

FLT3 and IRAK4 Inhibitor Emavusertib in Combination with BH3-Mimetics in the Treatment of Acute Myeloid Leukemia

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Cell Lines and Cell Culture
2.3. Cytotoxicity Assays
2.4. Imaging Cytometry
3. Results
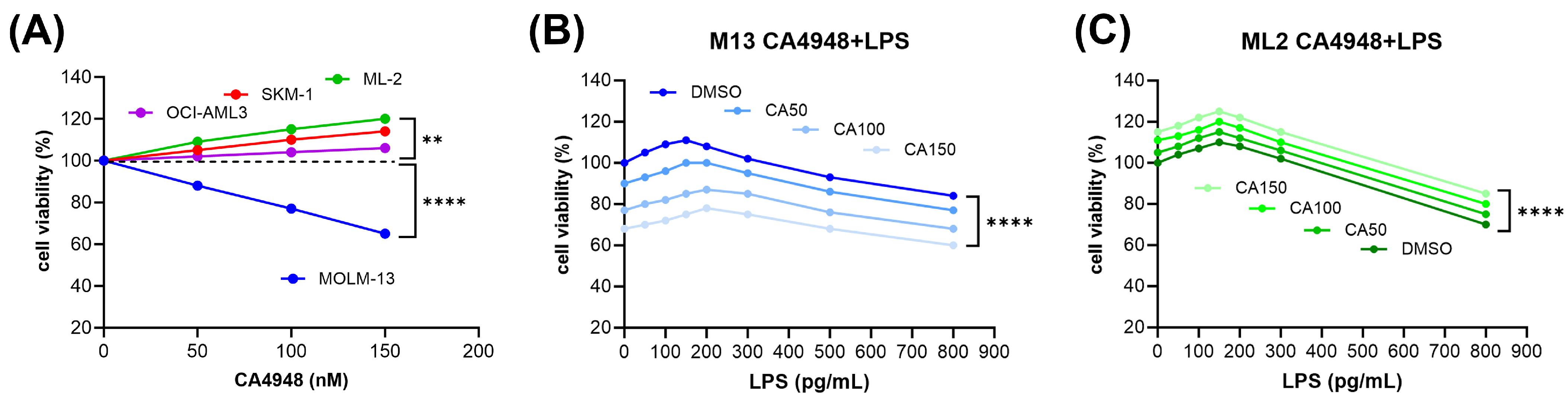
3.1. Inverse Response of AML Cell Lines to Emavusertib (CA4948) Monotherapy
3.2. Susceptibility of AML Cells to Emavusertib (CA4948) Treatment under Exposure to Lipopolysaccharide
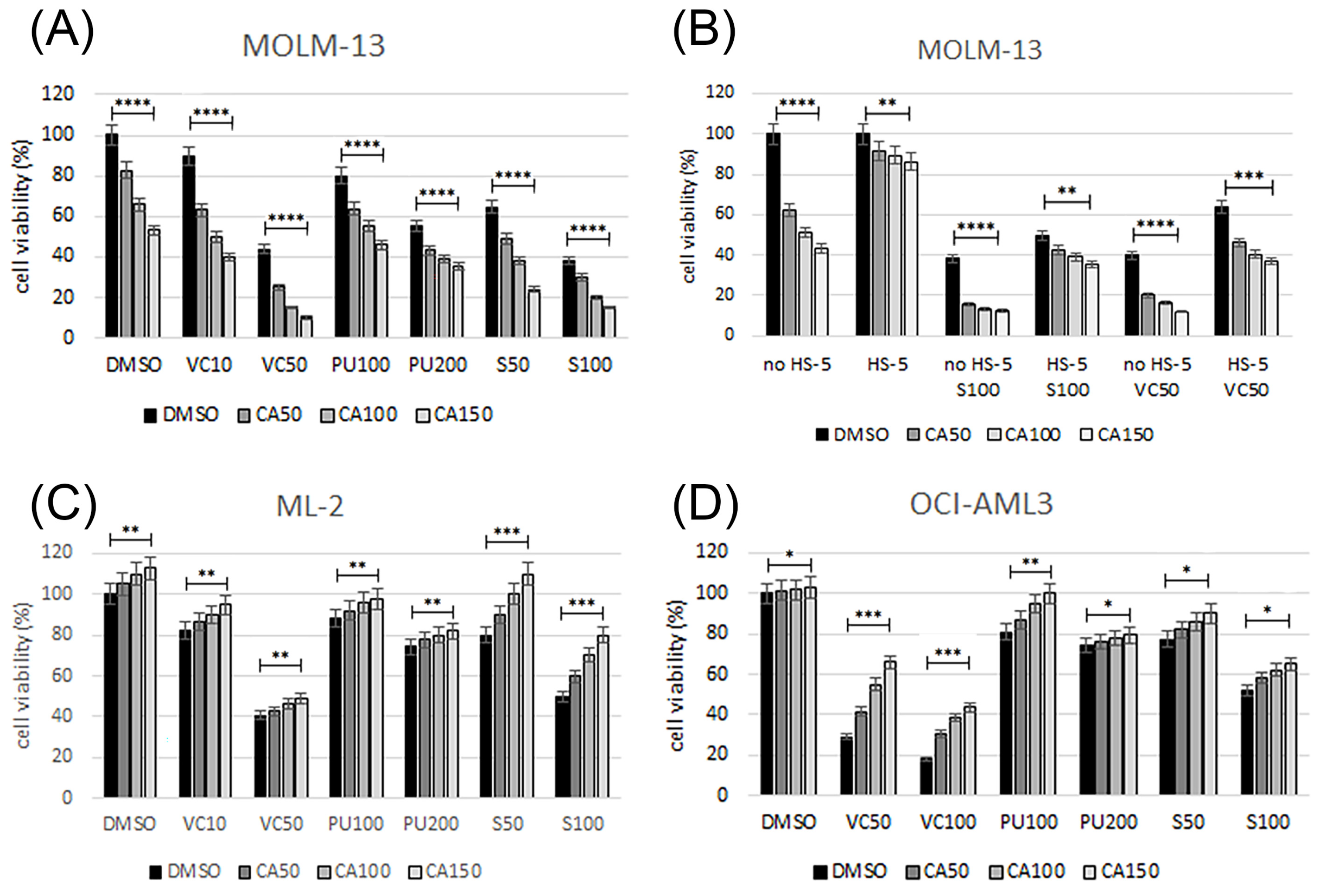
3.3. Emavusertib Combination Treatments in AML Cell Lines
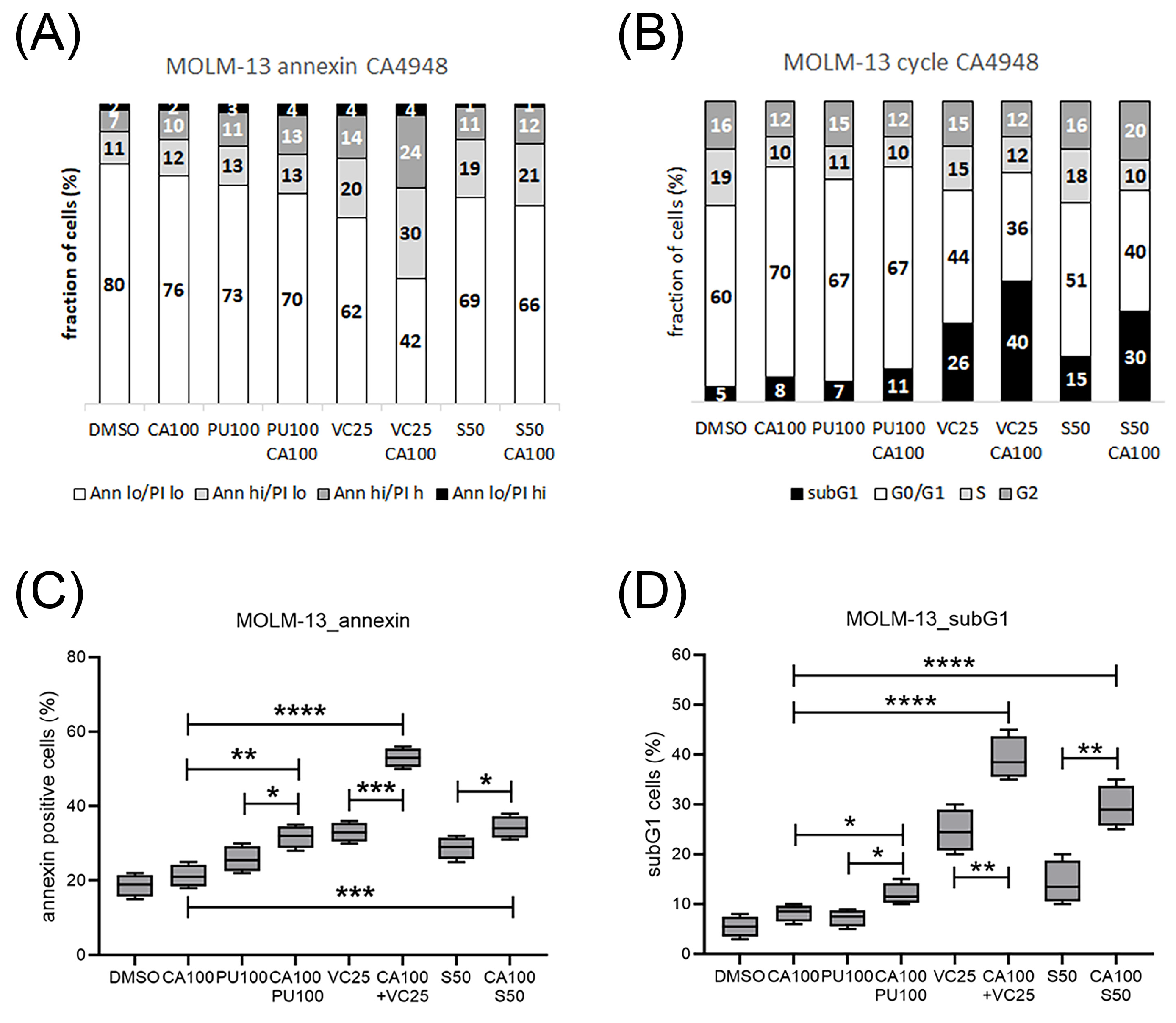
3.4. Emavusertib Treatment Induced Cell Cycle Arrest, Apoptosis and Cell Death
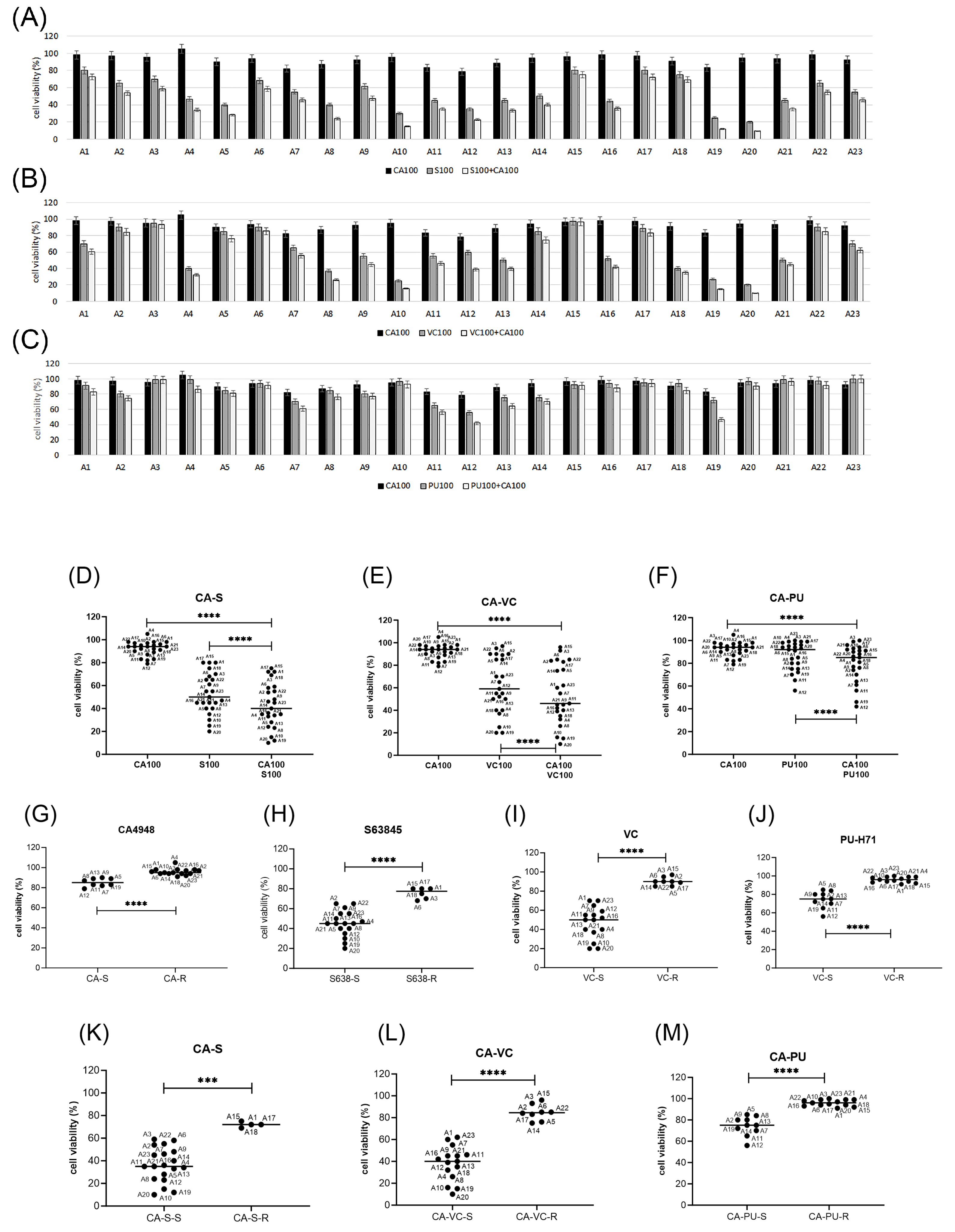
3.5. Emavusertib Combination Treatments in Primary AML Cells In Vitro
3.6. Biomarkers of Response to Emavusertib and Combination Treatments in Primary AML Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef]
- Zhao, J.C.; Agarwal, S.; Ahmad, H.; Amin, K.; Bewersdorf, J.P.; Zeidan, A.M. A Review of FLT3 Inhibitors in Acute Myeloid Leukemia. Blood Rev. 2022, 52, 100905. [Google Scholar] [CrossRef]
- Larrosa-Garcia, M.; Baer, M.R. FLT3 Inhibitors in Acute Myeloid Leukemia: Current Status and Future Directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Warmoes, M.; Lorenzi, P.L.; Mak, D.; Ruvolo, V.; Tan, L.; Cidado, J.; Drew, L.; et al. Targeting MCL-1 Dysregulates Cell Metabolism and Leukemia-Stroma Interactions and Resensitizes Acute Myeloid Leukemia to BCL-2 Inhibition. Haematologica 2022, 107, 58–76. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax Combined with Decitabine or Azacitidine in Treatment-Naive, Elderly Patients with Acute Myeloid Leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef]
- Seipel, K.; Schmitter, K.; Bacher, U.; Pabst, T. Rationale for a Combination Therapy Consisting of MCL1- and MEK-Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1779. [Google Scholar] [CrossRef]
- Seipel, K.; Kopp, B.; Bacher, U.; Pabst, T. BMI1-Inhibitor PTC596 in Combination with MCL1 Inhibitor S63845 or MEK Inhibitor Trametinib in the Treatment of Acute Leukemia. Cancers 2021, 13, 581. [Google Scholar] [CrossRef]
- Seipel, K.; Kohler, S.; Bacher, U.; Pabst, T. HSP90 Inhibitor PU-H71 in Combination with BH3-Mimetics in the Treatment of Acute Myeloid Leukemia. Curr. Issues Mol. Biol. 2023, 45, 7011–7026. [Google Scholar] [CrossRef]
- Bennett, J.; Starczynowski, D.T. IRAK1 and IRAK4 as Emerging Therapeutic Targets in Hematologic Malignancies. Curr. Opin. Hematol. 2022, 29, 8–19. [Google Scholar] [CrossRef]
- Pereira, M.; Gazzinelli, R.T. Regulation of Innate Immune Signaling by IRAK Proteins. Front. Immunol. 2023, 14, 1133354. [Google Scholar] [CrossRef]
- Suzuki, N.; Suzuki, S.; Duncan, G.S.; Millar, D.G.; Wada, T.; Mirtsos, C.; Takada, H.; Wakeham, A.; Itie, A.; Li, S.; et al. Severe Impairment of Interleukin-1 and Toll-like Receptor Signalling in Mice Lacking IRAK-4. Nature 2002, 416, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Wang, R.; Hua, M.; Zhang, C.; Han, F.; Xu, M.; Yang, X.; Li, G.; Hu, X.; Sun, T.; et al. NLRP3 Inflammasome Promotes the Progression of Acute Myeloid Leukemia via IL-1β Pathway. Front. Immunol. 2021, 12, 661939. [Google Scholar] [CrossRef] [PubMed]
- Rhyasen, G.W.; Starczynowski, D.T. IRAK Signalling in Cancer. Br. J. Cancer 2015, 112, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.D.; Manning-Bennett, A.T.; Abuhelwa, A.Y. Investigational IRAK-4 Inhibitors for the Treatment of Rheumatoid Arthritis. Expert. Opin. Investig. Drugs 2020, 29, 475–482. [Google Scholar] [CrossRef]
- Melgar, K.; Walker, M.M.; Jones, L.M.; Bolanos, L.C.; Hueneman, K.; Wunderlich, M.; Jiang, J.-K.; Wilson, K.M.; Zhang, X.; Sutter, P.; et al. Overcoming Adaptive Therapy Resistance in AML by Targeting Immune Response Pathways. Sci. Transl. Med. 2019, 11, eaaw8828. [Google Scholar] [CrossRef]
- Hosseini, M.M.; Kurtz, S.E.; Abdelhamed, S.; Mahmood, S.; Davare, M.A.; Kaempf, A.; Elferich, J.; McDermott, J.E.; Liu, T.; Payne, S.H.; et al. Inhibition of Interleukin-1 Receptor-Associated Kinase-1 Is a Therapeutic Strategy for Acute Myeloid Leukemia Subtypes. Leukemia 2018, 32, 2374–2387. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.Y.; Zhao, Q.; Buelow, D.R.; Phelps, M.; Walker, A.R.; Mims, A.S.; Vasu, S.; Behbehani, G.; Blachly, J.; Blum, W.; et al. Preclinical Activity and a Pilot Phase I Study of Pacritinib, an Oral JAK2/FLT3 Inhibitor, and Chemotherapy in FLT3-ITD-Positive AML. Investig. New Drugs 2020, 38, 340–349. [Google Scholar] [CrossRef]
- Parrondo, R.D.; Iqbal, M.; Von Roemeling, R.; Von Roemeling, C.; Tun, H.W. IRAK-4 Inhibition: Emavusertib for the Treatment of Lymphoid and Myeloid Malignancies. Front. Immunol. 2023, 14, 1239082. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Winer, E.S.; DeAngelo, D.J.; Tarantolo, S.; Sallman, D.A.; Dugan, J.; Groepper, S.; Giagounidis, A.; Götze, K.; Metzeler, K.H.; et al. S129: Takeaim Leukemia—A Phase 1/2A Study of the IRAK4 Inhibitor Emavusertib (CA-4948) AS Monotherapy or in Combination with Azacitidine or Venetoclax in Relapsed/Refractory AML or MDS. HemaSphere 2022, 6, 30. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Platzbecker, U.; Lim, K.-H.; Nowakowski, G.; Abdel-Wahab, O.; Kantarjian, H.; Verma, A.; Starczynowski, D.T. Research and Clinical Updates on IRAK4 and Its Roles in Inflammation and Malignancy: Themes and Highlights from the 1st Symposium on IRAK4 in Cancer. Front. Hematol. 2024, 3, 1339870. [Google Scholar] [CrossRef]
- Albakova, Z.; Mangasarova, Y.; Albakov, A.; Gorenkova, L. HSP70 and HSP90 in Cancer: Cytosolic, Endoplasmic Reticulum and Mitochondrial Chaperones of Tumorigenesis. Front. Oncol. 2022, 12, 829520. [Google Scholar] [CrossRef] [PubMed]
- Cabaud-Gibouin, V.; Durand, M.; Quéré, R.; Girodon, F.; Garrido, C.; Jego, G. Heat-Shock Proteins in Leukemia and Lymphoma: Multitargets for Innovative Therapeutic Approaches. Cancers 2023, 15, 984. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, C.; Song, C. Pan- and Isoform-Specific Inhibition of Hsp90: Design Strategy and Recent Advances. Eur. J. Med. Chem. 2022, 238, 114516. [Google Scholar] [CrossRef]
- Ren, X.; Li, T.; Zhang, W.; Yang, X. Targeting Heat-Shock Protein 90 in Cancer: An Update on Combination Therapy. Cells 2022, 11, 2556. [Google Scholar] [CrossRef] [PubMed]
- Shumilov, E.; Flach, J.; Kohlmann, A.; Banz, Y.; Bonadies, N.; Fiedler, M.; Pabst, T.; Bacher, U. Current Status and Trends in the Diagnostics of AML and MDS. Blood Rev. 2018, 32, 508–519. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s That Spur Autophagy and Immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Chaiwut, R.; Kasinrerk, W. Very Low Concentration of Lipopolysaccharide Can Induce the Production of Various Cytokines and Chemokines in Human Primary Monocytes. BMC Res. Notes 2022, 15, 42. [Google Scholar] [CrossRef]
- Smith, M.A.; Choudhary, G.S.; Pellagatti, A.; Choi, K.; Bolanos, L.C.; Bhagat, T.D.; Gordon-Mitchell, S.; Von Ahrens, D.; Pradhan, K.; Steeples, V.; et al. U2AF1 Mutations Induce Oncogenic IRAK4 Isoforms and Activate Innate Immune Pathways in Myeloid Malignancies. Nat. Cell Biol. 2019, 21, 640–650. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Pellagatti, A.; Agianian, B.; Smith, M.A.; Bhagat, T.D.; Gordon-Mitchell, S.; Sahu, S.; Pandey, S.; Shah, N.; Aluri, S.; et al. Activation of Targetable Inflammatory Immune Signaling Is Seen in Myelodysplastic Syndromes with SF3B1 Mutations. Elife 2022, 11, e78136. [Google Scholar] [CrossRef]
- Bennett, J.; Ishikawa, C.; Agarwal, P.; Yeung, J.; Sampson, A.; Uible, E.; Vick, E.; Bolanos, L.C.; Hueneman, K.; Wunderlich, M.; et al. Paralog-Specific Signaling by IRAK1/4 Maintains MyD88-Independent Functions in MDS/AML. Blood 2023, 142, 989–1007. [Google Scholar] [CrossRef]
- Wang, R.; Yang, X.; Liu, J.; Zhong, F.; Zhang, C.; Chen, Y.; Sun, T.; Ji, C.; Ma, D. Gut Microbiota Regulates Acute Myeloid Leukaemia via Alteration of Intestinal Barrier Function Mediated by Butyrate. Nat. Commun. 2022, 13, 2522. [Google Scholar] [CrossRef]
- Venet, F.; Demaret, J.; Gossez, M.; Monneret, G. Myeloid Cells in Sepsis-Acquired Immunodeficiency. Ann. N. Y. Acad. Sci. 2021, 1499, 3–17. [Google Scholar] [CrossRef]
- Schrijver, I.T.; Théroude, C.; Roger, T. Myeloid-Derived Suppressor Cells in Sepsis. Front. Immunol. 2019, 10, 447308. [Google Scholar] [CrossRef]
- Seipel, K.; Brügger, Y.; Mandhair, H.; Bacher, U.; Pabst, T. Rationale for Combining the BCL2 Inhibitor Venetoclax with the PI3K Inhibitor Bimiralisib in the Treatment of IDH2- and FLT3-Mutated Acute Myeloid Leukemia. Int. J. Mol. Sci. 2022, 23, 12587. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Speranza, G.; Anderson, L.; Chen, A.P.; Do, K.; Eugeni, M.; Weil, M.; Rubinstein, L.; Majerova, E.; Collins, J.; Horneffer, Y.; et al. First-in-Human Study of the Epichaperome Inhibitor PU-H71: Clinical Results and Metabolic Profile. Investig. New Drugs 2018, 36, 230–239. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.-N.; Moujalled, D.M.; et al. The MCL1 Inhibitor S63845 Is Tolerable and Effective in Diverse Cancer Models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Parry, N.; Wheadon, H.; Copland, M. The Application of BH3 Mimetics in Myeloid Leukemias. Cell Death Dis. 2021, 12, 222. [Google Scholar] [CrossRef]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. NPM1-Mutated Acute Myeloid Leukemia: From Bench to Bedside. Blood 2020, 136, 1707–1721. [Google Scholar] [CrossRef]
- Seipel, K.; Marques, M.A.T.; Sidler, C.; Mueller, B.U.; Pabst, T. MDM2- and FLT3-Inhibitors in the Treatment of FLT3-ITD Acute Myeloid Leukemia, Specificity and Efficacy of NVP-HDM201 and Midostaurin. Haematologica 2018, 103, 1862–1872. [Google Scholar] [CrossRef]
- Brenner, A.K.; Bruserud, Ø. Functional Toll-Like Receptors (TLRs) Are Expressed by a Majority of Primary Human Acute Myeloid Leukemia Cells and Inducibility of the TLR Signaling Pathway Is Associated with a More Favorable Phenotype. Cancers 2019, 11, 973. [Google Scholar] [CrossRef]
- Nakatomi, K.; Ueno, H.; Ishikawa, Y.; Salim, R.C.; Mori, Y.; Kanemoto, I.; Tancharoen, S.; Kikuchi, K.; Miura, N.; Omori, T.; et al. TLR4/MD-2 Is a Receptor for Extracellular Nucleophosmin 1. Biomed. Rep. 2021, 14, 21. [Google Scholar] [CrossRef]
- Radu, P.; Zurzu, M.; Paic, V.; Bratucu, M.; Garofil, D.; Tigora, A.; Georgescu, V.; Prunoiu, V.; Pasnicu, C.; Popa, F.; et al. CD34—Structure, Functions and Relationship with Cancer Stem Cells. Medicina 2023, 59, 938. [Google Scholar] [CrossRef]
- Vergez, F.; Green, A.S.; Tamburini, J.; Sarry, J.-E.; Gaillard, B.; Cornillet-Lefebvre, P.; Pannetier, M.; Neyret, A.; Chapuis, N.; Ifrah, N.; et al. High Levels of CD34+CD38low/−CD123+ Blasts Are Predictive of an Adverse Outcome in Acute Myeloid Leukemia: A Groupe Ouest-Est Des Leucémies Aiguës et Maladies Du Sang (GOELAMS) Study. Haematologica 2011, 96, 1792–1798. [Google Scholar] [CrossRef]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Acute Myeloid Leukemia Patients. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef]
- Rahmani, N.E.; Ramachandra, N.; Sahu, S.; Gitego, N.; Lopez, A.; Pradhan, K.; Bhagat, T.D.; Gordon-Mitchell, S.; Pena, B.R.; Kazemi, M.; et al. ASXL1 Mutations Are Associated with Distinct Epigenomic Alterations That Lead to Sensitivity to Venetoclax and Azacytidine. Blood Cancer J. 2021, 11, 157. [Google Scholar] [CrossRef]
- Feld, J.; Tremblay, D.; Dougherty, M.; Czaplinska, T.; Sanchez, G.; Brady, C.; Kremyanskaya, M.; Bar-Natan, M.; Keyzner, A.; Marcellino, B.K.; et al. Safety and Efficacy: Clinical Experience of Venetoclax in Combination With Hypomethylating Agents in Both Newly Diagnosed and Relapsed/Refractory Advanced Myeloid Malignancies. Hemasphere 2021, 5, e549. [Google Scholar] [CrossRef]
- Seipel, K.; Graber, C.; Flückiger, L.; Bacher, U.; Pabst, T. Rationale for a Combination Therapy with the STAT5 Inhibitor AC-4-130 and the MCL1 Inhibitor S63845 in the Treatment of FLT3-Mutated or TET2-Mutated Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 8092. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Disease | Mutation Profile (VAF) | Karyotype | Source | CD34 % | CD117 % | Blast (%) | BM Inf (%) | CD11b % |
|---|---|---|---|---|---|---|---|---|---|
| A1 | AML-M0 | TP53 G245S (92%) | complex | BM | 97 | 93 | 74 | 80 | 5 |
| A2 | AML-M4 | FLT3-TKD (0.565), NPM1 (19%), SRSF2 (47%), TET2 Q1357fs (42%), TET2 L1816fs (35%), TP53 G245D (5%) | triple 8 | PB | 1 | 1 | 46 | 50 | 96 |
| A3 | AML-M5 | ASXL1 G646fs (41%), KRAS Q61R (39%), SH2B3 L14P (17%), TET2 Y1294H (47%), U2AF1 Q157R (41%) | mono 7 | BM | 1 | 86 | 53 | 80 | <1 |
| A4 | AML-M1/2 | CEBPA K313del (49%), GATA2 A318G (14%), TET2 H436fs (47%) | normal | BM | 6 | 86 | 80 | 90 | 1 |
| A5 | AML-M4 | NPM1 (42%), PTPN11 E69K (40%) | na | BM | 6 | 16 | 65 | 65 | 86 |
| A6 | AML-M4 | ASXL1 G646fs (42%), KRAS G12D (64%), TET2 R1214W (32%) | normal | PB | 1 | 1 | na | 80 | 100 |
| A7 | AML-M1 | FLT3-ITD (0,783), BCOR G1382fs (79%), TET2 L1420* (39%), U2AF1 S34F (40%) | triple 8 | BM | 18 | 30 | na | na | 12 |
| A8 | AML-M5 | FLT3-ITD (0.5), DNMT3A R882H (48%), NPM1 (49%), IDH2 R140Q (49%) | normal | BM | 1 | 86 | 87 | 90 | <1 |
| A9 | AML-M1/2 | FLT3-ITD (1.1), NPM1 (43%) | normal | BM | 4 | 86 | 50 | 90 | 8 |
| A10 | AML-NOS | FLT3-ITD (0.45), TET2 R1261H (47%), TET2 H1904R (48%), SRSF2 (54%) | normal | PB | 97 | 99 | 61 | 80 | 3 |
| A11 | AML-M1 | FLT3-ITD (1.2), NPM1 (35%), WT1 R462P (47%) | normal | PB | 19 | 90 | 86 | 90 | 10 |
| A12 | AML-M1/2 | FLT3-ITD (1.0), DNMT3A R882C (50%), NPM1 (39%), RUNX1 P263S (51%) | normal | PB | 10 | 84 | 92 | 90 | 1 |
| A13 | AML_M1 | FLT3-ITD (0.56) | na | PB | <1 | 80 | 92 | na | 5 |
| A14 | AML-M4 | FLT3-ITD (0.86), DNMT3A (45%), NPM1 (36%), SUZ12 (51%) | normal | PB | <1 | 84 | 21 | 20 | 7 |
| A15 | MDS-AML | TET2 Q278* (42%), TET2 M1701fs (35%), NPM1 (31%), ASXL1 (38%), SRSF2 (42%) | triple 8 | PB | 51 | 50 | 69 | 80 | 35 |
| A16 | AML-M1 | NRAS (45%), DNMT3A (46%), NPM1 (22%), RAD21 (44%) | + 21 | PB | <1 | 97 | 89 | 90 | <1 |
| A17 | AML-M4 | NRAS (32%), PTPN11 F285I (46%), DNMT3A S243fs (45%), DNMT3A M880V (48%) | der (7;14) | PB | 87 | 20 | 8 | 25 | 12 |
| A18 | AML-M0 | ASXL1 (48 %), IDH2 (45 %), RUNX1 (43%), SRSF2 (34 %), STAG2 (9 %) | +13 | PB | 98 | 94 | 64 | 80 | 1 |
| A19 | AML-M1 | FLT3-ITD (1.0), IDH2 (47%), NPM1 (48%) | normal | PB | 20 | 95 | 94 | 90 | 40 |
| A20 | AML-M1 | normal | normal | PB | 16 | 20 | 89 | 90 | <1 |
| A21 | PV-AML | FLT3-TKD (0.16), PTPN11 Y62D (18%), IDH1 (42%), NPM1 (38%), SRSF2 (40%) | t(8;21), -Y | PB | 78 | 37 | 83 | 90 | 32 |
| A22 | AML-M2 | SF3B1 (50%), TET2 S689fs*4 (50%), CBL (87%) | normal | BM | 76 | 64 | 69 | 80 | 25 |
| A23 | AML-M4 | DNMT3A V895M (46%), NPM1 (33%), IDH2 R140Q (46%) | normal | PB | <1 | 30 | 86 | 85 | 57 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seipel, K.; Mandhair, H.; Bacher, U.; Pabst, T. FLT3 and IRAK4 Inhibitor Emavusertib in Combination with BH3-Mimetics in the Treatment of Acute Myeloid Leukemia. Curr. Issues Mol. Biol. 2024, 46, 2946-2960. https://doi.org/10.3390/cimb46040184
Seipel K, Mandhair H, Bacher U, Pabst T. FLT3 and IRAK4 Inhibitor Emavusertib in Combination with BH3-Mimetics in the Treatment of Acute Myeloid Leukemia. Current Issues in Molecular Biology. 2024; 46(4):2946-2960. https://doi.org/10.3390/cimb46040184
Chicago/Turabian StyleSeipel, Katja, Harpreet Mandhair, Ulrike Bacher, and Thomas Pabst. 2024. "FLT3 and IRAK4 Inhibitor Emavusertib in Combination with BH3-Mimetics in the Treatment of Acute Myeloid Leukemia" Current Issues in Molecular Biology 46, no. 4: 2946-2960. https://doi.org/10.3390/cimb46040184