
Oxytocin Exhibits Neuroprotective Effects on Hippocampal Cultures under Severe Oxygen–Glucose Deprivation Conditions
 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
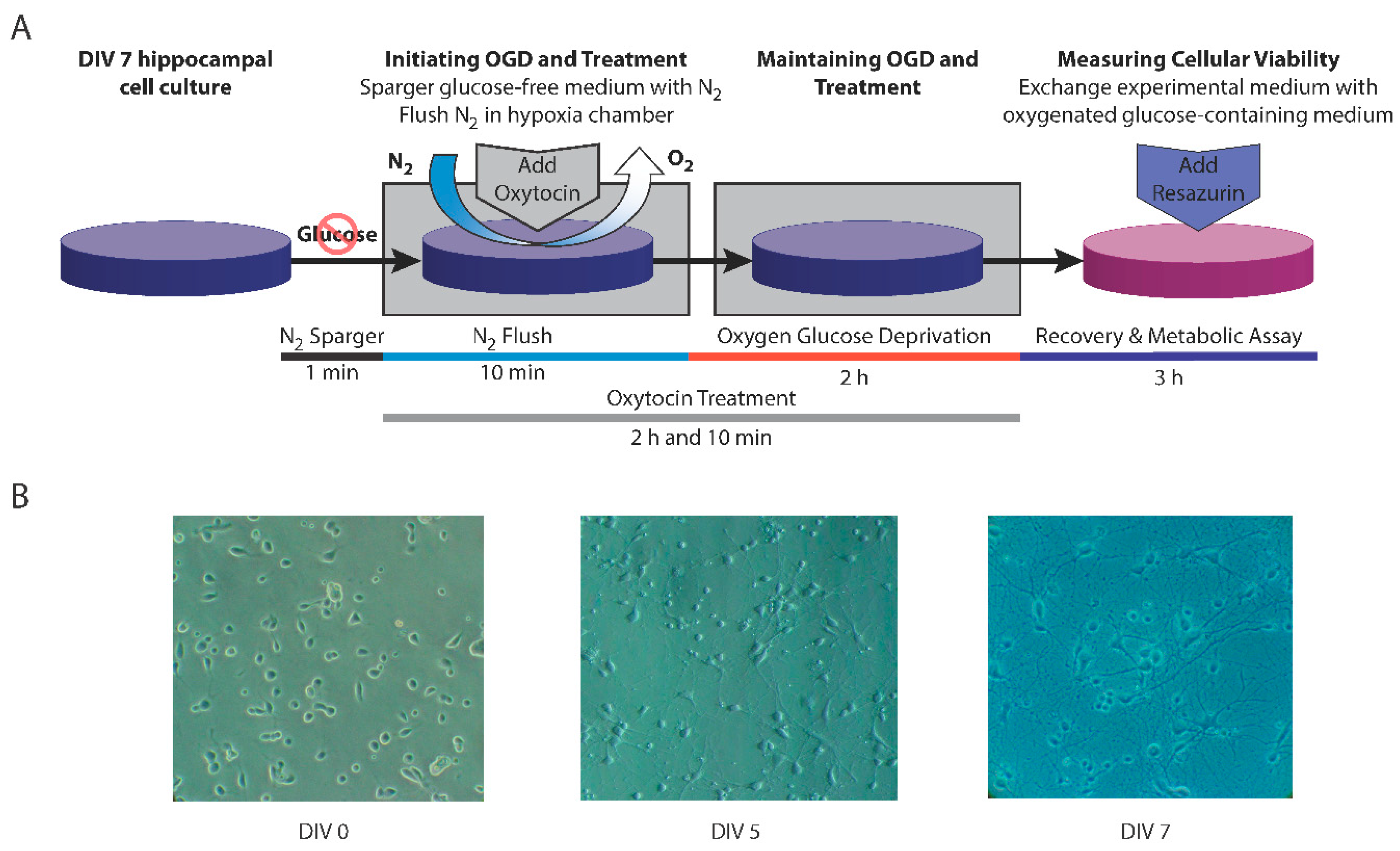
2.1. Primary Cultures of Rat Hippocampal Neurons
2.2. Exposure to Oxygen–Glucose Deprivation
2.3. Treatment with Oxytocin
2.4. Assessment of Cellular Metabolism and Viability
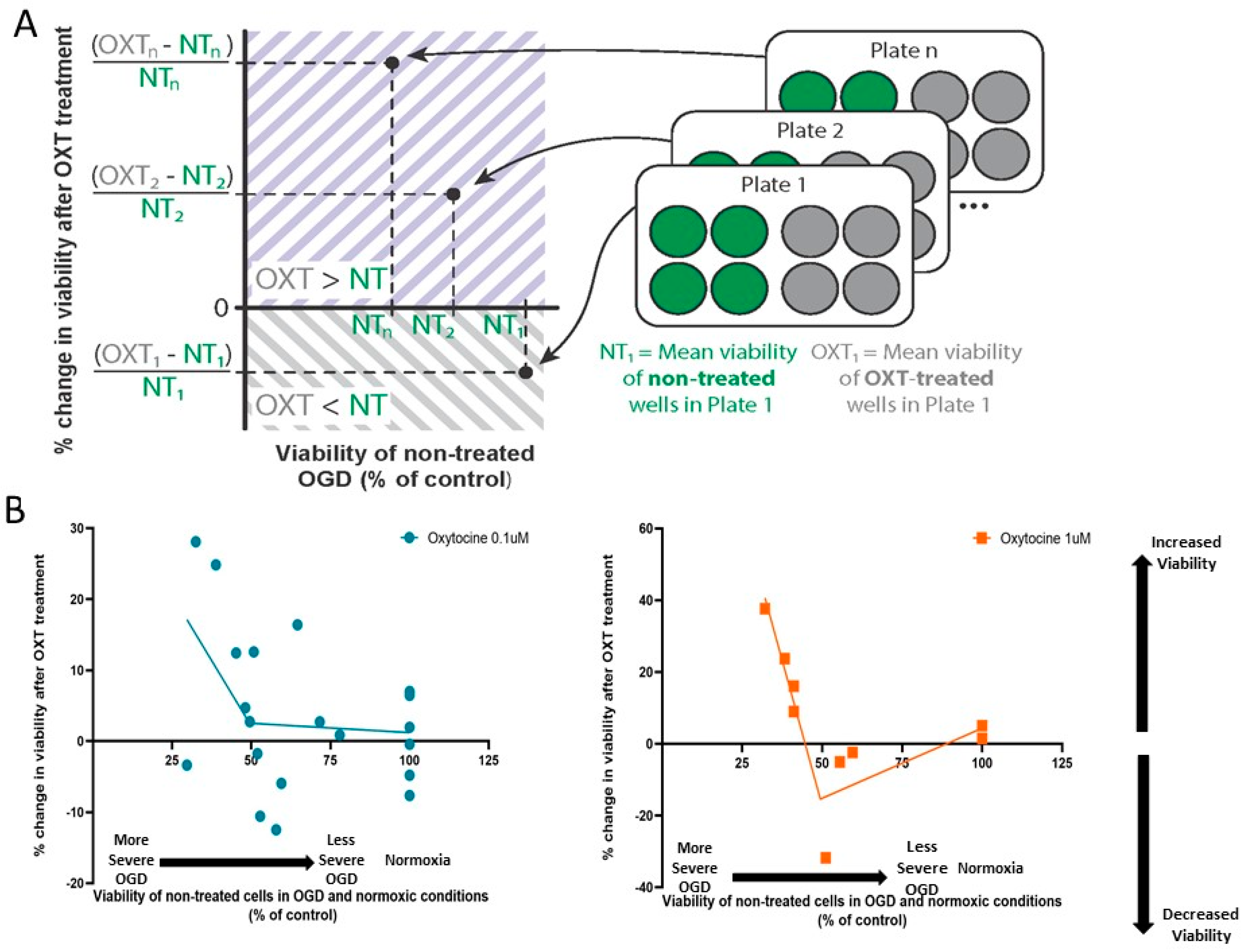
2.5. Data Analysis
3. Results
3.1. Oxytocin Did Not Affect the Viability of DIV7 Hippocampal Cell Cultures under Normoxic Conditions
3.2. Cellular Viability after 2 h of OGD Followed a Normal Distribution
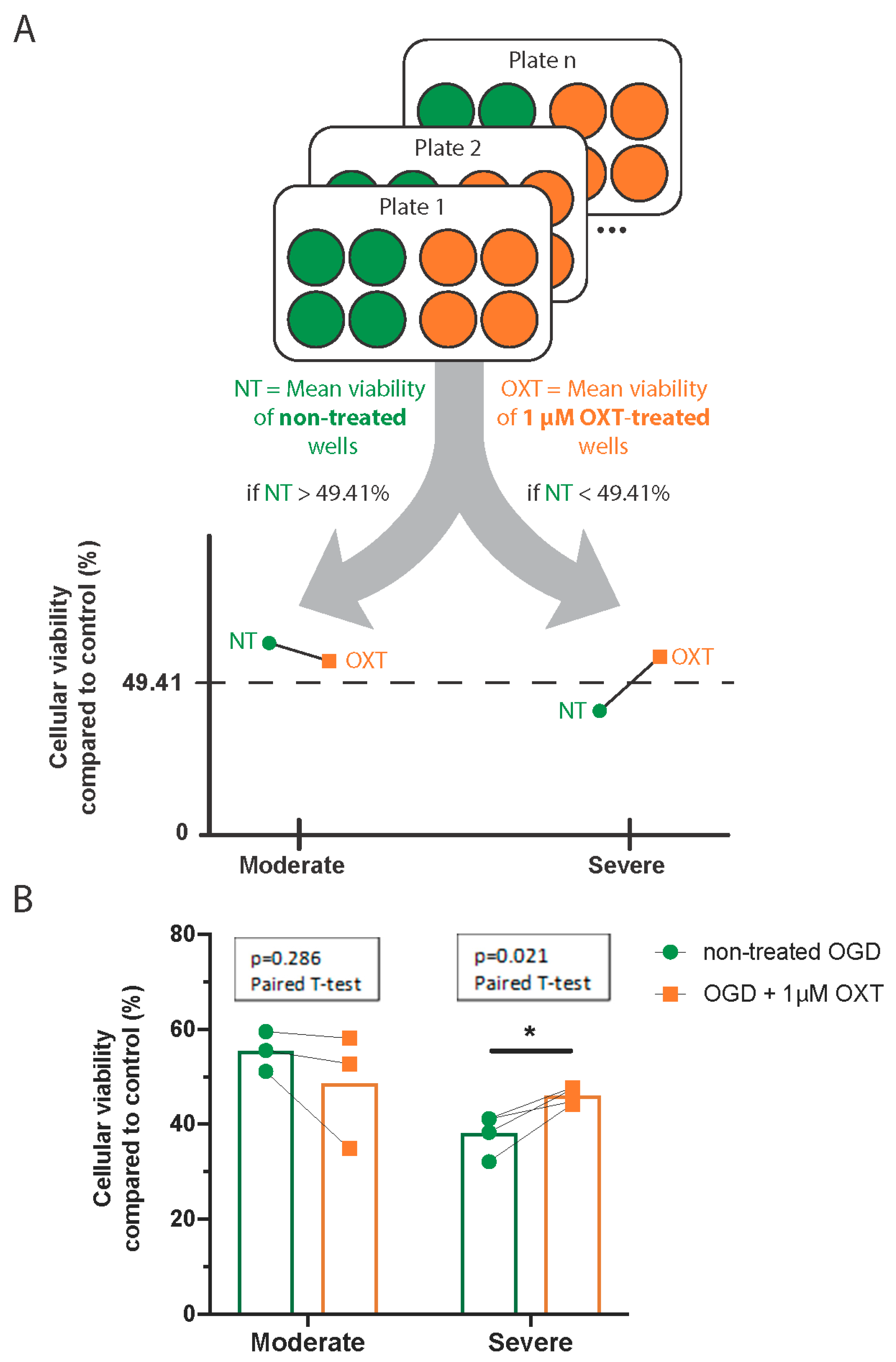
3.3. Oxytocin’s Effect on Cellular Viability Changed with OGD Severity
3.4. Administration of 1 µM Oxytocin Increased the Viability of DIV7 Hippocampal Neurons Exposed to Severe OGD but Not of Those Exposed to Moderate OGD
3.5. Administration of 0.1 µM Oxytocin Had No Significant Effect on the Viability of Cultures Exposed to OGD, Regardless of OGD Severity
4. Discussion
Study Limitations and Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gillam-Krakauer, M.; Gowen, C.W., Jr. Birth Asphyxia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Zagrean, A.-M.; Georgescu, I.-A.; Iesanu, M.I.; Ionescu, R.-B.; Haret, R.M.; Panaitescu, A.M.; Zagrean, L. Chapter Three—Oxytocin and Vasopressin in the Hippocampus. In Hormones, Regulators and Hippocampus; Vitamins and Hormones; Litwack, G., Ed.; Academic Press: Cambridge, MA, USA, 2022; Volume 118, pp. 83–127. [Google Scholar]
- Russell, J.A.; Leng, G.; Douglas, A.J. The Magnocellular Oxytocin System, the Fount of Maternity: Adaptations in Pregnancy. Front. Neuroendocrinol. 2003, 24, 27–61. [Google Scholar] [CrossRef]
- Krol, K.M.; Moulder, R.G.; Lillard, T.S.; Grossmann, T.; Connelly, J.J. Epigenetic Dynamics in Infancy and the Impact of Maternal Engagement. Sci. Adv. 2019, 5, eaay0680. [Google Scholar] [CrossRef]
- Besser, G.M.; Mortimer, C.H. Hypothalamic Regulatory Hormones: A Review. J. Clin. Pathol. 1974, 27, 173–184. [Google Scholar] [CrossRef]
- Tyzio, R.; Cossart, R.; Khalilov, I.; Minlebaev, M.; Hübner, C.A.; Represa, A.; Ben-Ari, Y.; Khazipov, R. Maternal Oxytocin Triggers a Transient Inhibitory Switch in GABA Signaling in the Fetal Brain during Delivery. Science 2006, 314, 1788–1792. [Google Scholar] [CrossRef]
- Walter, M.H.; Abele, H.; Plappert, C.F. The Role of Oxytocin and the Effect of Stress During Childbirth: Neurobiological Basics and Implications for Mother and Child. Front. Endocrinol. 2021, 12, 742236. [Google Scholar] [CrossRef]
- Wayock, C.P.; Meserole, R.L.; Saria, S.; Jennings, J.M.; Huisman, T.A.G.M.; Northington, F.J.; Graham, E.M. Perinatal Risk Factors for Severe Injury in Neonates Treated with Whole-Body Hypothermia for Encephalopathy. Am. J. Obstet. Gynecol. 2014, 211, 41.e1–41.e8. [Google Scholar] [CrossRef]
- Mairesse, J.; Zinni, M.; Pansiot, J.; Hassan-Abdi, R.; Demene, C.; Colella, M.; Charriaut-Marlangue, C.; Rideau Batista Novais, A.; Tanter, M.; Maccari, S.; et al. Oxytocin Receptor Agonist Reduces Perinatal Brain Damage by Targeting Microglia. Glia 2019, 67, 345–359. [Google Scholar] [CrossRef]
- Knoop, M.; Possovre, M.-L.; Jacquens, A.; Charlet, A.; Baud, O.; Darbon, P. The Role of Oxytocin in Abnormal Brain Development: Effect on Glial Cells and Neuroinflammation. Cells 2022, 11, 3899. [Google Scholar] [CrossRef]
- Ceanga, M.; Spataru, A.; Zagrean, A.-M. Oxytocin Is Neuroprotective against Oxygen-Glucose Deprivation and Reoxygenation in Immature Hippocampal Cultures. Neurosci. Lett. 2010, 477, 15–18. [Google Scholar] [CrossRef]
- Cao, W.; Pavlinec, C.; Gravenstein, N.; Seubert, C.N.; Martynyuk, A.E. Roles of Aldosterone and Oxytocin in Abnormalities Caused by Sevoflurane Anesthesia in Neonatal Rats. Anesthesiology 2012, 117, 791–800. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Oxytocin and Vasopressin, and the GABA Developmental Shift During Labor and Birth: Friends or Foes? Front. Cell. Neurosci. 2018, 12, 254. [Google Scholar] [CrossRef]
- Farrant, M.; Kaila, K. The Cellular, Molecular and Ionic Basis of GABA(A) Receptor Signalling. Prog. Brain Res. 2007, 160, 59–87. [Google Scholar] [CrossRef]
- Olsen, R.W.; Sieghart, W. GABA A Receptors: Subtypes Provide Diversity of Function and Pharmacology. Neuropharmacology 2009, 56, 141–148. [Google Scholar] [CrossRef]
- Watanabe, M.; Fukuda, A. Development and Regulation of Chloride Homeostasis in the Central Nervous System. Front. Cell Neurosci. 2015, 9, 371. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Gaiarsa, J.-L.; Tyzio, R.; Khazipov, R. GABA: A Pioneer Transmitter That Excites Immature Neurons and Generates Primitive Oscillations. Physiol. Rev. 2007, 87, 1215–1284. [Google Scholar] [CrossRef]
- Tyzio, R.; Represa, A.; Jorquera, I.; Ben-Ari, Y.; Gozlan, H.; Aniksztejn, L. The Establishment of GABAergic and Glutamatergic Synapses on CA1 Pyramidal Neurons is Sequential and Correlates with the Development of the Apical Dendrite. J. Neurosci. 1999, 19, 10372–10382. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Excitatory Actions of Gaba during Development: The Nature of the Nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef]
- Kahle, K.T.; Staley, K.J. The Bumetanide-Sensitive Na-K-2Cl Cotransporter NKCC1 as a Potential Target of a Novel Mechanism-Based Treatment Strategy for Neonatal Seizures. Neurosurg. Focus. 2008, 25, E22. [Google Scholar] [CrossRef]
- Dzhala, V.I.; Talos, D.M.; Sdrulla, D.A.; Brumback, A.C.; Mathews, G.C.; Benke, T.A.; Delpire, E.; Jensen, F.E.; Staley, K.J. NKCC1 Transporter Facilitates Seizures in the Developing Brain. Nat. Med. 2005, 11, 1205–1213. [Google Scholar] [CrossRef]
- Yamada, J.; Okabe, A.; Toyoda, H.; Kilb, W.; Luhmann, H.J.; Fukuda, A. Cl− Uptake Promoting Depolarizing GABA Actions in Immature Rat Neocortical Neurones Is Mediated by NKCC1. J. Physiol. 2004, 557, 829–841. [Google Scholar] [CrossRef]
- Leonzino, M.; Busnelli, M.; Antonucci, F.; Verderio, C.; Mazzanti, M.; Chini, B. The Timing of the Excitatory-to-Inhibitory GABA Switch Is Regulated by the Oxytocin Receptor via KCC2. Cell Rep. 2016, 15, 96–103. [Google Scholar] [CrossRef]
- Bos, R.; Sadlaoud, K.; Boulenguez, P.; Buttigieg, D.; Liabeuf, S.; Brocard, C.; Haase, G.; Bras, H.; Vinay, L. Activation of 5-HT2A Receptors Upregulates the Function of the Neuronal K-Cl Cotransporter KCC2. Proc. Natl. Acad. Sci. USA 2013, 110, 348–353. [Google Scholar] [CrossRef]
- Tasca, C.I.; Dal-Cim, T.; Cimarosti, H. In Vitro Oxygen-Glucose Deprivation to Study Ischemic Cell Death. Methods Mol. Biol. 2015, 1254, 197–210. [Google Scholar] [CrossRef]
- Zăgrean, A.M.; Spataru, A.; Ceangă, M.; Zăgrean, L. The Single versus Combinatorial Effects of MK-801, CNQX, Nifedipine and AP-3 on Primary Cultures of Cerebellar Granule Cells in an Oxygen-Glucose Deprivation Model. Rom. J. Morphol. Embryol. 2014, 55, 811–816. [Google Scholar]
- Spataru, A.; Le Duc, D.; Zagrean, L.; Zagrean, A.-M. Ethanol Exposed Maturing Rat Cerebellar Granule Cells Show Impaired Energy Metabolism and Increased Cell Death after Oxygen-Glucose Deprivation. Neural Regen. Res. 2019, 14, 485–490. [Google Scholar] [CrossRef]
- Panaitescu, A.M.; Isac, S.; Pavel, B.; Ilie, A.S.; Ceanga, M.; Totan, A.; Zagrean, L.; Peltecu, G.; Zagrean, A.M. Oxytocin Reduces Seizure Burden and Hippocampal Injury in a Rat Model of Perinatal Asphyxia. Acta Endocrinol. 2018, 14, 315–319. [Google Scholar] [CrossRef]
- Peerboom, C.; Wierenga, C.J. The Postnatal GABA Shift: A Developmental Perspective. Neurosci. Biobehav. Rev. 2021, 124, 179–192. [Google Scholar] [CrossRef]
- Brewer, G.J.; Torricelli, J.R. Isolation and Culture of Adult Neurons and Neurospheres. Nat. Protoc. 2007, 2, 1490–1498. [Google Scholar] [CrossRef]
- Zagrean, A.-M.; Grigoras, I.-F.; Iesanu, M.I.; Ionescu, R.-B.; Chitimus, D.M.; Haret, R.M.; Ianosi, B.; Ceanga, M.; Zagrean, L. Neuronal Transmembrane Chloride Transport Has a Time-Dependent Influence on Survival of Hippocampal Cultures to Oxygen-Glucose Deprivation. Brain Sci. 2019, 9, 360. [Google Scholar] [CrossRef]
- Brewer, G.J.; Torricelli, J.R.; Evege, E.K.; Price, P.J. Optimized Survival of Hippocampal Neurons in B27-Supplemented NeurobasalTM, a New Serum-Free Medium Combination. J. Neurosci. Res. 1993, 35, 567–576. [Google Scholar] [CrossRef]
- Sahu, M.P.; Nikkilä, O.; Lågas, S.; Kolehmainen, S.; Castrén, E. Culturing Primary Neurons from Rat Hippocampus and Cortex. Neuronal Signal. 2019, 3, NS20180207. [Google Scholar] [CrossRef]
- Le Duc, D.; Spataru, A.; Ceanga, M.; Zagrean, L.; Schöneberg, T.; Toescu, E.C.; Zagrean, A.-M. Developmental Exposure to Ethanol Increases the Neuronal Vulnerability to Oxygen-Glucose Deprivation in Cerebellar Granule Cell Cultures. Brain Res. 2015, 1614, 1–13. [Google Scholar] [CrossRef]
- Lyu, H.; Sun, D.M.; Ng, C.P.; Chen, J.F.; He, Y.Z.; Lam, S.Y.; Zheng, Z.Y.; Askarifirouzjaei, H.; Wang, C.C.; Young, W.; et al. A New Hypoxic Ischemic Encephalopathy Model in Neonatal Rats. Heliyon 2021, 7, e08646. [Google Scholar] [CrossRef]
- Wu, Z.; Xie, C.; Kuang, H.; Wu, J.; Chen, X.; Liu, H.; Liu, T. Oxytocin Mediates Neuroprotection against Hypoxic-Ischemic Injury in Hippocampal CA1 Neuron of Neonatal Rats. Neuropharmacology 2021, 187, 108488. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain Development in Rodents and Humans: Identifying Benchmarks of Maturation and Vulnerability to Injury across Species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef]
- Ripamonti, S.; Ambrozkiewicz, M.C.; Guzzi, F.; Gravati, M.; Biella, G.; Bormuth, I.; Hammer, M.; Tuffy, L.P.; Sigler, A.; Kawabe, H.; et al. Transient Oxytocin Signaling Primes the Development and Function of Excitatory Hippocampal Neurons. Elife 2017, 6, e22466. [Google Scholar] [CrossRef]
- Kaneko, Y.; Pappas, C.; Tajiri, N.; Borlongan, C.V. Oxytocin Modulates GABAAR Subunits to Confer Neuroprotection in Stroke In Vitro. Sci. Rep. 2016, 6, 35659. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Chen, C.-C.; Huang, C.-C.; Nishimori, K.; Hsu, K.-S. Oxytocin Stimulates Hippocampal Neurogenesis via Oxytocin Receptor Expressed in CA3 Pyramidal Neurons. Nat. Commun. 2017, 8, 537. [Google Scholar] [CrossRef]
- Haugstad, T.S.; Karlsen, H.E.; Krajtči, P.; Due-Tønnessen, B.; Larsen, M.; Sandberg, C.; Sand, O.; Brandtzaeg, P.; Langmoen, I.A. Efflux of Gamma-Aminobutyric Acid Caused by Changes in Ion Concentrations and Cell Swelling Simulating the Effect of Cerebral Ischaemia. Acta Neurochir. 1997, 139, 453–463. [Google Scholar] [CrossRef]
- Annink, K.V.; de Vries, L.S.; Groenendaal, F.; van den Heuvel, M.P.; van Haren, N.E.M.; Swaab, H.; van Handel, M.; Jongmans, M.J.; Benders, M.J.; van der Aa, N.E. The Long-Term Effect of Perinatal Asphyxia on Hippocampal Volumes. Pediatr. Res. 2019, 85, 43–49. [Google Scholar] [CrossRef]
- Valeeva, G.; Valiullina, F.; Khazipov, R. Excitatory Actions of GABA in the Intact Neonatal Rodent Hippocampus In Vitro. Front. Cell. Neurosci. 2013, 7, 20. [Google Scholar] [CrossRef]
- Moutin, E.; Hemonnot, A.-L.; Seube, V.; Linck, N.; Rassendren, F.; Perroy, J.; Compan, V. Procedures for Culturing and Genetically Manipulating Murine Hippocampal Postnatal Neurons. Front. Synaptic Neurosci. 2020, 12, 19. [Google Scholar] [CrossRef]
- Grabrucker, A.; Vaida, B.; Bockmann, J.; Boeckers, T.M. Synaptogenesis of Hippocampal Neurons in Primary Cell Culture. Cell Tissue Res. 2009, 338, 333–341. [Google Scholar] [CrossRef]
- Cheyne, J.E.; Grant, L.; Butler-Munro, C.; Foote, J.W.; Connor, B.; Montgomery, J.M. Synaptic Integration of Newly Generated Neurons in Rat Dissociated Hippocampal Cultures. Mol. Cell Neurosci. 2011, 47, 203–214. [Google Scholar] [CrossRef]
- Biffi, E.; Regalia, G.; Menegon, A.; Ferrigno, G.; Pedrocchi, A. The Influence of Neuronal Density and Maturation on Network Activity of Hippocampal Cell Cultures: A Methodological Study. PLoS ONE 2013, 8, e83899. [Google Scholar] [CrossRef]
- Myers, M.M.; Grieve, P.G.; Izraelit, A.; Fifer, W.P.; Isler, J.R.; Darnall, R.A.; Stark, R.I. Developmental Profiles of Infant EEG: Overlap with Transient Cortical Circuits. Clin. Neurophysiol. 2012, 123, 1502–1511. [Google Scholar] [CrossRef]
- Chung, H.; Kim, E.; Lee, D.H.; Seo, S.; Ju, S.; Lee, D.; Kim, H.; Park, S. Ghrelin Inhibits Apoptosis in Hypothalamic Neuronal Cells during Oxygen-Glucose Deprivation. Endocrinology 2007, 148, 148–159. [Google Scholar] [CrossRef]
- Ye, R.; Li, N.; Han, J.; Kong, X.; Cao, R.; Rao, Z.; Zhao, G. Neuroprotective Effects of Ginsenoside Rd against Oxygen-Glucose Deprivation in Cultured Hippocampal Neurons. Neurosci. Res. 2009, 64, 306–310. [Google Scholar] [CrossRef]
- Iijima, T.; Mishima, T.; Akagawa, K.; Iwao, Y. Mitochondrial Hyperpolarization after Transient Oxygen-Glucose Deprivation and Subsequent Apoptosis in Cultured Rat Hippocampal Neurons. Brain Res. 2003, 993, 140–145. [Google Scholar] [CrossRef]
- Culmsee, C.; Zhu, C.; Landshamer, S.; Becattini, B.; Wagner, E.; Pellecchia, M.; Blomgren, K.; Plesnila, N. Apoptosis-Inducing Factor Triggered by Poly(ADP-Ribose) Polymerase and Bid Mediates Neuronal Cell Death after Oxygen-Glucose Deprivation and Focal Cerebral Ischemia. J. Neurosci. 2005, 25, 10262–10272. [Google Scholar] [CrossRef]
- Limatola, C.; Lauro, C.; Catalano, M.; Ciotti, M.T.; Bertollini, C.; Di Angelantonio, S.; Ragozzino, D.; Eusebi, F. Chemokine CX3CL1 Protects rat Hippocampal Neurons against Glutamate-Mediated Excitotoxicity. J. Neuroimmunol. 2005, 166, 19–28. [Google Scholar] [CrossRef]
- Wu, H.; Che, X.; Tang, J.; Ma, F.; Pan, K.; Zhao, M.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. The K(+)-Cl(−) Cotransporter KCC2 and Chloride Homeostasis: Potential Therapeutic Target in Acute Central Nervous System Injury. Mol. Neurobiol. 2016, 53, 2141–2151. [Google Scholar] [CrossRef]
- Galeffi, F.; Sah, R.; Pond, B.B.; George, A.; Schwartz-Bloom, R.D. Changes in Intracellular Chloride after Oxygen–Glucose Deprivation of the Adult Hippocampal Slice: Effect of Diazepam. J. Neurosci. 2004, 24, 4478–4488. [Google Scholar] [CrossRef]
- Jaenisch, N.; Witte, O.W.; Frahm, C. Downregulation of Potassium Chloride Cotransporter KCC2 after Transient Focal Cerebral Ischemia. Stroke 2010, 41, e151–e159. [Google Scholar] [CrossRef]
- Inglefield, J.R.; Schwartz-Bloom, R.D. Optical Imaging of Hippocampal Neurons with a Chloride-Sensitive Dye: Early Effects of In Vitro Ischemia. J. Neurochem. 1998, 70, 2500–2509. [Google Scholar] [CrossRef]
- van den Pol, A.N.; Obrietan, K.; Chen, G. Excitatory Actions of GABA after Neuronal Trauma. J. Neurosci. 1996, 16, 4283–4292. [Google Scholar] [CrossRef]
- Schwartz-Bloom, R.D.; Sah, R. Gamma-Aminobutyric Acid(A) Neurotransmission and Cerebral Ischemia. J. Neurochem. 2001, 77, 353–371. [Google Scholar] [CrossRef]
- Toda, T.; Ishida, K.; Kiyama, H.; Yamashita, T.; Lee, S. Down-Regulation of KCC2 Expression and Phosphorylation in Motoneurons, and Increases the Number of in Primary Afferent Projections to Motoneurons in Mice with Post-Stroke Spasticity. PLoS ONE 2014, 9, e114328. [Google Scholar] [CrossRef]
- Pond, B.B.; Berglund, K.; Kuner, T.; Feng, G.; Augustine, G.J.; Schwartz-Bloom, R.D. The Chloride Transporter Na+-K+-Cl− Cotransporter Isoform-1 Contributes to Intracellular Chloride Increases after In Vitro Ischemia. J. Neurosci. 2006, 26, 1396–1406. [Google Scholar] [CrossRef]
- Mittmann, T.; Qü, M.; Zilles, K.; Luhmann, H.J. Long-Term Cellular Dysfunction after Focal Cerebral Ischemia: In Vitro Analyses. Neuroscience 1998, 85, 15–27. [Google Scholar] [CrossRef]
- Katchman, A.N.; Vicini, S.; Hershkowitz, N. Mechanism of Early Anoxia-Induced Suppression of the GABAA-Mediated Inhibitory Postsynaptic Current. J. Neurophysiol. 1994, 71, 1128–1138. [Google Scholar] [CrossRef]
- Antonucci, R.; Porcella, A.; Pilloni, M.D. Perinatal Asphyxia in the Term Newborn. J. Pediatr. Neonatal Individ. Med. (JPNIM) 2014, 3, e030269. [Google Scholar] [CrossRef]
- McGuire, W. Perinatal Asphyxia. BMJ Clin. Evid. 2007, 2007, 0320. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean Viability | SD | Number of Plates | Total Number of Wells | p Value Paired t-Test | |
|---|---|---|---|---|---|
| Non-treated normoxia | 100% | 0 | 17 | 30 | |
| 0.1 µM OXT | 101.0% | 5.532% | 7 | 28 | 0.605 |
| 1 µM OXT | 104.3% | 0.870% | 2 | 7 | 0.341 |
| Mean Cellular Viability | SD | Number of Plates | Total Number of Wells | p Value Paired t-Test | ||
|---|---|---|---|---|---|---|
| Moderate OGD | No treatment | 55.39% | 4.2% | 3 | 19 | |
| 1 µM OXT | 48.58% | 12.15% | 3 | 15 | 0.286 | |
| Severe OGD | No treatment | 38.17% | 4.25% | 4 | 21 | |
| 1 µM OXT | 46.05% | 1.78% | 4 | 23 | 0.021 |
| Mean Cellular Viability | SD | Number of Plates | Total Number of Wells | p Value Paired t-Test | ||
|---|---|---|---|---|---|---|
| Moderate OGD | No treatment | 60.89% | 9.85% | 8 | 27 | |
| 0.1 µM OXT | 61.18% | 12.57% | 8 | 22 | 0.893 | |
| Severe OGD | No treatment | 40.64% | 8.31% | 6 | 22 | |
| 0.1 µM OXT | 45.13% | 8.8% | 6 | 26 | 0.051 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ionescu, M.I.; Grigoras, I.-F.; Ionescu, R.-B.; Chitimus, D.M.; Haret, R.M.; Ianosi, B.; Ceanga, M.; Zagrean, A.-M. Oxytocin Exhibits Neuroprotective Effects on Hippocampal Cultures under Severe Oxygen–Glucose Deprivation Conditions. Curr. Issues Mol. Biol. 2024, 46, 6223-6236. https://doi.org/10.3390/cimb46060371
Ionescu MI, Grigoras I-F, Ionescu R-B, Chitimus DM, Haret RM, Ianosi B, Ceanga M, Zagrean A-M. Oxytocin Exhibits Neuroprotective Effects on Hippocampal Cultures under Severe Oxygen–Glucose Deprivation Conditions. Current Issues in Molecular Biology. 2024; 46(6):6223-6236. https://doi.org/10.3390/cimb46060371
Chicago/Turabian StyleIonescu, Mara Ioana, Ioana-Florentina Grigoras, Rosana-Bristena Ionescu, Diana Maria Chitimus, Robert Mihai Haret, Bogdan Ianosi, Mihai Ceanga, and Ana-Maria Zagrean. 2024. "Oxytocin Exhibits Neuroprotective Effects on Hippocampal Cultures under Severe Oxygen–Glucose Deprivation Conditions" Current Issues in Molecular Biology 46, no. 6: 6223-6236. https://doi.org/10.3390/cimb46060371