
VEGF as a Key Actor in Recurrent Respiratory Papillomatosis: A Narrative Review


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
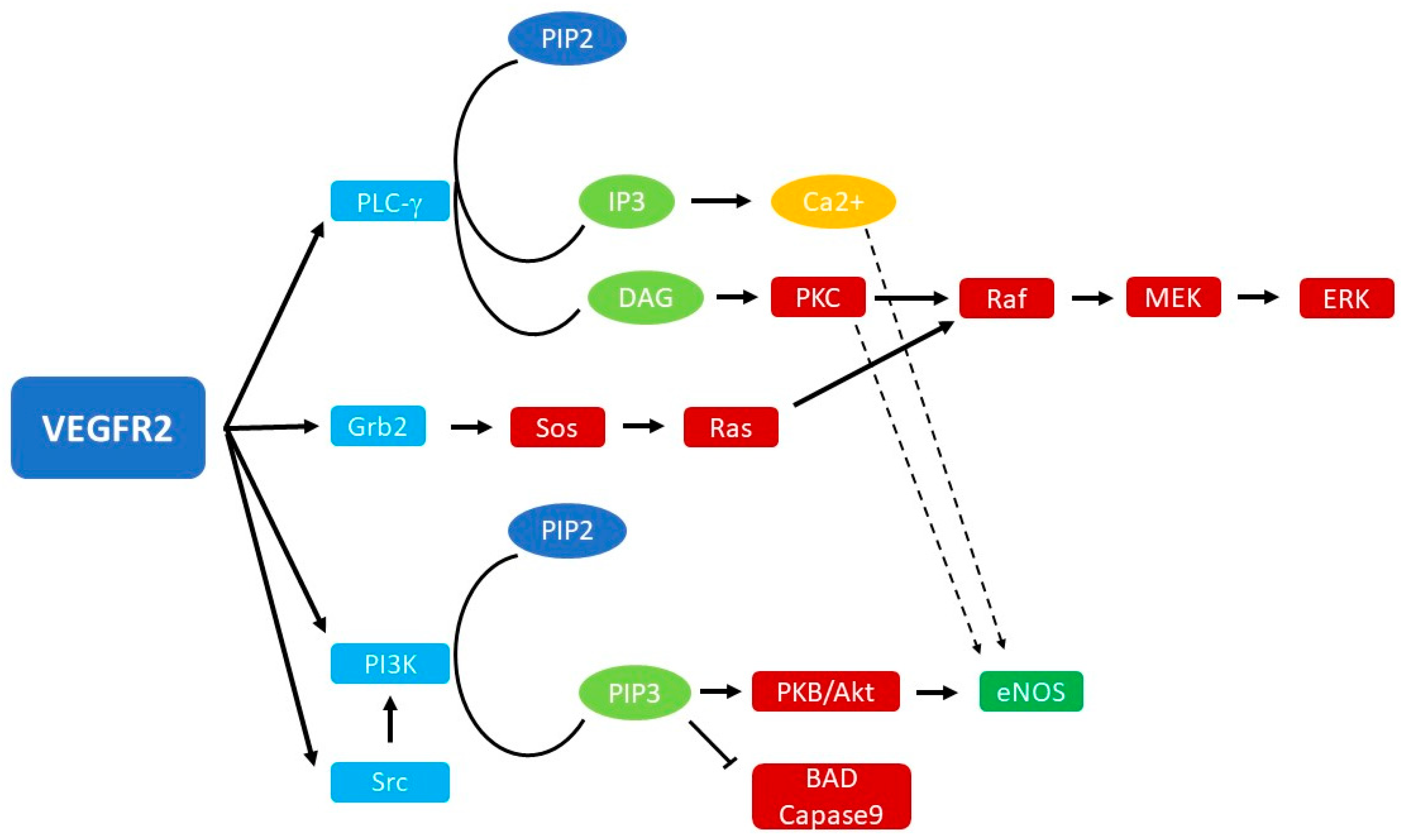
2. Biology of VEGF Signaling
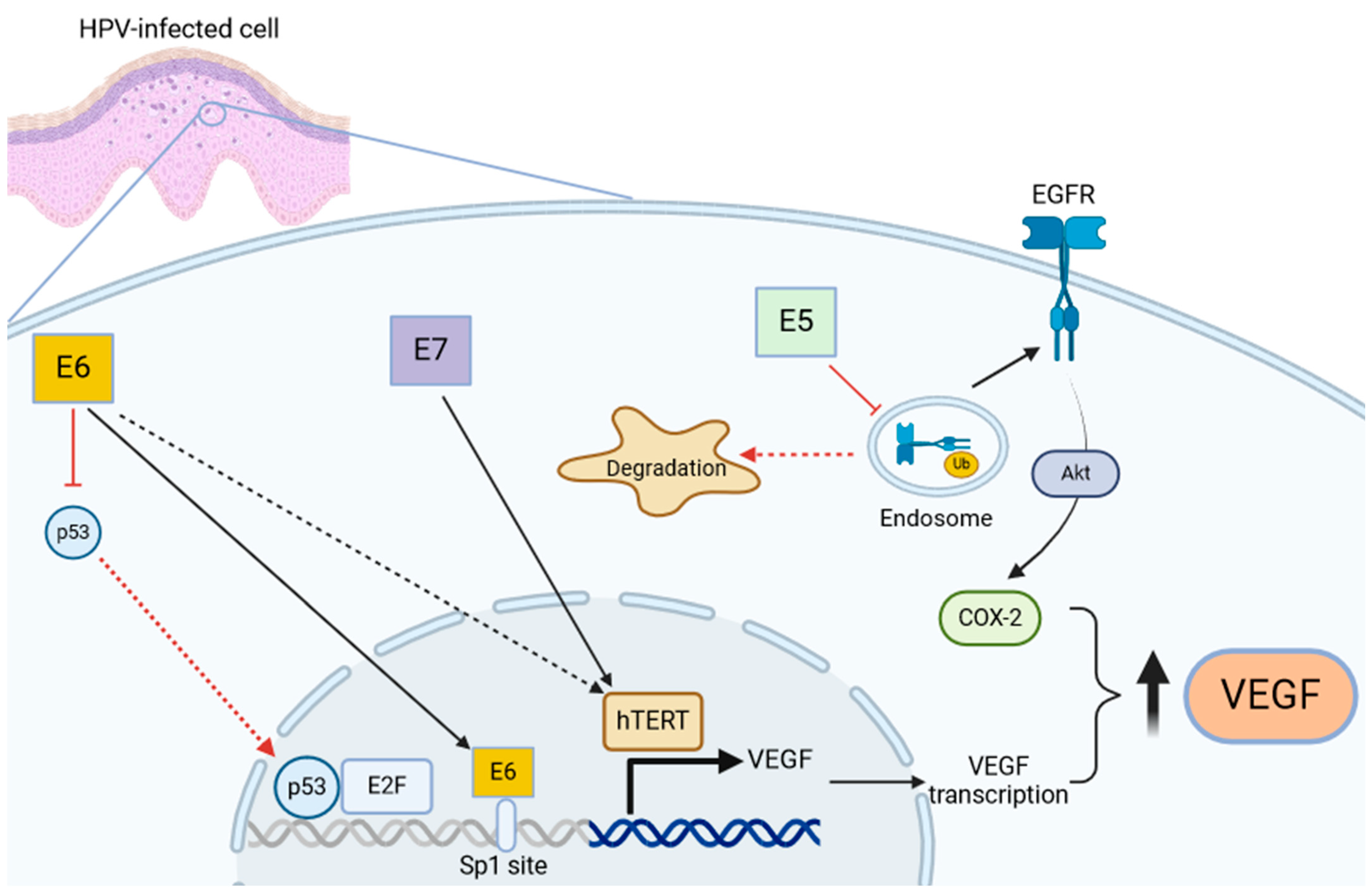
3. Role of VEGF in HPV-Mediated Diseases and RRP
4. Anti-Angiogenic Molecules in The Treatment of RRP: Present and Future
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Larson, D.A.; Derkay, C.S. Epidemiology of recurrent respiratory papillomatosis. APMIS 2010, 118, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Meites, E.; Stone, L.; Amiling, R.; Singh, V.; Unger, E.R.; Derkay, C.S.; Markowitz, L.E. Significant Declines in Juvenile-onset Recurrent Respiratory Papillomatosis Following Human Papillomavirus (HPV) Vaccine Introduction in the United States. Clin. Infect. Dis. 2021, 73, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. S1), 2–23. [Google Scholar] [CrossRef] [PubMed]
- Orosco, R.K.; Kedarisetty, S.; Hecht, A.S.; Chang, D.C.; Coffey, C.S.; Weissbrod, P.A. Predictors of high-risk and low-risk oral HPV infection in the U nited S tates. Laryngoscope 2016, 126, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Fortes, H.R.; Von Ranke, F.M.; Escuissato, D.L.; Araujo Neto, C.A.; Zanetti, G.; Hochhegger, B.; Souza, C.A.; Marchiori, E. Recurrent respiratory papillomatosis: A state-of-the-art review. Respir. Med. 2017, 126, 116–121. [Google Scholar] [CrossRef]
- Hartley, C.; Hamilton, J.; Birzgalis, A.R.; Farrington, W.T. Recurrent respiratory papillomatosis—The Manchester experience, 1974–1992. J. Laryngol. Otol. 1994, 108, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Goon, P.; Sauzet, O.; Schuermann, M.; Oppel, F.; Shao, S.; Scholtz, L.-U.; Sudhoff, H.; Goerner, M. Recurrent Respiratory Papillomatosis (RRP)—Meta-analyses on the use of the HPV vaccine as adjuvant therapy. npj Vaccines 2023, 8, 49. [Google Scholar] [CrossRef]
- Mau, T.; Amin, M.R.; Belafsky, P.C.; Best, S.R.; Friedman, A.D.; Klein, A.M.; Lott, D.G.; Paniello, R.C.; Pransky, S.M.; Saba, N.F.; et al. Interim Results of a Phase 1/2 Open-Label Study of INO-3107 for HPV-6 and/or HPV-11-Associated Recurrent Respiratory Papillomatosis. Laryngoscope 2023, 133, 3087–3093. [Google Scholar] [CrossRef] [PubMed]
- Norberg, S.M.; Bai, K.; Sievers, C.; Robbins, Y.; Friedman, J.; Yang, X.; Kenyon, M.; Ward, E.; Schlom, J.; Gulley, J.; et al. The tumor microenvironment state associates with response to HPV therapeutic vaccination in patients with respiratory papillomatosis. Sci. Transl. Med. 2023, 15, eadj0740. [Google Scholar] [CrossRef]
- Ouda, A.M.; Elsabagh, A.A.; Elmakaty, I.M.; Gupta, I.; Vranic, S.; Al-Thawadi, H.; Al Moustafa, A.-E. HPV and Recurrent Respiratory Papillomatosis: A Brief Review. Life 2021, 11, 1279. [Google Scholar] [CrossRef]
- Helt, A.-M. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 2003, 24, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Tuong, Z.K.; Frazer, I.H. Papillomavirus Immune Evasion Strategies Target the Infected Cell and the Local Immune System. Front. Oncol. 2019, 9, 682. [Google Scholar] [CrossRef]
- Oh, S.T.; Longworth, M.S.; Laimins, L.A. Roles of the E6 and E7 Proteins in the Life Cycle of Low-Risk Human Papillomavirus Type 11. J. Virol. 2004, 78, 2620–2626. [Google Scholar] [CrossRef]
- Soloperto, D.; Gazzini, S.; Cerullo, R. Molecular Mechanisms of Carcinogenesis in Pediatric Airways Tumors. Int. J. Mol. Sci. 2023, 24, 2195. [Google Scholar] [CrossRef] [PubMed]
- Toussaint-Smith, E.; Donner, D.B.; Roman, A. Expression of human papillomavirus type 16 E6 and E7 oncoproteins in primary foreskin keratinocytes is sufficient to alter the expression of angiogenic factors. Oncogene 2004, 23, 2988–2995. [Google Scholar] [CrossRef] [PubMed]
- Verma, H.; Chandran, A.; Shaktivel, P.; Singh, A.; Kaushal, S.; Sikka, K.; Thakar, A.; Irugu, D.V.K. The serum and tissue expression of vascular endothelial growth factor-in recurrent respiratory papillomatosis. Int. J. Pediatr. Otorhinolaryngol. 2021, 146, 110737. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Shing, Y. Angiogenesis. J. Biol. Chem. 1992, 267, 10931–10934. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Claesson-Welsh, L. Signal Transduction by Vascular Endothelial Growth Factor Receptors. Cold Spring Harb. Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-L.; Chen, H.-H.; Zheng, L.-L.; Sun, L.-P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef]
- Kerr, D.J. Targeting angiogenesis in cancer: Clinical development of bevacizumab. Nat. Rev. Clin. Oncol. 2004, 1, 39–43. [Google Scholar] [CrossRef]
- Nagel, S.; Busch, C.; Blankenburg, T.; Schütte, W. Behandlung der respiratorischen Papillomatose—Kasuistik zur systemischen Therapie mit Bevacizumab. Pneumologie 2009, 63, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, L.; Ziylan, F.; Smeeing, D.P.J.; Dikkers, F.G.; Rinkel, R.N.P.M. Bevacizumab as treatment option for recurrent respiratory papillomatosis: A systematic review. Eur. Arch. Otorhinolaryngol. 2022, 279, 4229–4240. [Google Scholar] [CrossRef] [PubMed]
- Evans, I. An Overview of VEGF-Mediated Signal Transduction. In VEGF Signaling; Fiedler, L., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; Volume 1332, pp. 91–120. [Google Scholar] [CrossRef]
- Roskoski, R. VEGF receptor protein–tyrosine kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2008, 375, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Vargas, S.O.; Healy, G.B.; Rahbar, R.; Folkman, J.; Tan, X.; McGill, T.J.; Brown, L.F. Role of Vascular Endothelial Growth Factor—A in Recurrent Respiratory Papillomatosis. Ann. Otol. Rhinol. Laryngol. 2005, 114, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- LeCouter, J.; Moritz, D.R.; Li, B.; Phillips, G.L.; Liang, X.H.; Gerber, H.-P.; Hillan, K.J.; Ferrara, N. Angiogenesis-Independent Endothelial Protection of Liver: Role of VEGFR-1. Science 2003, 299, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Barleon, B.; Sozzani, S.; Zhou, D.; Weich, H.; Mantovani, A.; Marme, D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 1996, 87, 3336–3343. [Google Scholar] [CrossRef] [PubMed]
- Simons, M. An Inside View: VEGF Receptor Trafficking and Signaling. Physiology 2012, 27, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Claesson-Welsh, L. VEGF Receptor Signal Transduction. Sci. STKE 2001, 2001, re21. [Google Scholar] [CrossRef]
- Peach, C.; Mignone, V.; Arruda, M.; Alcobia, D.; Hill, S.; Kilpatrick, L.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef]
- Takahashi, T.; Ueno, H.; Shibuya, M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene 1999, 18, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.-P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular Endothelial Growth Factor Regulates Endothelial Cell Survival through the Phosphatidylinositol 3′-Kinase/Akt Signal Transduction Pathway. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; García-Cardeña, G.; Madri, J.A.; Sessa, W.C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Investig. 1997, 100, 3131–3139. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Ellis, L.M. Role of the Vascular Endothelial Growth Factor Pathway in Tumor Growth and Angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, R.M.; Page, D.J.; Ostergaard, P.; Keavney, B.D. The physiological and pathological functions of VEGFR3 in cardiac and lymphatic development and related diseases. Cardiovasc. Res. 2021, 117, 1877–1890. [Google Scholar] [CrossRef]
- Matsumura, K.; Hirashima, M.; Ogawa, M.; Kubo, H.; Hisatsune, H.; Kondo, N.; Nishikawa, S.; Chiba, T.; Nishikawa, S.-I. Modulation of VEGFR-2–mediated endothelial-cell activity by VEGF-C/VEGFR-3. Blood 2003, 101, 1367–1374. [Google Scholar] [CrossRef]
- Irrthum, A.; Karkkainen, M.J.; Devriendt, K.; Alitalo, K.; Vikkula, M. Congenital Hereditary Lymphedema Caused by a Mutation That Inactivates VEGFR3 Tyrosine Kinase. Am. J. Hum. Genet. 2000, 67, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Brice, G.; Child, A.H.; Evans, A.; Bell, R.; Mansour, S.; Burnand, K.; Sarfarazi, M.; Jeffery, S.; Mortimer, P. Milroy disease and the VEGFR-3 mutation phenotype. J. Med. Genet. 2005, 42, 98–102. [Google Scholar] [CrossRef]
- Stacker, S.A.; Caesar, C.; Baldwin, M.E.; Thornton, G.E.; Williams, R.A.; Prevo, R.; Jackson, D.G.; Nishikawa, S.; Kubo, H.; Achen, M.G. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat. Med. 2001, 7, 186–191. [Google Scholar] [CrossRef]
- He, Y.; Kozaki, K.-I.; Karpanen, T.; Koshikawa, K.; Yla-Herttuala, S.; Takahashi, T.; Alitalo, K. Suppression of Tumor Lymphangiogenesis and Lymph Node Metastasis by Blocking Vascular Endothelial Growth Factor Receptor 3 Signaling. JNCI J. Natl. Cancer Inst. 2002, 94, 819–825. [Google Scholar] [CrossRef]
- Lam, B.; Miller, J.; Kung, Y.J.; Wu, T.C.; Hung, C.; Roden, R.B.S.; Best, S.R. Profiling of VEGF Receptors and Immune Checkpoints in Recurrent Respiratory Papillomatosis. Laryngoscope 2024, 134, 2819–2825. [Google Scholar] [CrossRef] [PubMed]
- Alkharsah, K. VEGF Upregulation in Viral Infections and Its Possible Therapeutic Implications. Int. J. Mol. Sci. 2018, 19, 1642. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Bi, N.; Gui, L.; Peng, Z. Elective neck dissection or “watchful waiting”: Optimal management strategy for early stage N0 tongue carcinoma using decision analysis techniques. Chin. Med. J. 2008, 121, 1646–1650. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 Oncoprotein Encoded by Human Papillomavirus Types 16 and 18 Promotes the Degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Crook, T.; Vousden, K.H. Interaction of HPV E6 with p53 and associated proteins. Biochem. Soc. Trans. 1994, 22, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Farhang Ghahremani, M.; Goossens, S.; Haigh, J.J. The p53 family and VEGF regulation: “It’s complicated”. Cell Cycle 2013, 12, 1331–1332. [Google Scholar] [CrossRef] [PubMed]
- Farhang Ghahremani, M.; Goossens, S.; Nittner, D.; Bisteau, X.; Bartunkova, S.; Zwolinska, A.; Hulpiau, P.; Haigh, K.; Haenebalcke, L.; Drogat, B.; et al. p53 promotes VEGF expression and angiogenesis in the absence of an intact p21-Rb pathway. Cell Death Differ. 2013, 20, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Gamell, C.; Wolyniec, K.; Haupt, Y. Interplay between p53 and VEGF: How to prevent the guardian from becoming a villain. Cell Death Differ. 2013, 20, 852–854. [Google Scholar] [CrossRef] [PubMed]
- López-Ocejo, O.; Viloria-Petit, A.; Bequet-Romero, M.; Mukhopadhyay, D.; Rak, J.; Kerbel, R.S. Oncogenes and tumor angiogenesis: The HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene 2000, 19, 4611–4620. [Google Scholar] [CrossRef]
- Zhou, L.; Zheng, D.; Wang, M.; Cong, Y.-S. Telomerase reverse transcriptase activates the expression of vascular endothelial growth factor independent of telomerase activity. Biochem. Biophys. Res. Commun. 2009, 386, 739–743. [Google Scholar] [CrossRef]
- Li, F.; Cui, J. Human telomerase reverse transcriptase regulates vascular endothelial growth factor expression via human papillomavirus oncogene E7 in HPV-18-positive cervical cancer cells. Med. Oncol. 2015, 32, 199. [Google Scholar] [CrossRef]
- Liu, X.; Yuan, H.; Fu, B.; Disbrow, G.L.; Apolinario, T.; Tomaić, V.; Kelley, M.L.; Baker, C.C.; Huibregtse, J.; Schlegel, R. The E6AP Ubiquitin Ligase Is Required for Transactivation of the hTERT Promoter by the Human Papillomavirus E6 Oncoprotein. J. Biol. Chem. 2005, 280, 10807–10816. [Google Scholar] [CrossRef]
- Straight, S.W.; Herman, B.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J. Virol. 1995, 69, 3185–3192. [Google Scholar] [CrossRef]
- Zhang, B.; Srirangam, A.; Potter, D.A.; Roman, A. HPV16 E5 protein disrupts the c-Cbl–EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 2005, 24, 2585–2588. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, Q.; Nishitani, J.; Brown, J.; Shi, S.; Le, A.D. Overexpression of Human Papillomavirus Type 16 Oncoproteins Enhances Hypoxia-Inducible Factor 1α Protein Accumulation and Vascular Endothelial Growth Factor Expression in Human Cervical Carcinoma Cells. Clin. Cancer Res. 2007, 13, 2568–2576. [Google Scholar] [CrossRef]
- Tilborghs, S.; Corthouts, J.; Verhoeven, Y.; Arias, D.; Rolfo, C.; Trinh, X.B.; Van Dam, P.A. The role of Nuclear Factor-kappa B signaling in human cervical cancer. Crit. Rev. Oncol./Hematol. 2017, 120, 141–150. [Google Scholar] [CrossRef]
- DeVoti, J.A.; Rosenthal, D.W.; Wu, R.; Abramson, A.L.; Steinberg, B.M.; Bonagura, V.R. Immune Dysregulation and Tumor-Associated Gene Changes in Recurrent Respiratory Papillomatosis: A Paired Microarray Analysis. Mol. Med. 2008, 14, 608–617. [Google Scholar] [CrossRef]
- Carifi, M.; Napolitano, D.; Morandi, M.; Dall’Olio, D. Recurrent respiratory papillomatosis: Current and future perspectives. Ther. Clin. Risk Manag. 2015, 11, 731–738. [Google Scholar] [CrossRef]
- Bertino, G.; Pedretti, F.; Mauramati, S.; Filauro, M.; Vallin, A.; Mora, F.; Crosetti, E.; Succo, G.; Peretti, G.; Benazzo, M. Recurrent laryngeal papillomatosis: Multimodal therapeutic strategies. Literature review and multicentre retrospective study. Acta Otorhinolaryngol. Ital. 2023, 43 (Suppl. S1), S111–S122. [Google Scholar] [CrossRef]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef]
- Mukherji, S.K. Bevacizumab (Avastin). AJNR Am. J. Neuroradiol. 2010, 31, 235–236. [Google Scholar] [CrossRef]
- Kazazi-Hyseni, F.; Beijnen, J.H.; Schellens, J.H.M. Bevacizumab. Oncologist 2010, 15, 819–825. [Google Scholar] [CrossRef]
- Madaki, S.; Habib, S.; Yero, M. Outcome of multiple retinal capillary hemangioma following focal laser and intravitreal bevacizumab injection: A case report. J. West. Afr. Coll. Surg. 2023, 13, 111–115. [Google Scholar] [CrossRef]
- Kini, S.D.; Yiu, D.W.; Weisberg, R.A.; Davila, J.F.; Chelius, D.C. Bevacizumab as Treatment for Epistaxis in Hereditary Hemorrhagic Telangiectasia: A Literature Review. Ann. Otol. Rhinol. Laryngol. 2019, 128, 467–471. [Google Scholar] [CrossRef]
- Raper, D.M.S.; Winkler, E.A.; Rutledge, W.C.; Cooke, D.L.; Abla, A.A. An Update on Medications for Brain Arteriovenous Malformations. Neurosurgery 2020, 87, 871–878. [Google Scholar] [CrossRef]
- Evers, G.; Schliemann, C.; Beule, A.; Schmidt, L.; Schulze, A.B.; Kessler, C.; Hoffmann, T.K.; Wiewrodt, R.; Groll, A.H.; Bleckmann, A.; et al. Long-Term Follow-Up on Systemic Bevacizumab Treatment in Recurrent Respiratory Papillomatosis. Laryngoscope 2021, 131, E1926–E1933. [Google Scholar] [CrossRef]
- Best, S.R.; Mohr, M.; Zur, K.B. Systemic bevacizumab for recurrent respiratory papillomatosis: A national survey. Laryngoscope 2017, 127, 2225–2229. [Google Scholar] [CrossRef]
- Best, S.R.; Friedman, A.D.; Landau-Zemer, T.; Barbu, A.M.; Burns, J.A.; Freeman, M.W.; Halvorsen, Y.-D.; Hillman, R.E.; Zeitels, S.M. Safety and Dosing of Bevacizumab (Avastin) for the Treatment of Recurrent Respiratory Papillomatosis. Ann. Otol. Rhinol. Laryngol. 2012, 121, 587–593. [Google Scholar] [CrossRef]
- Mohr, M.; Schliemann, C.; Biermann, C.; Schmidt, L.-H.; Kessler, T.; Schmidt, J.; Wiebe, K.; Müller, K.-M.; Hoffmann, T.K.; Groll, A.H.; et al. Rapid response to systemic bevacizumab therapy in recurrent respiratory papillomatosis. Oncol. Lett. 2014, 8, 1912–1918. [Google Scholar] [CrossRef]
- Hall, S.R.; Thiriveedi, M.; Yandrapalli, U.; Zhang, N.; Lott, D.G. Sublesional Bevacizumab Injection for Recurrent Respiratory Papillomatosis: Evaluation of Utility in a Typical Clinical Practice. Ann. Otol. Rhinol. Laryngol. 2021, 130, 1164–1170. [Google Scholar] [CrossRef]
- Willems, E.; Gerne, L.; George, C.; D’Hondt, M. Adverse effects of bevacizumab in metastatic colorectal cancer: A case report and literature review. Acta Gastro-Enterol. Belg. 2019, 82, 322–325. [Google Scholar]
- Ranpura, V.; Hapani, S.; Wu, S. Treatment-Related Mortality With Bevacizumab in Cancer Patients: A Meta-analysis. JAMA 2011, 305, 487. [Google Scholar] [CrossRef]
- Huang, H.; Zheng, Y.; Zhu, J.; Zhang, J.; Chen, H.; Chen, X. An Updated Meta-Analysis of Fatal Adverse Events Caused by Bevacizumab Therapy in Cancer Patients. PLoS ONE 2014, 9, e89960. [Google Scholar] [CrossRef]
- Syed, Y.Y. SB8: A Bevacizumab Biosimilar. Target. Oncol. 2020, 15, 787–790. [Google Scholar] [CrossRef]
- Goldschmidt, J.; Hanes, V. The Totality of Evidence and Use of ABP 215, a Biosimilar to Bevacizumab. Oncol. Ther. 2021, 9, 213–223. [Google Scholar] [CrossRef]
- Calleja, M.A.; Albanell, J.; Aranda, E.; García-Foncillas, J.; Feliu, A.; Rivera, F.; Oyagüez, I.; Salinas-Ortega, L.; Soto Alvarez, J. Budget impact analysis of bevacizumab biosimilars for cancer treatment in adult patients in Spain. Eur. J. Hosp. Pharm. 2023, 30, e40–e47. [Google Scholar] [CrossRef]
- Lacal, P.M.; Graziani, G. Therapeutic implication of vascular endothelial growth factor receptor-1 (VEGFR-1) targeting in cancer cells and tumor microenvironment by competitive and non-competitive inhibitors. Pharmacol. Res. 2018, 136, 97–107. [Google Scholar] [CrossRef]
- Paillasse, M.R.; Esquerré, M.; Bertrand, F.A.; Poussereau-Pomié, C.; Pichery, M.; Visentin, V.; Gueguen-Dorbes, G.; Gaujarengues, F.; Barron, P.; Badet, G.; et al. Targeting Tumor Angiogenesis with the Selective VEGFR-3 Inhibitor EVT801 in Combination with Cancer Immunotherapy. Cancer Res. Commun. 2022, 2, 1504–1519. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gazzini, S.; Cerullo, R.; Soloperto, D. VEGF as a Key Actor in Recurrent Respiratory Papillomatosis: A Narrative Review. Curr. Issues Mol. Biol. 2024, 46, 6757-6768. https://doi.org/10.3390/cimb46070403
Gazzini S, Cerullo R, Soloperto D. VEGF as a Key Actor in Recurrent Respiratory Papillomatosis: A Narrative Review. Current Issues in Molecular Biology. 2024; 46(7):6757-6768. https://doi.org/10.3390/cimb46070403
Chicago/Turabian StyleGazzini, Sandra, Raffaele Cerullo, and Davide Soloperto. 2024. "VEGF as a Key Actor in Recurrent Respiratory Papillomatosis: A Narrative Review" Current Issues in Molecular Biology 46, no. 7: 6757-6768. https://doi.org/10.3390/cimb46070403