.; Glavas, M.M.; Covey, S.D.; Kieffer, T.J. Tissue-specific effects of leptin on glucose and lipid metabolism. Endocr. Rev. 2021, 42, 1–28. [Google Scholar] [CrossRef]
- Hamamah, S.; Amin, A.; Al-Kassir, A.L.; Chuang, J.; Covasa, M. Dietary Fat Modulation of Gut Microbiota and Impact on Regulatory Pathways Controlling Food Intake. Nutrients 2023, 15, 3365. [Google Scholar] [CrossRef] [PubMed]
- Ullah, R.; Rauf, N.; Nabi, G.; Yi, S.; Yu-Dong, Z.; Fu, J. Mechanistic insight into high-fat diet-induced metabolic inflammation in the arcuate nucleus of the hypothalamus. Biomed. Pharmacother. 2021, 142, 112012. [Google Scholar] [CrossRef]
- Zhao, S.; Kusminski, C.M.; Elmquist, J.K.; Scherer, P.E. Leptin: Less Is More. Diabetes 2020, 69, 823–829. [Google Scholar] [CrossRef]
- do Carmo, J.M.; da Silva, A.A.; Cai, Z.; Lin, S.; Dubinion, J.H.; Hall, J.E. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptorsin proopiomelanocortin neurons. Hypertension 2011, 57, 918–926. [Google Scholar] [CrossRef] [PubMed]
- .Martins, F.F.; Santos-Reis, T.; Marinho, T.S.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Hypothalamic anorexigenic signaling pathways (leptin, amylin, and proopiomelanocortin) are semaglutide (GLP-1 analog) targets in obesity control in mice. Life Sci. 2023, 313, 121268. [Google Scholar] [CrossRef]
- Maruhashi, T.; Higashi, Y. Pathophysiological Association between Diabetes Mellitus and Endothelial Dysfunction. Antioxidants 2021, 10, 1306. [Google Scholar] [CrossRef] [PubMed]
- Colak, E.; Pap, D. The Role of Oxidative Stress in the Development of Obesity and Obesity-Related Metabolic Disorders. J. Med. Biochem. 2021, 40, 1–9. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- DeVallance, E.; Li, Y.; Jurczak, M.J.; Cifuentes-Pagano, E.; Pagano, P.J. The Role of NADPH Oxidases in the Etiology of Obesity and Metabolic Syndrome: Contribution of Individual Isoforms and Cell Biology. Antioxid. Redox Signal. 2019, 31, 687–709. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Höhn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef]
- Lee, J.-H.; Lee, J. Endoplasmic Reticulum (ER) Stress and Its Role in Pancreatic β-Cell Dysfunction and Senescence in Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 4843. [Google Scholar] [CrossRef]
- Hotamisligi, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Omar-Hmeadi, M.; Idevall-Hagren, O. Insulin granule biogenesis and exocytosis. Cell. Mol. Life Sci. 2021, 78, 1957–1970. [Google Scholar] [CrossRef] [PubMed]
- De Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Veluthakal, R.; Esparza, D.; Hoolachan, J.M.; Balakrishnan, R.; Ahn, M.; Oh, E.; Jayasena, C.S.; Thurmond, D.C. Mitochondrial Dysfunction, Oxidative Stress, and Inter-Organ Miscommunications in T2D Progression. Int. J. Mol. Sci. 2024, 25, 1504. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cuevas, J.; Sandoval-Rodriguez, A.; Meza-Rios, A.; Monroy-Ramírez, H.C.; Galicia-Moreno, M.; García-Bañuelos, J.; Santos, A.; Armendariz-Borunda, J. Molecular Mechanisms of Obesity-Linked Cardiac Dysfunction: An Up-Date on Current Knowledge. Cells 2021, 10, 629. [Google Scholar] [CrossRef] [PubMed]
- Zamora, M.; Villena, J.A. Contribution of Impaired Insulin Signaling to the Pathogenesis of Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 2833. [Google Scholar] [CrossRef] [PubMed]
- Armandi, A.; Rosso, C.; Caviglia, G.P.; Bugianesi, E. Insulin Resistance across the Spectrum of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 155. [Google Scholar] [CrossRef] [PubMed]
- Daffu, G.; del Pozo, C.H.; O’Shea, K.M.; Ananthakrishnan, R.; Ramasamy, R.; Schmidt, A.M. Radical roles for RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and beyond. Int. J. Mol. Sci. 2013, 14, 19891–19910. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Receptor for AGE (RAGE): Signaling mechanisms in the pathogenesis of diabetes and its complications. Ann. N. Y. Acad. Sci. 2011, 1243, 88–102. [Google Scholar] [CrossRef]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, Y.; Shi, L.; Li, L.; Zhang, D.; Gong, Z.; Wu, Q. Activation and modulation of the AGEs-RAGE axis: Implications for inflammatory pathologies and therapeutic interventions—A review. Pharmacol. Res. 2024, 206, 107282. [Google Scholar] [CrossRef]
- Lombardo, G.E.; Russo, C.; Maugeri, A.; Navarra, M. Sirtuins as Players in the Signal Transduction of Citrus Flavonoids. Int. J. Mol. Sci. 2024, 25, 1956. [Google Scholar] [CrossRef]
- Wensveen, F.M.; Valenti’c, S.; Šestan, M.; Turk Wensveen, T.; Poli’c, B. The “Big Bang” in obese fat: Events initiating obesity-induced adipose tissue inflammation. Eur. J. Immunol. 2015, 45, 2446–2456. [Google Scholar] [CrossRef]
- Yan, M.; Lin, K.; Huang, D.; Li, J.; Qu, X.; Chen, K. Semaglutide attenuates pathological electrophysiological remodeling in diabetic cardiomyopathy via restoring Cx43 expression. Endocrine 2024, 84, 969–979. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Q.; Feng, X.; Liu, Y.; Zhou, Y. Mitochondrial Dysfunction in Cardiovascular Diseases: Potential Targets for Treatment. Front. Cell Dev. Biol. 2022, 10, 841523. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.M.; Lombardo, P.S.; Malik, N.; Brun, S.N.; Hellberg, K.; Van Nostrand, J.L.; Garcia, D.; Baumgart, J.; Diffenderfer, K.; Asara, J.M.; et al. AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Sci. Adv. 2021, 7, eabg4544. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Tuo, X.; Li, B.; Deng, Z.; Qiu, Y.; Xie, H. Semaglutide attenuates excessive exercise-induced myocardial injury through inhibiting oxidative stress and inflammation in rats. Life Sci. 2020, 250, 117531. [Google Scholar] [CrossRef]
- Yamagishi, S.; Fukami, K.; Matsui, T. Crosstalk between advanced glycation end products (AGEs)-receptor RAGE axis and dipeptidyl peptidase-4-incretin system in diabetic vascular complications. Cardiovasc. Diabetol. 2015, 14, 2. [Google Scholar] [CrossRef]
- Alharbi, S.H. Anti-inflammatory role of glucagon-like peptide 1 receptor agonists and its clinical implications. Ther. Adv. Endocrinol. Metab. 2024, 15, 20420188231222367. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Chen, S.; Chen, X.; Niu, S.; Yue, L.; Pan, X.; Li, Z.; Chen, X. An Effective Glucagon-like Peptide-1 Receptor Agonists, Semaglutide, Improves Sarcopenic Obesity in Obese Mice by Modulating Skeletal Muscle Metabolism. Drug Des. Dev. Ther. 2022, 16, 3723–3735. [Google Scholar] [CrossRef] [PubMed]
- Sargeant, J.A.; Henson, J.; King, J.A.; Yates, T.; Khunti, K.; Davies, M.J. A Review of the Effects of Glucagon-like Peptide-1 Receptor Agonists and Sodium-Glucose Cotransporter 2 Inhibitors on Lean Body Mass in Humans. Endocrinol. Metab. 2019, 34, 247–262. [Google Scholar] [CrossRef]
- Magkos, F.; Fraterrigo, G.; Yoshino, J.; Luecking, C.; Kirbach, K.; Kelly, S.C.; de las Fuentes, L.; He, S.; Okunade, A.L.; Patterson, B.W.; et al. Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab. 2016, 23, 591–601. [Google Scholar] [CrossRef]
- Bikou, A.; Dermiki-Gkana, F.; Penteris, M.; Constantinides, T.K.; Kontogiorgis, C. A systematic review of the effect of semaglutide on lean mass: Insights from clinical trials. Expert Opin. Pharmacother. 2024, 25, 611–619. [Google Scholar] [CrossRef]
- Volpe, M.; Gallo, G. Obesity and cardiovascular disease: An executive document on pathophysiological and clinical links promoted by the Italian Society of Cardiovascular Prevention (SIPREC). Front. Cardiovasc. Med. 2023, 10, 1136340. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, S.; Sada, Y.; Mihara, S.; Sasaki, Y.; Sone, M.; Tanaka, Y. Oral Semaglutide Induces Loss of Body Fat Mass without Affecting Muscle Mass in Patients with Type 2 Diabetes. J. Clin. Med. Res. 2023, 15, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, Y.; Masaki, T.; Kamata, A.; Miyamoto, S.; Yoshida, Y.; Okamoto, M.; Gotoh, K.; Shibata, H. The Effectiveness of GLP-1 Receptor Agonist Semaglutide on Body Composition in Elderly Obese Diabetic Patients: A Pilot Study. Medicines 2022, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, J.C.; Costa, J.G.; Haynes, A.; Naylor, L.H.; Fegan, P.G.; Yeap, B.B.; Green, D.J. Incretin-Based Weight Loss Pharmacotherapy: Can Resistance Exercise Optimize Changes in Body Composition? Diabetes Care 2024, 47, 1718–1730. [Google Scholar] [CrossRef]
- Hulett, N.A.; Scalzo, R.L.; Reusch, J.E.B. Glucose Uptake by Skeletal Muscle within the Contexts of Type 2 Diabetes and Exercise: An Integrated Approach. Nutrients 2022, 14, 647. [Google Scholar] [CrossRef] [PubMed]
- Virmani, M.A.; Cirulli, M. The Role of l-Carnitine in Mitochondria, Prevention of Metabolic Inflexibility and Disease Initiation. Int. J. Mol. Sci. 2022, 23, 2717. [Google Scholar] [CrossRef]
- Hong, S.H.; Choi, K.M. Sarcopenic Obesity, Insulin Resistance, and Their Implications in Cardiovascular and Metabolic Consequences. Int. J. Mol. Sci. 2020, 21, 494. [Google Scholar] [CrossRef]
- Lisco, G.; Disoteo, O.E.; De Tullio, A.; De Geronimo, V.; Giagulli, V.A.; Monzani, F.; Jirillo, E.; Cozzi, R.; Guastamacchia, E.; De Pergola, G.; et al. Sarcopenia and Diabetes: A Detrimental Liaison of Advancing Age. Nutrients 2023, 16, 63. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Li, C.W.; Yu, K.; Shyh-Chang, N.; Jiang, Z.; Liu, T.; Ma, S.; Luo, L.; Guang, L.; Liang, K.; Ma, W.; et al. Pathogenesis of sarcopenia and the relationship with fat mass: Descriptive review. J. Cachexia Sarcopenia Muscle 2022, 13, 781–794. [Google Scholar] [CrossRef]
- Morozzi, G.; Beccafico, S.; Bianchi, R.; Riuzzi, F.; Bellezza, I.; Giambanco, I.; Arcuri, C.; Minelli, A.; Donato, R. Oxidative stress-induced S100B accumulation converts myoblasts into brown adipocytes via an NF-κB/YY1/miR-133 axis and NF-κB/YY1/BMP-7 axis. Cell Death Differ. 2017, 24, 2077–2088. [Google Scholar] [CrossRef]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef]
- Gellhaus, B.; Böker, K.O.; Schilling, A.F.; Saul, D. Therapeutic Consequences of Targeting the IGF-1/PI3K/AKT/FOXO3 Axis in Sarcopenia: A Narrative Review. Cells 2023, 12, 2787. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ji, Y.; Liu, R.; Zhu, X.; Wang, K.; Yang, X.; Liu, B.; Gao, Z.; Huang, Y.; Shen, Y.; et al. Mitochondrial dysfunction: Roles in skeletal muscle atrophy. J. Transl. Med. 2023, 21, 503. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.Y.; Choi, H.M.; Yang, H.I.; Kim, K.S. Dysregulated Autophagy Mediates Sarcopenic Obesity and Its Complications via AMPK and PGC1α Signaling Pathways: Potential Involvement of Gut Dysbiosis as a Pathological Link. Int. J. Mol. Sci. 2020, 21, 6887. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, M.; Li, T.; Dong, N.; Yi, L.; Zhang, Q.; Mi, M. GLP-1 regulates exercise endurance and skeletal muscle remodeling via GLP-1R/AMPK pathway. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2022, 1869, 119300. [Google Scholar] [CrossRef]
- Xiang, J.; Qin, L.; Zhong, J.; Xia, N.; Liang, Y. GLP-1RA Liraglutide and Semaglutide Improves Obesity-Induced Muscle Atrophy via SIRT1 Pathway. Diabetes Metab. Syndr. Obes. 2023, 16, 2433–2446. [Google Scholar] [CrossRef]
- Szekeres, Z.; Nagy, A.; Jahner, K.; Szabados, E. Impact of Selected Glucagon-like Peptide-1 Receptor Agonists on Serum Lipids, Adipose Tissue, and Muscle Metabolism-A Narrative Review. Int. J. Mol. Sci. 2024, 25, 8214. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Kaji, K.; Nishimura, N.; Kubo, T.; Tomooka, F.; Shibamoto, A.; Suzuki, J.; Tsuji, Y.; Fujinaga, Y.; Kitagawa, K.; et al. Glucagon-like peptide-1 receptor agonist, semaglutide attenuates chronic liver disease-induced skeletal muscle atrophy in diabetic mice. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2023, 1869, 166770. [Google Scholar] [CrossRef]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Zhou, R.; Sun, Z.F.; Long, J.W.; Gong, Y.Q. Novel Insights into the Roles and Mechanisms of GLP-1 Receptor Agonists against Aging-Related Diseases. Aging Dis. 2022, 13, 468–490. [Google Scholar] [CrossRef] [PubMed]
- Rajawat, Y.S.; Hilioti, Z.; Bossis, I. Aging: Central role for autophagy and the lysosomal degradative system. Ageing Res. Rev. 2009, 8, 199–213. [Google Scholar] [CrossRef]
- Camacho-Encina, M.; Booth, L.K.; Redgrave, R.E.; Folaranmi, O.; Spyridopoulos, I.; Richardson, G.D. Cellular Senescence, Mitochondrial Dysfunction, and Their Link to Cardiovascular Disease. Cells 2024, 13, 353. [Google Scholar] [CrossRef] [PubMed]
- Wrona, M.V.; Ghosh, R.; Coll, K.; Chun, C.; Yousefzadeh, M.J. The 3 I’s of immunity and aging: Immunosenescence, inflammaging, and immune resilience. Front. Aging 2024, 5, 1490302. [Google Scholar] [CrossRef]
- Narasimhan, A.; Flores, R.R.; Robbins, P.D.; Niedernhofer, L.J. Role of Cellular Senescence in Type II Diabetes. Endocrinology 2021, 162, bqab136. [Google Scholar] [CrossRef]
- Ge, Y.; Zhou, M.; Chen, C.; Wu, X.; Wang, X. Role of AMPK mediated pathways in autophagy and aging. Biochimie 2022, 195, 100–113. [Google Scholar] [CrossRef]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The Senescence-Associated Secretory Phenotype (SASP) in the Challenging Future of Cancer Therapy and Age-Related Diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Yoshino, M.; Yoshino, J.; Smith, G.I.; Stein, R.I.; Bittel, A.J.; Bittel, D.C.; Reeds, D.N.; Sinacore, D.R.; Cade, W.T.; Patterson, B.W.; et al. Worksite-based intensive lifestyle therapy has profound cardiometabolic benefits in people with obesity and type 2 diabetes. Cell Metab. 2022, 34, 1431–1441.e5. [Google Scholar] [CrossRef] [PubMed]
- Pereira, Q.C.; Dos Santos, T.W.; Fortunato, I.M.; Ribeiro, M.L. The Molecular Mechanism of Polyphenols in the Regulation of Ageing Hallmarks. Int. J. Mol. Sci. 2023, 24, 5508. [Google Scholar] [CrossRef]
- Cheng, Y.; Pitoniak, A.; Wang, J.; Bohmann, D. Preserving transcriptional stress responses as an anti-aging strategy. Aging Cell 2021, 20, e13297. [Google Scholar] [CrossRef]
- Fernando, R.; Drescher, C.; Nowotny, K.; Grune, T.; Castro, J.P. Impaired proteostasis during skeletal muscle aging. Free Radic. Biol. Med. 2019, 132, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Jun, H.-S. Anti-diabetic actions of glucagon-like peptide-1 on pancreatic beta-cells. Metabolism 2014, 63, 9–19. [Google Scholar] [CrossRef]
- Vaiserman, A.; De Falco, E.; Koliada, A.; Maslova, O.; Balistreri, C.R. Anti-ageing gene therapy: Not so far away? Ageing Res. Rev. 2019, 56, 100977. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.J.; Sharma, A.; Mair, W.B. Metabolic Communication and Healthy Aging: Where Should We Focus Our Energy? Dev. Cell 2020, 54, 196–211. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; McLean, B.A.; Baggio, L.L.; Koehler, J.A.; Hammoud, R.; Rittig, N.; Yabut, J.M.; Seeley, R.J.; Brown, T.J.; Drucker, D.J. Central glucagon-like peptide 1 receptor activation inhibits Toll-like receptor agonist-induced inflammation. Cell Metab. 2024, 36, 130–143.e5. [Google Scholar] [CrossRef] [PubMed]
- de Vos, W.M.; Tilg, H.; Van Hul, M.; Cani, P.D. Gut microbiome and health: Mechanistic insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef]
- Wang, K.; Lai, W.; Min, T.; Wei, J.; Bai, Y.; Cao, H.; Guo, J.; Su, Z. The Effect of Enteric-Derived Lipopolysaccharides on Obesity. Int. J. Mol. Sci. 2024, 25, 4305. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zeng, F.; Luo, X.; Lei, Y.; Li, J.; Lu, S.; Huang, X.; Lan, Y.; Liu, R. GLP-1 Receptor: A New Target for Sepsis. Front. Pharmacol. 2021, 12, 706908. [Google Scholar] [CrossRef] [PubMed]
- Gabery, S.; Salinas, C.G.; Paulsen, S.J.; Ahnfelt-Rønne, J.; Alanentalo, T.; Baquero, A.F.; Buckley, S.T.; Farkas, E.; Fekete, C.; Frederiksen, K.S.; et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight 2020, 5, e133429. [Google Scholar] [CrossRef]
- Grieco, M.; Giorgi, A.; Gentile, M.C.; d’Erme, M.; Morano, S.; Maras, B.; Filardi, T. Glucagon-like Peptide-1: A Focus on Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1112. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, J. Semaglutide Pretreatment Induces Cardiac Autophagy to Reduce Myocardial Injury in Septic Mice. Discov. Med. 2023, 35, 853–860. [Google Scholar] [CrossRef]
- Mazzieri, A.; Basta, G.; Calafiore, R.; Luca, G. GLP-1 RAs and SGLT2i: Two antidiabetic agents associated with immune and inflammation modulatory properties through the common AMPK pathway. Front. Immunol. 2023, 14, 1163288. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Semaglutide | Biological Effects | Molecular Mechanisms | References |
|---|---|---|---|
| Pancreatic GLP-1 and insulin related actions | Insulin biosynthesis and secretion | cAMP-dependent-PKA/PI3K/mTOR cAMP/PKA/PI3K/GLUT4 transporter | [20,21,40,44,202] |
| Improves insulin sensitivity (in adipose tissue and muscles) | AMPK/SIRT1/GLUT4 transporter Upregulation of phosphorylated IRS-1 | [44,120,181] [46] | |
| Stimulates β-cell proliferation Inhibits β-cell apoptosis | PKA/PI3K downstream FOXO1 regulation AMPK, cAMP-activated CREB | [40] | |
| Inhibits glucagon release | [40,47] | ||
| Brain and appetite regulation actions | Appetite and food intake reduction, satiety inducer Increase energy expenditure | Direct stimulation of anorexigenic melanocortin, POMC/CART neurons Indirect inhibition of orexigenic NPY/AgRP | [17,125,131,203] [114,115,116,131] |
| Improves leptin sensitivity | GLP-1 interaction with anorexic leptin signaling | [131] | |
| Inhibition of suppressor PTP1B/SOCS3 | [125,129,131] | ||
| Inhibition of PTP1B counteracts leptin resistance | [116] | ||
| Neuroprotection | Hypothalamic AMPK/SIRT1 | [80,120,121] | |
| Adipose tissue responce | Enhances BAT activation via transcriptional regulators Enhances WAT browning | Upregulation of AMPK/SIRT1, UCP1, PRDM16, PGC-1α, CIDEA, FGF21, FNDC5 | [1,44,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,154] |
| Inhibits WAT inflammation | AMPK/SIRT1 NF-κB deacetylation S100a8, S100a9, Cxcl2 inhibition | [22,35,80,204] [110,111] | |
| WAT reduction | FGF21 up-regulation Lowering ANGPTL4 and LPL activity | [90,112] | |
| Muscle function | Improve skeletal muscle atrophy in obesity | Direct GLP-1R stimulation in myocytes cAMP-mediated PKA/AKT activation | [181,182,183] |
| Regulates autophagy | AMPK/SIRT1/PGC-1α activation | [40,83,177,179,180] | |
| Reduction in FOXO transcription, atrogin1, MuRF1 | [170,173,176,182,183] | ||
| Inhibition of NF-κB Inhibition of UPS-mediated skeletal muscle proteolysis | [181,183] | ||
| May inhibit the transition of myoblasts into adipocytes | May inhibit the anti-myogenic S100B | [110,173,174] | |
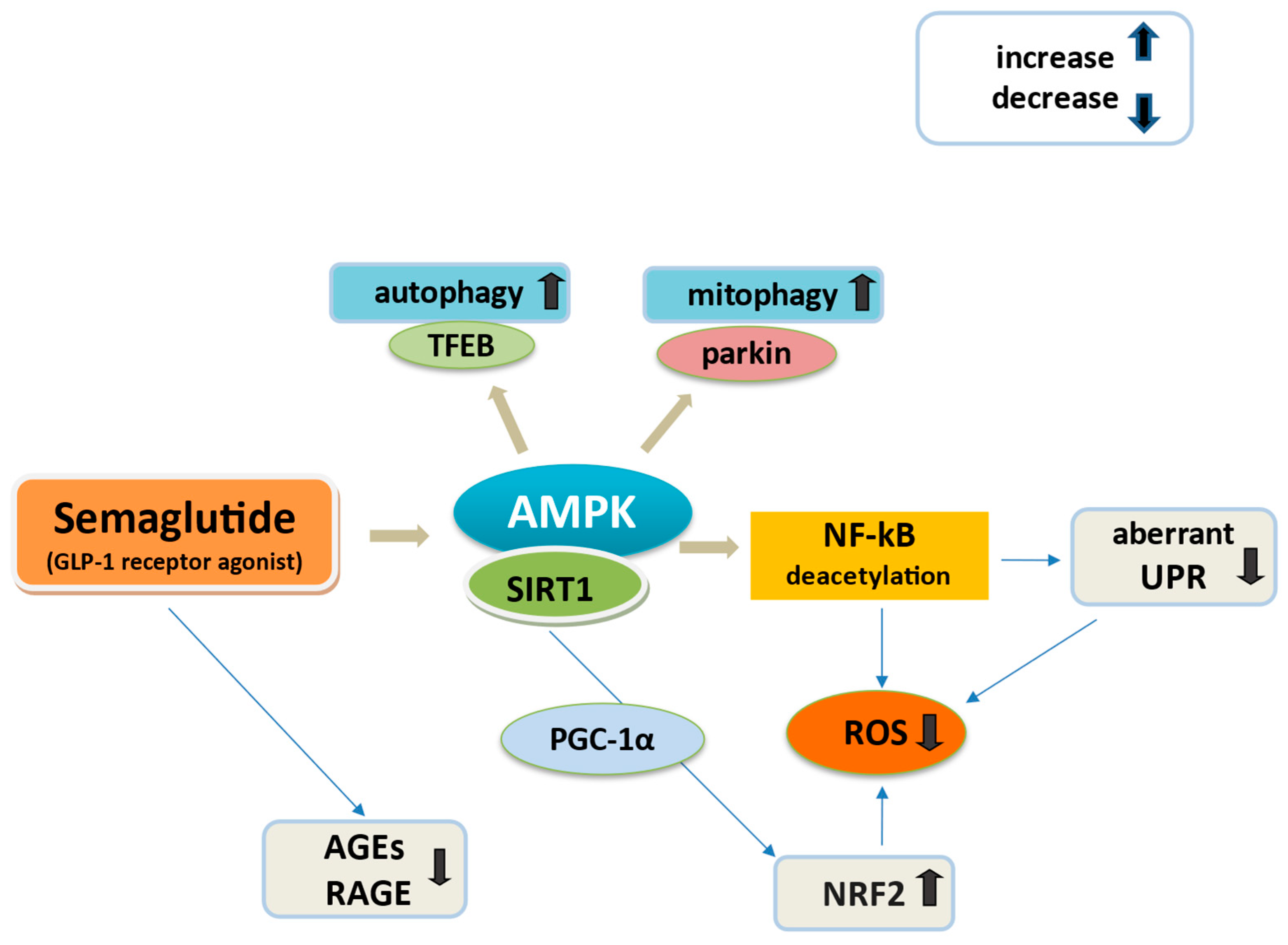
| Oxidative stress | Ameliorates mitochondrial function Mitigates mitochondrial and ER oxidative stress Regulates excessive ROS | SIRT1/AMPK activation SIRT1/NRF2 signaling ROS reduction SIRT1/AMPK mediated reduction of JNK/NF-kB ROS production and aberrant UPR activation NF-kB downregulation RAGE-RAGE ROS reduction | [4,35,137,152,154] [157,183,204] [89,158,159] |
| Activates autophagy And mitophagy | AMPK pathway activation AMPK increase in TFEB and parkin | [157] [77,156] | |
| Upregulation of antiapoptotic proteins Bcl-2, Bcl-xl Downregulation of proapoptotic Bax, Bad and caspases | [196,197] | ||
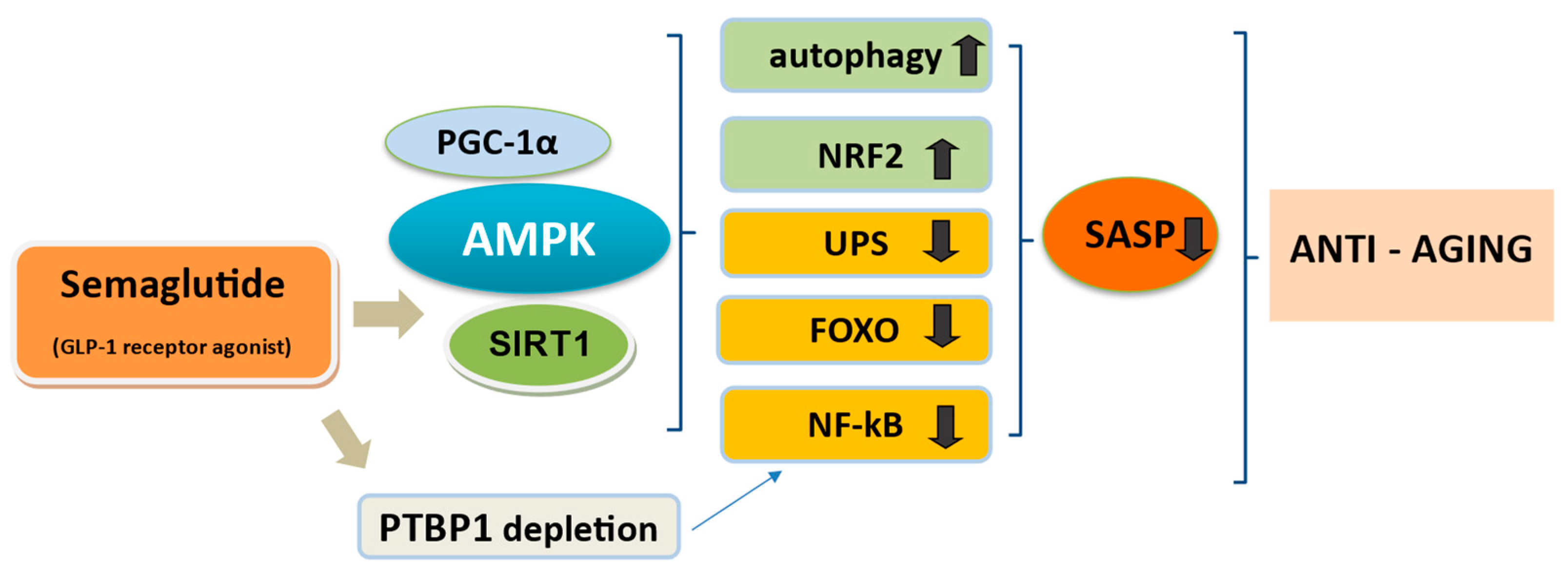
| Ageing and age-linked disorders | Activation of signaling, compromised during ageing | AMPK/SIRT1/ PGC-1α activation Blockade of NF-kB and FOXO transcription factors | [177,178,187] [192,193] |
| May deplete PTBP1 and downmodulate NF-kB pathway to mitigate the inflammatory effects of SASP SASP modulation by targeting the JAK/STAT pathway | [116,191] | ||
| Upregulation of NRF2 | [137,194] | ||
| May ameliorate UPS and autophagy-lysosomal system | [139,182,195] | ||
| Upregulation of antiapoptotic proteins Bcl-2 and Bcl-xl Downregulation of proapoptotic Bax, Bad and caspases | [46,196,197] | ||
| Infections/ Sepsis | Attenuate polymicrobial inflammation and sepsis-associated detrimental responses Suppress peripheral inflammation | Central neuronal GLP-1 effects controlled by the endogenous opioid system Regulation of NF-κB mediated inflammation control of receptors TLR4 AMPK activation GLP-1/AMPK FOXP3 immunoregulation | [199] [19,159,201,202] [202,205,206] |
| Study Ref. | Study Details Type of Mice, Aims, Methods, Interventions | Experimental Procedures | Results | Concluding Points |
|---|---|---|---|---|
| [46] 2019 Yang et al. | Sprague–Dawley (SD) male rats weighing 250–300 g aged 3 months Aim: Protective effects of semaglutide against middle cerebral artery occlusion injury in rats In vivo experiment: Randomly divided into three groups: Group 1: rats that received equal volume saline (Sham); group2: vehicle controls that received equal volume saline (Vehicle); group 3: semaglutide rats that received semaglutide at 10 nmol/kg i.p. observed for 21 days | Permanent middle cerebral artery occlusion (pMCAO) model for cerebral ischemia Tissue sampling Western blot analysis of the extracted proteins Immunofluorescence staining to detect DCX + cells in the hippocampal dentate gyrus at 1, 7, 14 and 21 days after MCAO | Semaglutide significantly reduced neurological impairments, increased hippocampal neuronal survival post ischemia Semaglutide can re-sensitize insulin signaling and normalize IRS1 activity Semaglutide reduces apoptosis signaling Bcl-2/BAX, Caspase-3 | Semaglutide normalize insulin signaling and activity of the IRS1 Reduces apoptosis Neuroprotection |
| [90] 2022 Feng et al. | Six-week-old male C57BL/6J mice Aim: Effect of semaglutide on FGF21 in high-fat diet mice In vivo experiment: For 13 weeks either fed with low-fat diet (LFD) or HFD HFD randomly divided for daily i.p. semaglutide at high dosage (600 μg/kg) weight) or control PBS injection for 1 week | Plasma: mouse adiponectin immunoassay kit, leptin Tissues: liver, WAT, BAT RT-PCR to determine effects of hFGF21 Quantitative reverse transcription PCR for RNA extraction Western blotting antibodies on prepared whole-cell lysates from liver, adipose to determine effects of hFGF21 | In HFD-semaglutide mice: Profound body weight lowering effect possibly inducing a “fasting-like” state in HFD Semaglutide: Reversed hypeleptinemia Elevated FGF21 Stimulated hepatic FGF21 Reduced and reversed HFD alterations in WAT Reduced WAT FGF21 resistance Maintained BAT | Confirmed that semaglutide can upregulate hepatic FGF21 production, restore FGF21 sensitivity, reverse HFD alterations in WAT and maintain BAT |
| [95] 2022 Martins et al. | C57BL/6 male mice Aim: Evaluation of response of adipocytes to semaglutide In vivo experiment: Two groups (a) control diet (C group) (b) high-fat diet (HF group) for 16 weeks After, each group was randomly Separated into two groups adding semaglutide s.c at 40 μg/kg once every 3 days and studied for an additional four weeks: (a) C, (b) control diet and semaglutide (CS), (c) HF diet, and (d) high-fat diet and semaglutide (HFS) Untreated groups were given sterile s.c saline. | Histology, fat pad fragments UCP1 staining, immunofluorescence Immunohistochemistry qRT-PCR mRNA expression | In obese mice semaglutide: Reduced WAT, proinflammatory markers, leptin (−80%) Lessened ER stress, enhanced UCP1 labeling, PPAR-α (+560%), PPAR-γ (+150%), NRF-1 (+260%) Increased thermogenetic gene expressions for the browning phenotype maintenance: beta-3 adrenergic receptor (+520%), Ucp1 (+110%) | Semaglutide lessens ER stress in WAT Semaglutide mitigates adipocyte hypertrophy Semaglutide enhances browning mediators and mitochondrial biogenesis in WAT |
| [110] 2024 Pan et al. | Six-week-old C57bl6 mice Aim: Uncover the cardioprotective impact of semaglutide In vivo experiment: Randomly assigned into normal chow diet (NCD), high-fat group (HFD) After 12 weeks, the HFD group was divided into HFD continued to eat high-fat diet and the semaglutide intervention group (Sema group) high-fat diet and semaglutide 30 nmol/kg/day i.p. for 12 weeks | Serum tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and malonic dialdehyde (MDA) levels were detected by ELISA Heart tissue ROS content measured by ROS staining of frozen sections Fluorescent microscopy Single-cell RNA sequencing screening for differentially expressed genes (DEGs) | Semaglutide reduced levels of ROS, IL-6, MDA, and TNF in cardiac tissue and serum Semaglutide significantly decreased the highly expressed in neutrophils S100a8, S100a9, and Cxcl2 | Semaglutide reduces body weight, improves inflammation and oxidative stress Inhibits of the expression of neutrophil inflammatory factors Whether semaglutide alleviates cardiac inflammation, and oxidative stress independently of body weight reduction; needs further study |
| [112] 2023 Zhu et al. | C57BL/6JC male mice, 7-week-old, 16–20 g Aim: Role of semaglutide in WAT In vivo experiment: Randomly divided to normal chow diet group (NCD) and high-fat diet (HFD) After 12 weeks of feeding Randomly divided to HFD+saline group (HFD) and HFD+semaglutide i.p. (30 nmol/kg/d) group (HFD-Sema) for 12 weeks | Serum analysis Histopathological analysis of epididymal WAT and interscapular BAT Paraffin blocks for hematoxylin and eosin (H&E) stain Peptide labeling Proteomic analysis Quantitation analysis Bioinformatics | In Semaglutide group vs HFD: (H&E) staining: diameter of adipocytes in WAT and BAT was markedly decreased Proteomics: 640 differentially expressed proteins (DEPs), 292 up-regulated and 348 down-regulated Bioinformatics: reduction of ANGPTL4, LPL | Semaglutide group: Reduction of 10 proteins involved in fatty acid transport and among them, adipogenic ANGPTL4, LPL Significant weight-loss effect Decreased WAT Increased BAT |
| [120] 2023 Wang et al. | 12-month-old male APP/PS1/Tau transgenic mice (3xTg) and C57B6/129 wild type mice (WT) Aim: Effect of semaglutide on glucose metabolism dysfunction in Alzheimer disease (AD) mice and cells In vivo experiment: Randomly assigned to four groups: WT + Saline, 3xTg + Saline, 3xTg + semaglutide, and 3xTg + semaglutide + EX527 (SIRT1 inhibitor) 15 mice in each group were injected with semaglutide (0.1 mg/kg, i. p. or saline (0.9%, i. p.) every other day for 30 days | Micro-PET/CT scanner Immunohistochemistry for brain tissue sections Immunofluorescence with DAPI staining Western blot and protein quantification performed by BCA protein assay Cell immunofluorescence with SIRT1 primary antibody Cell culture of HT22 cells | Semaglutide promoted the expression of SIRT1 and GLUT4 in 3xTg mice Semaglutide alleviated the Aβ and tau pathology in the hippocampus CA3 region of 3xTg mice | Semaglutide reduces mediated by GLP-1/SIRT1/GLUT4 pathway Semaglutide reduces pathology of AD animals |
| [131] 2023 Martins et al. | DIO male mice (C57BL/6J) Aim: Effect of semaglutide on the neuropeptide signaling implicated in hypothalamic energy metabolism in obede mice In vivo experiment: control diet (C) and high-fat diet (HF) for 16 weeks then re-divided in control diet (C), control plus semaglutides (CS), high-fat (HF), high-fat plus semaglutide (HFS) and high fat pair-feeding groups (HFPF) for 4 weeks | Plasma analysis to determine -active GLP-1 by multiplex biomarker immunoassays -leptin concentration by enzyme immune-assay kit The hypothalamus was stereotaxically sectioned and prepared for biochemical and molecular analysis Immunofluorescence qRT-PCR for hypothalamic mRNA gene expression Confocal laser scanning microscopy and DAPI staining for POMC and NPY labeling | DIO mice showed increased energy intake and body weight linked to leptin resistance Semaglutide: Improved GLP-1 in HFS vs. HF (+500%) Improved leptin in HF vs. C (+95%), but lessened in HFS vs. HF (−46%) Increased Socs3 in HF vs. C (+300%) but diminished in HFS vs. HF (70%) Augmented Pomc in the ARC (HFS vs. HF, +138%) Increased POMC labeling Reduced NPY labeling | Semaglutide treatment restored GLP-1 levels, regulated Socs3 expression in ARC, improved leptin sensitivity and the hypothalamic anorexigenic signaling (POMC/MC4R) for obesity control in DIO mice |
| [154] 2024 Yan et al. | Six-week-old male C57BL/6J mice Aim: Mechanisms and effects of semaglutide on myocardium injury and cardiac function in diabetic cardiomyopathy mice In vivo experiment: Randomly divided into four groups: control group, semaglutide group, diabetes group and diabetes + semaglutide treatment group Type 1 diabetes induced by i.p. streptozotocin Mice in the semaglutide intervention group were injected s.c semaglutide (0.15 mg/kg) every week for 8 week | Myocardial tissues sampling Western blot analysis RNA isolation and qRT- PCR Immunofluorescence staining Incubation with the primary antibody Cx43, nuclei were stained with DAPI | In the semaglutide-treated group when compared to the untreated: Semaglutide Significantly increased antioxidant enzymes (SOD) Significantly increased SIRT1/AMPK Normalized Cx43 protein expression Reversed RR, QRS, QT, and QTc intervals prolongation in ECG | SIRT1/AMPK signaling pathways may mediate the cardioprotective effects of semaglutide |
| [157] 2020 Li et al. | Male 4–6 weeks old SD rats, weighting 200–250 g Aim: Effects and mechanism of semaglutide on exercise-induced myocardial injury In vivo experiment: Divided randomly into normal rat group and overtraining rats total training 10 weeks then overtraining rats randomly divided as high dose of semaglutide treated group, medium dose semaglutide treated group, low dose semaglutide treated group and control rats without semaglutide treatment for 8-week | Cardiac tissues and blood samples LPS-induced oxidative stress injuries and inflammatory Response, assessed in H9c2 cell via MTT assay and Western blot Protein preparations, primary antibodies, H9c2 embryonic rat heart-derived (ventricular) cells Apoptosis detection kit and ROS detection kit | Semaglutide improve the viability and apoptosis of LPS treated myocardial H9C2 cells Semaglutide activate AMPK pathway, improve autophagy and inhibit ROS production in LPS treated H9C2 cells Semaglutide ameliorates myocardial injury markers in excessive exercise rat model | Semaglutide may reduce the inflammatory response by activating the AMPK pathway, inhibiting oxidative stress (ROS), improve autophagy and downregulate inflammatory cytokines (NF-kB) |
| [181] 2023 Xiang et al. | Male C57BL/6 mice (8 weeks old) weighing 20.1 ± 1.1 g Aim: Effect and molecular mechanisms of GLP-1R agonists liraglutide and semaglutide on obesity-induced muscle atrophy In vivo experiment: Mice randomly divided into regular diet and a high-fat diet group for 18 weeks After modeling obesity, mice were further divided into control group, liraglutide (LIRA) group, semaglutide (SEMA) group, high-fat diet (HFD) group, HFD + LIRA group, HFD + SEMA group, Semaglutide (60 ug/kg/d) s.c for 4 weeks | Histological analysis of gastrocnemius muscle C2C12 cells culture C2C12 myotubes were incubated with palmitic acid to induce obesity and skeletal muscle atrophy Immunofluorescence, nuclei were stained with DAPI qRT-PCR Western Blotting | HFD up-regulated the expression of muscle atrophy factor Atrogin-1 and suppressed the expression of myogenic factor Myogenin and SIRT1 Semaglutide: Activated SIRT1 Reduced atrogin-1 expression Increased myogenin expression Alleviated the decrease in GLUT4 expression induced by HFD | Semaglutide activation of SIRT-1 may be important to ameliorate insulin resistance upregulating GLUT4 and to alleviate obesity induced muscle atrophy |
| [183] 2023 Iwai et al. | Male diabetic KK-Ay mice aged 10 weeks old Aim: Effect of semaglutide on skeletal muscle wasting and dysfunction in liver disease-related skeletal muscle atrophy under diabetic conditions In vivo experiment: four groups treated for six weeks as follows: (i) normal diet (ND-Veh) (ii) ND and s.c.semaglutide (3 nmol/kg) every 3 days (ND-Sem group) (iii) DDC diet (iv) DDC diet plus s.c semaglutide every three days (DDC-Sem group) Vehicle: saline | Histological and immunofluorescent analyses of gastrocnemius muscle tissue RNA isolation and qRT-PCR Western blotting assay gel electrophoresis and primary antibodies incubation Tissue and whole cell lysates were extracted from gastrocnemius muscle tissues Cultured C2C12 myotubes Antibodies for western blotting were atrogin-1, MuRF1, myogenin, PGC-1α, SIRT1 | No significant differences in body length or weight between the ND-Veh and ND-Sem groups for six weeks treatment Intramuscular atrogin-1 and MuRF-1 levels increased in the DDC-Veh group, with this difference significantly attenuated in the DDC-Semaglutide group Semaglutide increased PGC-1α, SIRT1 Reduced NF-kB | Limited effect of semaglutide on physiological status Semaglutide: Directly activates GLP-1R in skeletal muscle to increase mitochondrial biogenesis and antioxidant activity Inhibits UPS Suppresses protein degradation Promotes myogenesis in mice Improve skeletal muscle atrophy in obesity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papakonstantinou, I.; Tsioufis, K.; Katsi, V. Spotlight on the Mechanism of Action of Semaglutide. Curr. Issues Mol. Biol. 2024, 46, 14514-14541. https://doi.org/10.3390/cimb46120872
Papakonstantinou I, Tsioufis K, Katsi V. Spotlight on the Mechanism of Action of Semaglutide. Current Issues in Molecular Biology. 2024; 46(12):14514-14541. https://doi.org/10.3390/cimb46120872
Chicago/Turabian StylePapakonstantinou, Ilias, Konstantinos Tsioufis, and Vasiliki Katsi. 2024. "Spotlight on the Mechanism of Action of Semaglutide" Current Issues in Molecular Biology 46, no. 12: 14514-14541. https://doi.org/10.3390/cimb46120872
APA StylePapakonstantinou, I., Tsioufis, K., & Katsi, V. (2024). Spotlight on the Mechanism of Action of Semaglutide. Current Issues in Molecular Biology, 46(12), 14514-14541. https://doi.org/10.3390/cimb46120872