
Unlocking the Potential: Semaglutide’s Impact on Alzheimer’s and Parkinson’s Disease in Animal Models

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. SEM Mechanism of Action
3. Therapeutic Potential of SEM in Neurodegenerative Diseases
4. SEM Pharmacokinetics in Animal Models
5. SEM Potential Mechanism of Action in AD
5.1. Cell Viability
5.2. Apoptosis Inhibition
5.3. Enhanced Autophagy
5.4. SIRT1/GLUT4 Pathway
5.5. Improved Aβ and Tau Pathology
5.6. Reduction of Neuroinflammation
6. SEM Potential Mechanism of Action in PD
6.1. Inhibition of Cell Death and Neurodegeneration
6.2. GSK3β Levels
6.3. CREB Gene Expression
6.4. Inflammation Response
6.5. Oxidative Stress
6.6. Autophagy Regulation
6.7. Effects on Mitochondrial Membrane Potential (ΔΨm)
7. SEM Effects on Motor Recovery
7.1. AD
7.1.1. Open Field Test
7.1.2. Novel Object Recognition Test
7.1.3. Y-Maze Task
7.2. PD
7.2.1. Rotarod Test
7.2.2. Vertical Pole Test
7.2.3. Open Field Test
7.2.4. Footprint Test (Motor Coordination)
7.2.5. Grip Strength Test
7.2.6. Apomorphine-Induced Rotation
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Su, M.; Nizamutdinov, D.; Liu, H.; Huang, J.H. Recent Mechanisms of Neurodegeneration and Photobiomodulation in the Context of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 9272. [Google Scholar] [CrossRef]
- Bano, D.; Ehninger, D.; Bagetta, G. Decoding metabolic signatures in Alzheimer’s disease: A mitochondrial perspective. Cell Death Discov. 2023, 9, 432. [Google Scholar] [CrossRef]
- Tahami Monfared, A.A.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer’s Disease: Epidemiology and Clinical Progression. Neurol. Ther. 2022, 11, 553–569. [Google Scholar] [CrossRef]
- Ferrer, I. Hypothesis review: Alzheimer’s overture guidelines. Brain Pathol. 2023, 33, e13122. [Google Scholar] [CrossRef] [PubMed]
- Theron, D.; Hopkins, L.N.; Sutherland, H.G.; Griffiths, L.R.; Fernandez, F. Can Genetic Markers Predict the Sporadic Form of Alzheimer’s Disease? An Updated Review on Genetic Peripheral Markers. Int. J. Mol. Sci. 2023, 24, 13480. [Google Scholar] [CrossRef] [PubMed]
- Wei, P. Ultra-Early Screening of Cognitive Decline Due to Alzheimer’s Pathology. Biomedicines 2023, 11, 1423. [Google Scholar] [CrossRef]
- Prajjwal, P.; Flores Sanga, H.S.; Acharya, K.; Tango, T.; John, J.; Rodriguez, R.S.C.; Dheyaa Marsool Marsool, M.; Sulaimanov, M.; Ahmed, A.; Hussin, O.A. Parkinson’s disease updates: Addressing the pathophysiology, risk factors, genetics, diagnosis, along with the medical and surgical treatment. Ann. Med. Surg. 2023, 85, 4887–4902. [Google Scholar] [CrossRef]
- Adam, H.; Gopinath, S.C.B.; Md Arshad, M.K.; Adam, T.; Parmin, N.A.; Husein, I.; Hashim, U. An update on pathogenesis and clinical scenario for Parkinson’s disease: Diagnosis and treatment. 3 Biotech 2023, 13, 142. [Google Scholar] [CrossRef]
- Smits, M.M.; Van Raalte, D.H. Safety of Semaglutide. Front. Endocrinol. 2021, 12, 645563, Erratum in: Front. Endocrinol. 2021, 12, 786732. [Google Scholar] [CrossRef]
- Meier, J.J. Efficacy of Semaglutide in a Subcutaneous and an Oral Formulation. Front. Endocrinol. 2021, 12, 645617. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes—State-of-the-art. Mol. Metab. 2021, 46, 101102. [Google Scholar] [CrossRef] [PubMed]
- Vilsbøll, T.; Bain, S.C.; Leiter, L.A.; Lingvay, I.; Matthews, D.; Simó, R.; Helmark, I.C.; Wijayasinghe, N.; Larsen, M. Semaglutide, Reduction in Glycated Haemoglobin and the Risk of Diabetic Retinopathy. Diabetes Obes. Metab. 2018, 20, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, B.; Julla, J.B.; Besbes, S.; Proust, M.; Vincentelli, C.; Alos, B.; Ancel, P.; Alzaid, F.; Garcia, R.; Mailly, P.; et al. Glucagon-Like Peptide 1 Receptor Agonists, Diabetic Retinopathy and Angiogenesis: The AngioSafe Type 2 Diabetes Study. J. Clin. Endocrinol. Metab. 2020, 105, e1549-60. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chen, S.; Si, A.; Luo, Y.; Hu, W.; Zhang, Y.; Ma, J. The long-term trend of Parkinson’s disease incidence and mortality in China and a Bayesian projection from 2020 to 2030. Front. Aging Neurosci. 2022, 14, 973310. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Arachchige, A.S.P.M. Depletion of dopamine in Parkinson’s disease and relevant therapeutic options: A review of the literature. AIMS Neurosci. 2023, 10, 200–231. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Yi, L.X.; Wang, D.Q.; Lim, T.M.; Tan, E.K. Role of dopamine in the pathophysiology of Parkinson’s disease. Transl. Neurodegener. 2023, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Cramb, K.M.L.; Beccano-Kelly, D.; Cragg, S.J.; Wade-Martins, R. Impaired dopamine release in Parkinson’s disease. Brain 2023, 146, 3117–3132. [Google Scholar] [CrossRef] [PubMed]
- Salameh, T.S.; Rhea, E.M.; Talbot, K.; Banks, W.A. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer’s and Parkinson’s disease therapeutics. Biochem Pharmacol. 2020, 180, 114187, Erratum in: Biochem Pharmacol. 2023, 210, 115474. https://doi.org/10.1016/j.bcp.2023.115474. [Google Scholar] [CrossRef]
- Cornell, S. A review of GLP-1 receptor agonists in type 2 diabetes: A focus on the mechanism of action of once-weekly agents. J. Clin. Pharm. Ther. 2020, 45 (Suppl. S1), 17–27. [Google Scholar] [CrossRef]
- Zhang, L.; Li, C.; Zhang, Z.; Zhang, Z.; Jin, Q.Q.; Li, L.; Hölscher, C. DA5-CH and Semaglutide Protect against Neurodegeneration and Reduce α-Synuclein Levels in the 6-OHDA Parkinson’s Disease Rat Model. Park. Dis. 2022, 2022, 1428817. [Google Scholar] [CrossRef]
- FDA Drug Approvals and Databases. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trial-snapshot-ozempic (accessed on 15 February 2024).
- FDA Medicines. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/ozempic (accessed on 15 February 2024).
- Liu, D.X.; Zhao, C.S.; Wei, X.N.; Ma, Y.P.; Wu, J.K. Semaglutide Protects against 6-OHDA Toxicity by Enhancing Autophagy and Inhibiting Oxidative Stress. Park. Dis. 2022, 2022, 6813017. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Chuang, Y.C.; Lin, T.K.; Yang, J.L. Alternative role of glucagon-like Peptide-1 receptor agonists in neurodegenerative diseases. Eur. J. Pharmacol. 2023, 938, 175439. [Google Scholar] [CrossRef] [PubMed]
- Laurindo, L.F.; Barbalho, S.M.; Guiguer, E.L.; da Silva Soares de Souza, M.; de Souza, G.A.; Fidalgo, T.M.; Araújo, A.C.; de Souza Gonzaga, H.F.; de Bortoli Teixeira, D.; de Oliveira Silva Ullmann, T.; et al. GLP-1a: Going beyond Traditional Use. Int. J. Mol. Sci. 2022, 23, 739. [Google Scholar] [CrossRef] [PubMed]
- Kabahizi, A.; Wallace, B.; Lieu, L.; Chau, D.; Dong, Y.; Hwang, E.S.; Williams, K.W. Glucagon-like peptide-1 (GLP-1) signalling in the brain: From neural circuits and metabolism to therapeutics. Br. J. Pharmacol. 2022, 179, 600–624. [Google Scholar] [CrossRef] [PubMed]
- McLean, B.A.; Wong, C.K.; Campbell, J.E.; Hodson, D.J.; Trapp, S.; Drucker, D.J. Revisiting the Complexity of GLP-1 Action from Sites of Synthesis to Receptor Activation. Endocr. Rev. 2021, 42, 101–132. [Google Scholar] [CrossRef] [PubMed]
- Mayendraraj, A.; Rosenkilde, M.M.; Gasbjerg, L.S. GLP-1 and GIP receptor signaling in beta cells—A review of receptor interactions and co-stimulation. Peptides 2022, 151, 170749. [Google Scholar] [CrossRef] [PubMed]
- Kopp, K.O.; Glotfelty, E.J.; Li, Y.; Greig, N.H. Glucagon-like peptide-1 (GLP-1) receptor agonists and neuroinflammation: Implications for neurodegenerative disease treatment. Pharmacol. Res. 2022, 186, 106550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, W.; Tian, X. The pleiotropic of GLP-1/GLP-1R axis in central nervous system diseases. Int. J. Neurosci. 2023, 133, 473–491. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, M.K.; Karuppasamy, M.; Sahoo, B.M. Therapeutic Potential of Semaglutide, a Newer GLP-1 Receptor Agonist, in Abating Obesity, Non-Alcoholic Steatohepatitis and Neurodegenerative diseases: A Narrative Review. Pharm. Res. 2022, 39, 1233–1248. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Li, L.; Hölscher, C. Neuroprotective effects of the novel GLP-1 long acting analogue semaglutide in the MPTP Parkinson’s disease mouse model. Neuropeptides 2018, 71, 70–80. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, H.; Liu, X.; Guo, X. Hypoglycemic medicines in the treatment of Alzheimer’s disease: Pathophysiological links between AD and glucose metabolism. Front. Pharmacol. 2023, 14, 1138499. [Google Scholar] [CrossRef]
- Mulvaney, C.A.; Duarte, G.S.; Handley, J.; Evans, D.J.; Menon, S.; Wyse, R.; Emsley, H.C. GLP-1 receptor agonists for Parkinson’s disease. Cochrane Database Syst. Rev. 2020, 7, CD012990. [Google Scholar] [CrossRef]
- Chang, Y.F.; Zhang, D.; Hu, W.M.; Liu, D.X.; Li, L. Semaglutide-mediated protection against Aβ correlated with enhancement of autophagy and inhibition of apotosis. J. Clin. Neurosci. 2020, 81, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Li, X.R.; Chai, S.F.; Li, W.R.; Li, S.; Hou, M.; Li, J.L.; Ye, Y.C.; Cai, H.Y.; Hölscher, C.; et al. Semaglutide ameliorates cognition and glucose metabolism dysfunction in the 3xTg mouse model of Alzheimer’s disease via the GLP-1R/SIRT1/GLUT4 pathway. Neuropharmacology 2023, 240, 109716. [Google Scholar] [CrossRef]
- Dai, C.; Tan, C.; Zhao, L.; Liang, Y.; Liu, G.; Liu, H.; Zhong, Y.; Liu, Z.; Mo, L.; Liu, X.; et al. Glucose metabolism impairment in Parkinson’s disease. Brain Res. Bull. 2023, 199, 110672. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.J.; Ma, D.; Song, L.L.; Zhai, Z.N.; Tao, Y.; Zhang, Y.; Cai, L.Y.; Hou, Y.H.; Chen, H.Y.; Wang, L.; et al. The role of GLP-1/GIP receptor agonists in Alzheimer’s disease. Adv. Clin. Exp. Med. 2020, 29, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Gouveri, E.; Popovic, D.S.; Papanas, N. Potential New Therapeutic Implications of Semaglutide: New Colours of the Rainbow? Diabetes Ther. 2024, 15, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Nowell, J.; Blunt, E.; Edison, P. Incretin and insulin signaling as novel therapeutic targets for Alzheimer’s and Parkinson’s disease. Mol. Psychiatry 2023, 28, 217–229. [Google Scholar] [CrossRef]
- Reich, N.; Hölscher, C. The neuroprotective effects of glucagon-like peptide 1 in Alzheimer’s and Parkinson’s disease: An in-depth review. Front Neurosci. 2022, 16, 970925. [Google Scholar] [CrossRef]
- Sadek, M.A.; Kandil, E.A.; El Sayed, N.S.; Sayed, H.M.; Rabie, M.A. Semaglutide, a novel glucagon-like peptide-1 agonist, amends experimental autoimmune encephalomyelitis-induced multiple sclerosis in mice: Involvement of the PI3K/Akt/GSK-3β pathway. Int. Immunopharmacol. 2023, 115, 109647. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Exploring Molecular Targets for Mitochondrial Therapies in Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 12486. [Google Scholar] [CrossRef] [PubMed]
- Tipa, R.O.; Balan, D.G.; Georgescu, M.T.; Ignat, L.A.; Vacaroiu, I.A.; Georgescu, D.E.; Raducu, L.; Mihai, D.A.; Chiperi, L.V.; Balcangiu-Stroescu, A.E. A Systematic Review of Semaglutide’s Influence on Cognitive Function in Preclinical Animal Models and Cell-Line Studies. Int. J. Mol. Sci. 2024, 25, 4972. [Google Scholar] [CrossRef]
- Chen, X.; Chen, S.; Li, Z.; Zhu, R.; Jia, Z.; Ban, J.; Zhen, R.; Chen, X.; Pan, X.; Ren, Q.; et al. Effect of semaglutide and empagliflozin on cognitive function and hippocampal phosphoproteomic in obese mice. Front. Pharmacol. 2023, 14, 975830. [Google Scholar] [CrossRef] [PubMed]
- Rebosio, C.; Balbi, M.; Passalacqua, M.; Ricciarelli, R.; Fedele, E. Presynaptic GLP-1 receptors enhance the depolarization-evoked release of glutamate and GABA in the mouse cortex and hippocampus. Biofactors 2018, 44, 148–157. [Google Scholar] [CrossRef]
- Park, J.S.; Kam, T.I.; Lee, S.; Park, H.; Oh, Y.; Kwon, S.H.; Song, J.J.; Kim, D.; Kim, H.; Jhaldiyal, A.; et al. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Day, C.M.; Abdella, S.; Garg, S. Alzheimer’s disease current therapies, novel drug delivery systems and future directions for better disease management. J. Control. Release 2024, 367, 402–424. [Google Scholar] [CrossRef] [PubMed]
- Lisco, G.; De Tullio, A.; Iovino, M.; Disoteo, O.; Guastamacchia, E.; Giagulli, V.A.; Triggiani, V. Dopamine in the Regulation of Glucose Homeostasis, Pathogenesis of Type 2 Diabetes, and Chronic Conditions of Impaired Dopamine Activity/Metabolism: Implication for Pathophysiological and Therapeutic Purposes. Biomedicines 2023, 11, 2993. [Google Scholar] [CrossRef]
- Gentilella, R.; Pechtner, V.; Corcos, A.; Consoli, A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: Are they all the same? Diabetes Metab. Res. Rev. 2019, 35, e3070. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Choi, H.I.; Wang, Y.; Luo, Y.; Hoffer, B.J.; Greig, N.H. A New Treatment Strategy for Parkinson’s Disease through the Gut-Brain Axis: The Glucagon-Like Peptide-1 Receptor Pathway. Cell Transplant. 2017, 26, 1560–1571. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Duda, P.; Wiśniewski, J.; Wójtowicz, T.; Wójcicka, O.; Jaśkiewicz, M.; Drulis-Fajdasz, D.; Rakus, D.; McCubrey, J.A.; Gizak, A. Targeting GSK3 signaling as a potential therapy of neurodegenerative diseases and aging. Expert. Opin. Ther. Targets. 2018, 10, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Kalinderi, K.; Papaliagkas, V.; Fidani, L. GLP-1 Receptor Agonists: A New Treatment in Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 3812. [Google Scholar] [CrossRef] [PubMed]
- Diz-Chaves, Y.; Maastor, Z.; Spuch, C.; Lamas, J.A.; González-Matías, L.C.; Mallo, F. Glucagon-like peptide 1 receptor activation: Anti-inflammatory effects in the brain. Neural Regen. Res. 2024, 19, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.M.; Tronieri, J.S.; Amaro, A.; Wadden, T.A. Semaglutide for the treatment of obesity. Trends Cardiovasc. Med. 2023, 33, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, N.C.; Davies, M.J.; Lingvay, I.; Knop, F.K. Semaglutide for the treatment of overweight and obesity: A review. Diabetes Obes. Metab. 2023, 25, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Park, E.J.; Choi, M.; Oh, H.S.; An, Y.; Kim, T.; Kim, T.H.; Shin, B.S.; Shin, S. Novel LC-MS/MS analysis of the GLP-1 analog semaglutide with its application to pharmacokinetics and brain distribution studies in rats. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2023, 1221, 123688. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.; Isaacs, D.; Clements, J.N. Pharmacokinetics and Clinical Implications of Semaglutide: A New Glucagon-Like Peptide (GLP)-1 Receptor Agonist. Clin. Pharmacokinet. 2018, 57, 1529–1538. [Google Scholar] [CrossRef]
- Secher, A.; Jelsing, J.; Baquero, A.F.; Hecksher-Sørensen, J.; Cowley, M.A.; Dalbøge, L.S.; Hansen, G.; Grove, K.L.; Pyke, C.; Raun, K.; et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J. Clin. Investig. 2014, 124, 4473–4488. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.B.; Pyke, C.; Rasch, M.G.; Dahl, A.B.; Knudsen, L.B.; Secher, A. Characterization of the Glucagonlike Peptide-1 Receptor in Male Mouse Brain Using a Novel Antibody and In Situ Hybridization. Endocrinology 2018, 159, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Kapitza, C.; Nosek, L.; Jensen, L.; Hartvig, H.; Jensen, C.B.; Flint, A. Semaglutide, a once-weekly human GLP-1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J. Clin. Pharmacol. 2015, 55, 497–504. [Google Scholar] [CrossRef]
- Smith, B.; Ownby, R.L. Disease-Modifying Treatments and Their Future in Alzheimer’s Disease Management. Cureus. 2024, 16, e56105. [Google Scholar] [CrossRef] [PubMed]
- Marbury, T.C.; Flint, A.; Jacobsen, J.B.; Derving Karsbøl, J.; Lasseter, K. Pharmacokinetics and Tolerability of a Single Dose of Semaglutide, a Human Glucagon-Like Peptide-1 Analog, in Subjects With and Without Renal Impairment. Clin. Pharmacokinet. 2017, 56, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Adem, M.A.; Decourt, B.; Sabbagh, M.N. Pharmacological Approaches Using Diabetic Drugs Repurposed for Alzheimer’s Disease. Biomedicines 2024, 12, 99. [Google Scholar] [CrossRef]
- Gabery, S.; Salinas, C.G.; Paulsen, S.J.; Ahnfelt-Rønne, J.; Alanentalo, T.; Baquero, A.F.; Buckley, S.T.; Farkas, E.; Fekete, C.; Frederiksen, K.S.; et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. 2020, 5, e133429. [Google Scholar] [CrossRef] [PubMed]
- Złotek, M.; Kurowska, A.; Herbet, M.; Piątkowska-Chmiel, I. GLP-1 Analogs, SGLT-2, and DPP-4 Inhibitors: A Triad of Hope for Alzheimer’s Disease Therapy. Biomedicines 2023, 11, 3035. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yu, F.; Lyu, Y.; Lu, X. Promising candidates from drug clinical trials: Implications for clinical treatment of Alzheimer’s disease in China. Front. Neurol. 2022, 13, 1034243. [Google Scholar] [CrossRef] [PubMed]
- Vejandla, B.; Savani, S.; Appalaneni, R.; Veeravalli, R.S.; Gude, S.S. Alzheimer’s Disease: The Past, Present, and Future of a Globally Progressive Disease. Cureus 2024, 16, e51705. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.-C.S.; Jennings, G.T.; Mohsen, M.O.; Vogel, M.; Bachmann, M.F. Alzheimer’s Disease: A Brief History of Immunotherapies Targeting Amyloid β. Int. J. Mol. Sci. 2023, 24, 3895. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.K.; Das, S.; Joseph, A.; Shenoy, G.G.; Alex, A.T.; Mudgal, J. Neurodegenerative Pathways in Alzheimer’s Disease: A Review. Curr. Neuropharmacol. 2021, 19, 679–692. [Google Scholar] [CrossRef]
- Wetering, J.V.; Geut, H.; Bol, J.J.; Galis, Y.; Timmermans, E.; Twisk, J.W.R.; Hepp, D.H.; Morella, M.L.; Pihlstrom, L.; Lemstra, A.W.; et al. Neuroinflammation is associated with Alzheimer’s disease co-pathology in dementia with Lewy bodies. Acta Neuropathol. Commun. 2024, 12, 73. [Google Scholar] [CrossRef]
- Subramanian, J.; Savage, J.C.; Tremblay, M.È. Synaptic Loss in Alzheimer’s Disease: Mechanistic Insights Provided by Two-Photon in vivo Imaging of Transgenic Mouse Models. Front. Cell. Neurosci. 2020, 14, 592607. [Google Scholar] [CrossRef] [PubMed]
- Manuello, J.; Min, J.; McCarthy, P.; Alfaro-Almagro, F.; Lee, S.; Smith, S.; Elliott, L.T.; Winkler, A.M.; Douaud, G. The effects of genetic and modifiable risk factors on brain regions vulnerable to ageing and disease. Nat. Commun. 2024, 15, 2576. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Liu, H.; Liu, X.; Zhang, W.; Zhang, S.; Jiao, B. APP, PSEN1, and PSEN2 Variants in Alzheimer’s Disease: Systematic Re-evaluation According to ACMG Guidelines. Front. Aging Neurosci. 2021, 13, 695808. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, H.S. Alzheimer’s Disease: A Decreased Cerebral Blood Flow to Critical Intraneuronal Elements Is the Cause. J. Alzheimers Dis. 2022, 85, 1419–1422. [Google Scholar] [CrossRef] [PubMed]
- Tamagno, E.; Guglielmotto, M.; Vasciaveo, V.; Tabaton, M. Oxidative Stress and Beta Amyloid in Alzheimer’s Disease. Which Comes First: The Chicken or the Egg? Antioxidants 2021, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative Stress Occurs Prior to Amyloid Aβ Plaque Formation and Tau Phosphorylation in Alzheimer’s Disease: Role of Glutathione and Metal Ions. ACS Chem. Neurosci. 2023, 14, 2944–2954. [Google Scholar] [CrossRef]
- Xia, W.; Yu, H.; Wen, P. Meta-analysis on GLP-1 mediated modulation of autophagy in islet β-cells: Prospectus for improved wound healing in type 2 diabetes. Int. Wound, J. 2024, 21, e14841. [Google Scholar] [CrossRef]
- Dave, B.P.; Shah, Y.B.; Maheshwari, K.G.; Mansuri, K.A.; Prajapati, B.S.; Postwala, H.I.; Chorawala, M.R. Pathophysiological Aspects and Therapeutic Armamentarium of Alzheimer’s Disease: Recent Trends and Future Development. Cell. Mol. Neurobiol. 2023, 43, 3847–3884. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef]
- Currais, A.; Prior, M.; Lo, D.; Jolivalt, C.; Schubert, D.; Maher, P. Diabetes exacerbates amyloid and neurovascular pathology in aging-accelerated mice. Aging Cell 2012, 11, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.; Bose, N.; Nisenbaum, L.; Partrick, K.A.; Fillit, H.M. The Critical Role of Biomarkers for Drug Development Targeting the Biology of Aging. J. Prev. Alzheimers Dis. 2023, 10, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Nørgaard, C.H.; Friedrich, S.; Hansen, C.T.; Gerds, T.; Ballard, C.; Møller, D.V.; Knudsen, L.B.; Kvist, K.; Zinman, B.; Holm, E.; et al. Treatment with glucagon-like peptide-1 receptor agonists and incidence of dementia: Data from pooled double-blind randomized controlled trials and nationwide disease and prescription registers. Alzheimers Dement. 2022, 8, e12268. [Google Scholar] [CrossRef]
- Muriach, M.; Flores-Bellver, M.; Romero, F.J.; Barcia, J.M. Diabetes and the brain: Oxidative stress, inflammation, and autophagy. Oxid. Med. Cell Longev. 2014, 2014, 102158. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Soga, T.; Okano, H.J.; Parhar, I. α-Synuclein-mediated neurodegeneration in Dementia with Lewy bodies: The pathobiology of a paradox. Cell Biosci. 2021, 11, 196. [Google Scholar] [CrossRef] [PubMed]
- Borghammer, P.; Okkels, N.; Weintraub, D. Parkinson’s Disease and Dementia with Lewy Bodies: One and the Same. J. Park. Dis. 2024, 14, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Kouli, A.; Spindler, L.R.B.; Fryer, T.D.; Hong, Y.T.; Malpetti, M.; Aigbirhio, F.I.; White, S.R.; Camacho, M.; O’Brien, J.T.; Williams-Gray, C.H. Neuroinflammation is linked to dementia risk in Parkinson’s disease. Brain 2024, 147, 923–935. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, X.; Yin, Y.; Yang, F.; Zhang, Y.; He, X.; Wen, D.; Li, B.X.; Ma, K. Association Between Neuroinflammation and Parkinson’s Disease: A Comprehensive Mendelian Randomization Study. Mol. Neurobiol. 2024, 1–11. [Google Scholar] [CrossRef]
- Payne, T.; Burgess, T.; Bradley, S.; Roscoe, S.; Sassani, M.; Dunning, M.J.; Hernandez, D.; Scholz, S.; McNeill, A.; Taylor, R.; et al. Multimodal assessment of mitochondrial function in Parkinson’s disease. Brain 2024, 147, 267–280. [Google Scholar] [CrossRef]
- Blagov, A.; Postnov, A.; Sukhorukov, V.; Popov, M.; Uzokov, J.; Orekhov, A. Significance of Mitochondrial Dysfunction in the Pathogenesis of Parkinson’s Disease. Front. Biosci. (Landmark Ed.) 2024, 29, 36. [Google Scholar] [CrossRef]
- Bhore, N.; Bogacki, E.C.; O’Callaghan, B.; Plun-Favreau, H.; Lewis, P.A.; Herbst, S. Common genetic risk for Parkinson’s disease and dysfunction of the endo-lysosomal system. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2024, 379, 20220517. [Google Scholar] [CrossRef]
- Hull, A.; Atilano, M.L.; Gergi, L.; Kinghorn, K.J. Lysosomal storage, impaired autophagy and innate immunity in Gaucher and Parkinson’s diseases: Insights for drug discovery. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2024, 379, 20220381. [Google Scholar] [CrossRef]
- Volta, M. Roles of neuronal lysosomes in the etiology of Parkinson’s disease. Neural Regen. Res. 2024, 19, 1981–1983. [Google Scholar] [CrossRef]
- Poupon-Bejuit, L.; Hughes, M.P.; Liu, W.; Geard, A.; Faour-Slika, N.; Whaler, S.; Massaro, G.; Rahim, A.A. A GLP1 receptor agonist diabetes drug ameliorates neurodegeneration in a mouse model of infantile neurometabolic disease. Sci. Rep. 2022, 12, 13825. [Google Scholar] [CrossRef]
- Cerroni, C.; Steiner, A.; Seanez, L.; Kwon, S.; Lewis, A.S. Effects of repeated developmental GLP-1R agonist exposure on young adult behavior and hippocampal structure in mice. Neurosci. Lett. 2023, 808, 137299. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.S.; Mak, W.Q.; Tan, L.K.S.; Ng, C.X.; Chan, H.H.; Yeow, S.H.; Foo, J.B.; Ong, Y.S.; How, C.W.; Khaw, K.Y. Microbiota-gut-brain axis and its therapeutic applications in neurodegenerative diseases. Signal Transduct. Target. Ther. 2024, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Battini, V.; Barbieri, M.A.; Carnovale, C.; Spina, E.; Clementi, E.; Sessa, M. Comparing major and mild cognitive impairment risks in older type-2 diabetic patients: A Danish register-based study on dipeptidyl peptidase-4 inhibitors vs. glucagon-like peptide-1 analogues. J. Neurol. 2024, 271, 3417–3425. [Google Scholar] [CrossRef] [PubMed]
- Dei Cas, A.; Micheli, M.M.; Aldigeri, R.; Gardini, S.; Ferrari-Pellegrini, F.; Perini, M.; Messa, G.; Antonini, M.; Spigoni, V.; Cinquegrani, G.; et al. Long-acting exenatide does not prevent cognitive decline in mild cognitive impairment: A proof-of-concept clinical trial. J. Endocrinol. Investig. 2024, 1–11. [Google Scholar] [CrossRef]
- Husain, M.; Bain, S.C.; Jeppesen, O.K.; Lingvay, I.; Sørrig, R.; Treppendahl, M.B.; Vilsbøll, T. Semaglutide (SUSTAIN and PIONEER) Reduces Cardiovascular Events in Type 2 Diabetes Across Varying Cardiovascular Risk. Diabetes Obes. Metab. 2020, 22, 442–451. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Li, L.; Hölscher, C. Semaglutide is Neuroprotective and Reduces α-Synuclein Levels in the Chronic MPTP Mouse Model of Parkinson’s Disease. J. Park. Dis. 2019, 9, 157–171. [Google Scholar] [CrossRef] [PubMed]

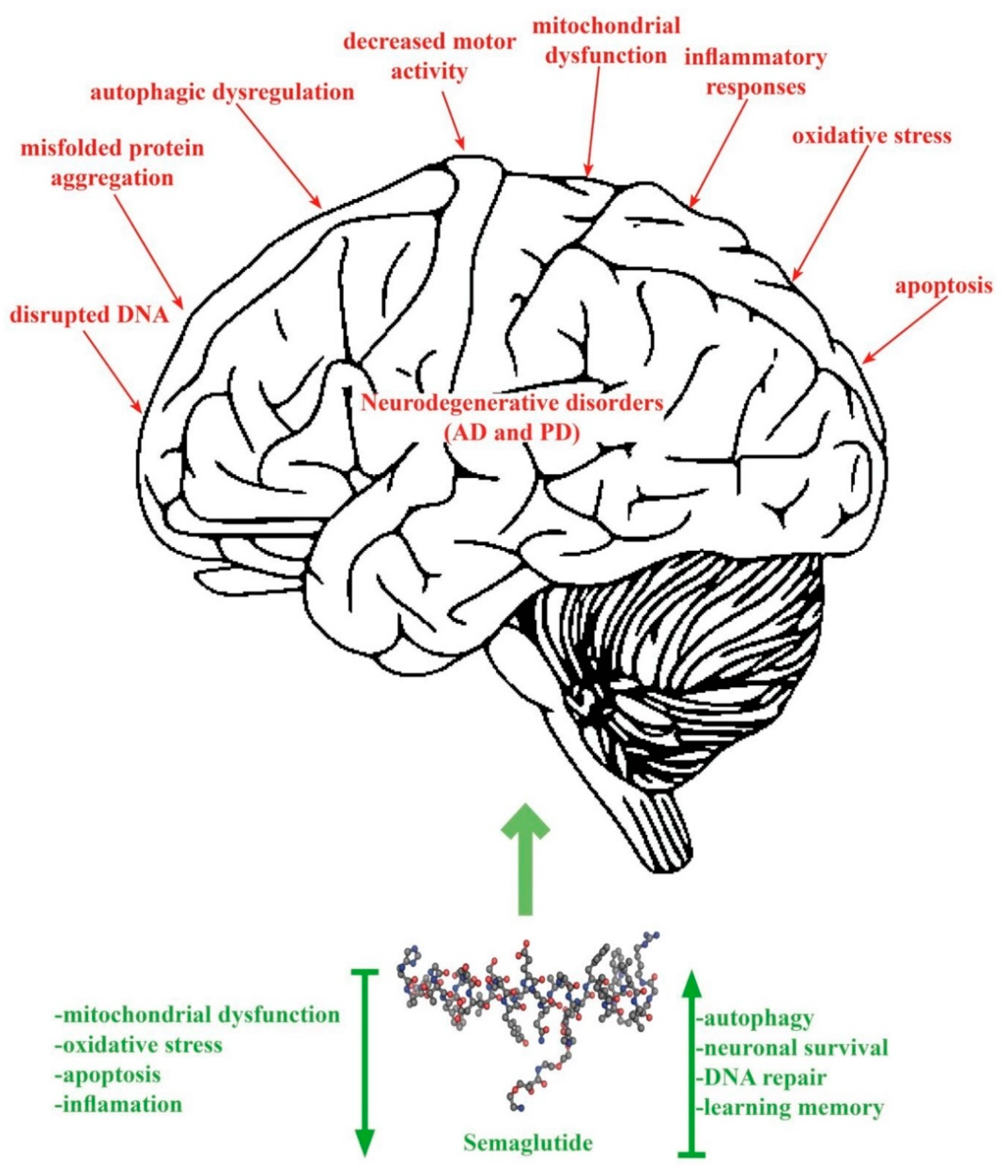
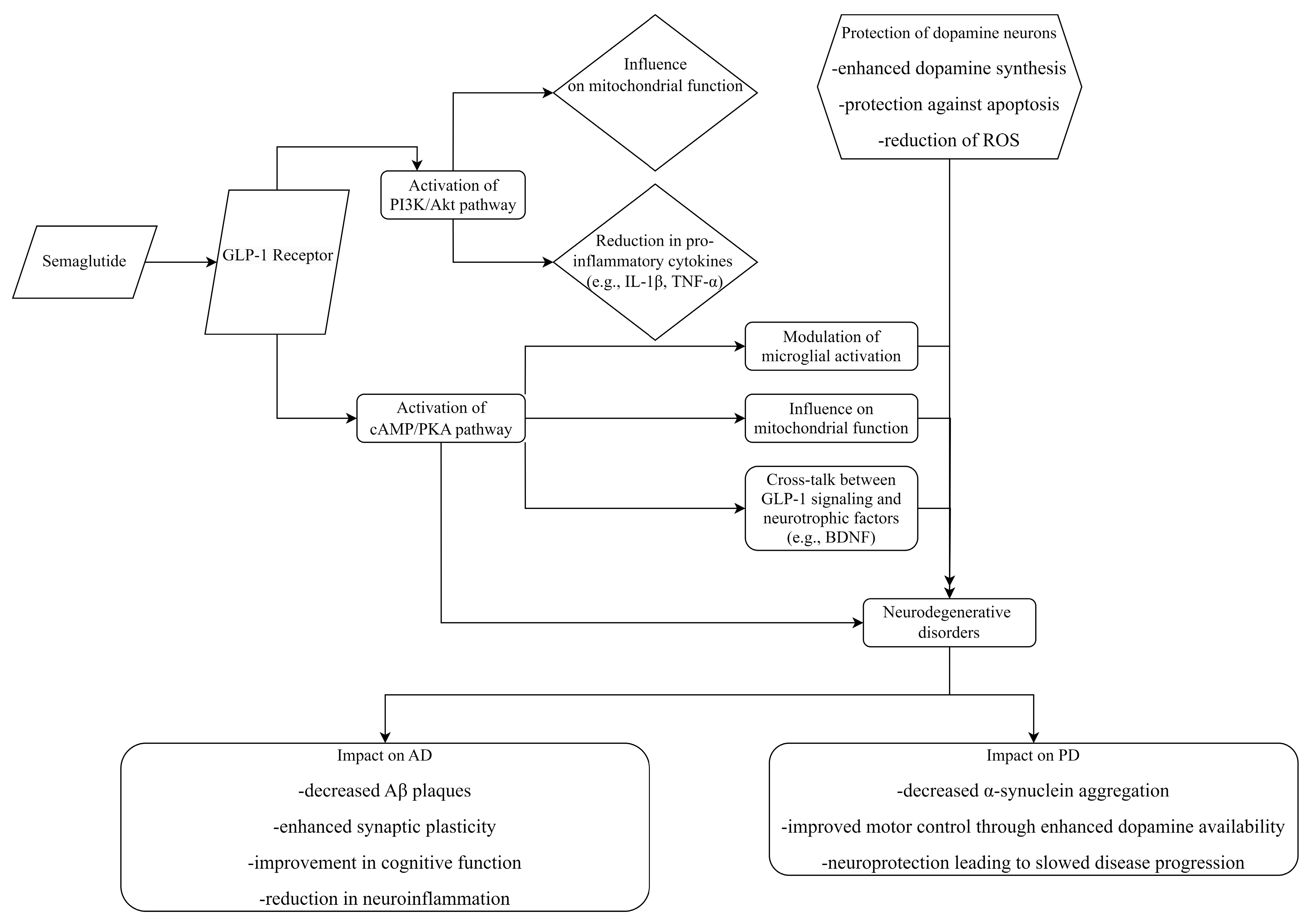

{kind=link}
{kind=link}
{kind=link}
| Article | Animals/ Cells | Study Design (Randomization, Blind) | Pathology Model | Biochemical Tests | Behavior Tests | SEM (dose, Administration, Period of Time) | Other Drugs |
|---|---|---|---|---|---|---|---|
| Poupon-Bejuit 2022 [97] | Mice | Randomized, double-blind | Pla2g6−/− transgenic mice |
| Open field Rotarod Vertical pole | 0.5, 0.25, 0.15 μg/g, i.p. injected once a week until the end of the experiment | No |
| Zhang 2018 [32] | Mice | Randomized | MPTP 20 mg/kg i.p. once daily for 7 days |
| Open field Rotarod Footprint | 25 nmol/kg, i.p., once daily for 7 days | Liraglutide 25 nmol/kg i.p. once daily for 7 days |
| Zhang 2019 [103] | Mice | In vivo, randomized | MPTP 20 mg/kg i.p. once daily for 30 days |
| Open field Rotarod Footprint gait Grip strength | 25 nmol/kg, i.p., once daily for 30 days | Liraglutide 25 nmol/kg i.p. once daily for 30 days |
| Zhang 2022 [20] | Rats | Randomized, blind | 6-OHDA |
| Apomorphine Rotation | 25 nmol/kg, i.p., once daily for 30 days postlesion | DA5-CH 25 nmol/kg, i.p., once daily for 30 days postlesion |
| Liu 2022 [23] | SH-SY5Y cells | In vitro | 6-OHDA |
| Not applicable | 0, 1, 10, 100 nmol/L semaglutide | Liraglutide 0, 1, 10, 100 nmol/L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meca, A.D.; Boboc, I.K.S.; Mititelu-Tartau, L.; Bogdan, M. Unlocking the Potential: Semaglutide’s Impact on Alzheimer’s and Parkinson’s Disease in Animal Models. Curr. Issues Mol. Biol. 2024, 46, 5929-5949. https://doi.org/10.3390/cimb46060354
Meca AD, Boboc IKS, Mititelu-Tartau L, Bogdan M. Unlocking the Potential: Semaglutide’s Impact on Alzheimer’s and Parkinson’s Disease in Animal Models. Current Issues in Molecular Biology. 2024; 46(6):5929-5949. https://doi.org/10.3390/cimb46060354
Chicago/Turabian StyleMeca, Andreea Daniela, Ianis Kevyn Stefan Boboc, Liliana Mititelu-Tartau, and Maria Bogdan. 2024. "Unlocking the Potential: Semaglutide’s Impact on Alzheimer’s and Parkinson’s Disease in Animal Models" Current Issues in Molecular Biology 46, no. 6: 5929-5949. https://doi.org/10.3390/cimb46060354
APA StyleMeca, A. D., Boboc, I. K. S., Mititelu-Tartau, L., & Bogdan, M. (2024). Unlocking the Potential: Semaglutide’s Impact on Alzheimer’s and Parkinson’s Disease in Animal Models. Current Issues in Molecular Biology, 46(6), 5929-5949. https://doi.org/10.3390/cimb46060354