
Secondary Analysis of Human Bulk RNA-Seq Dataset Suggests Potential Mechanisms for Letrozole Resistance in Estrogen-Positive (ER+) Breast Cancer


Abstract
:1. Introduction
2. Materials and Methods
2.1. Retrieving Fastq Files, Preprocessing, and Enriching RNA-Sequencing Data
2.2. Target and Mechanistic Marker Prediction
2.3. Protein Network Analysis
3. Results
3.1. The Majority of Differentially Expressed Genes in ER+ Treatment Resistance Are Upregulated
3.2. The Signaling Pathway Impact Analysis Identified Four Significantly Affected Pathways
3.3. Targets Prioritized for Repurposing from the Identified Pathways
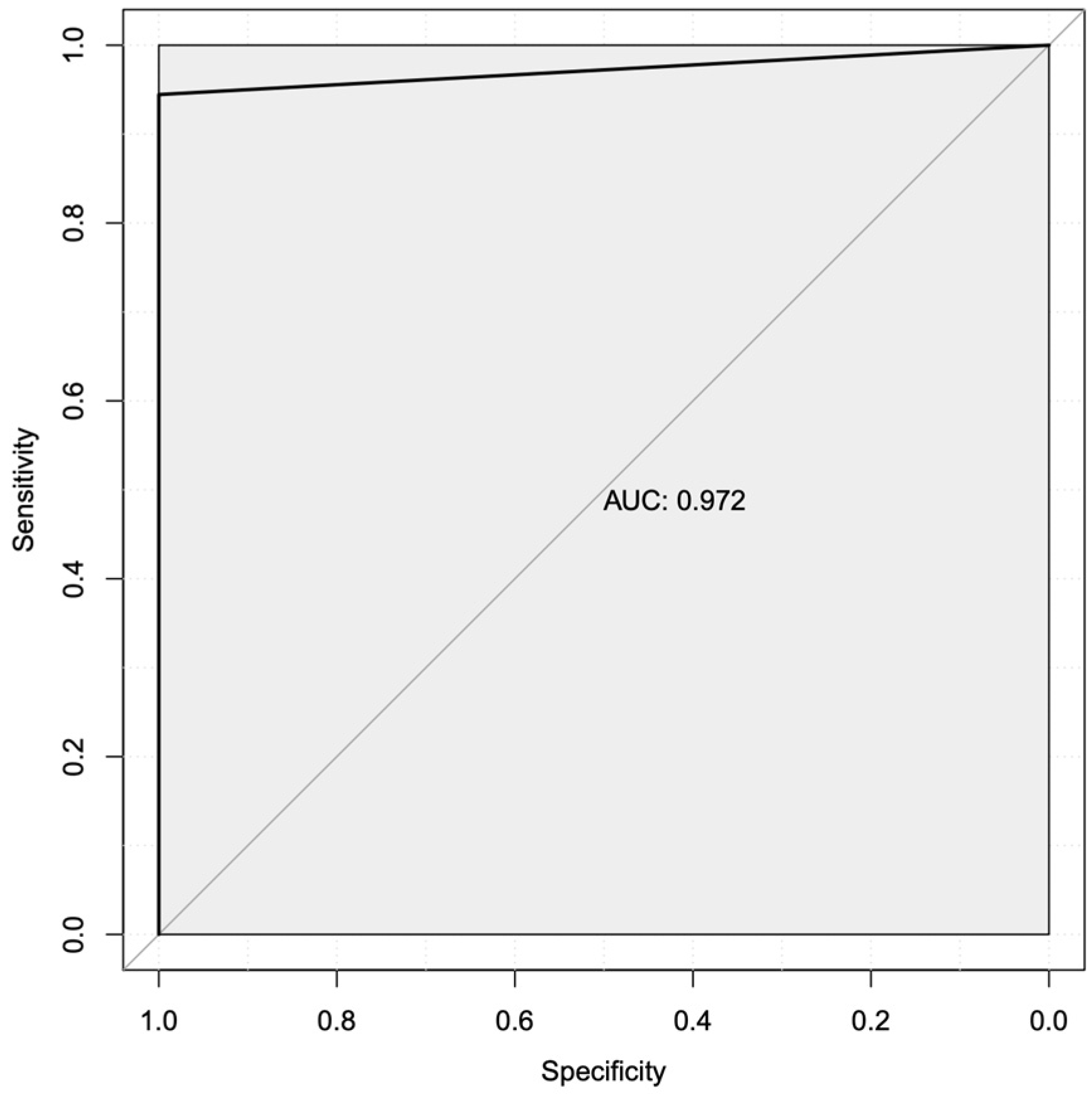
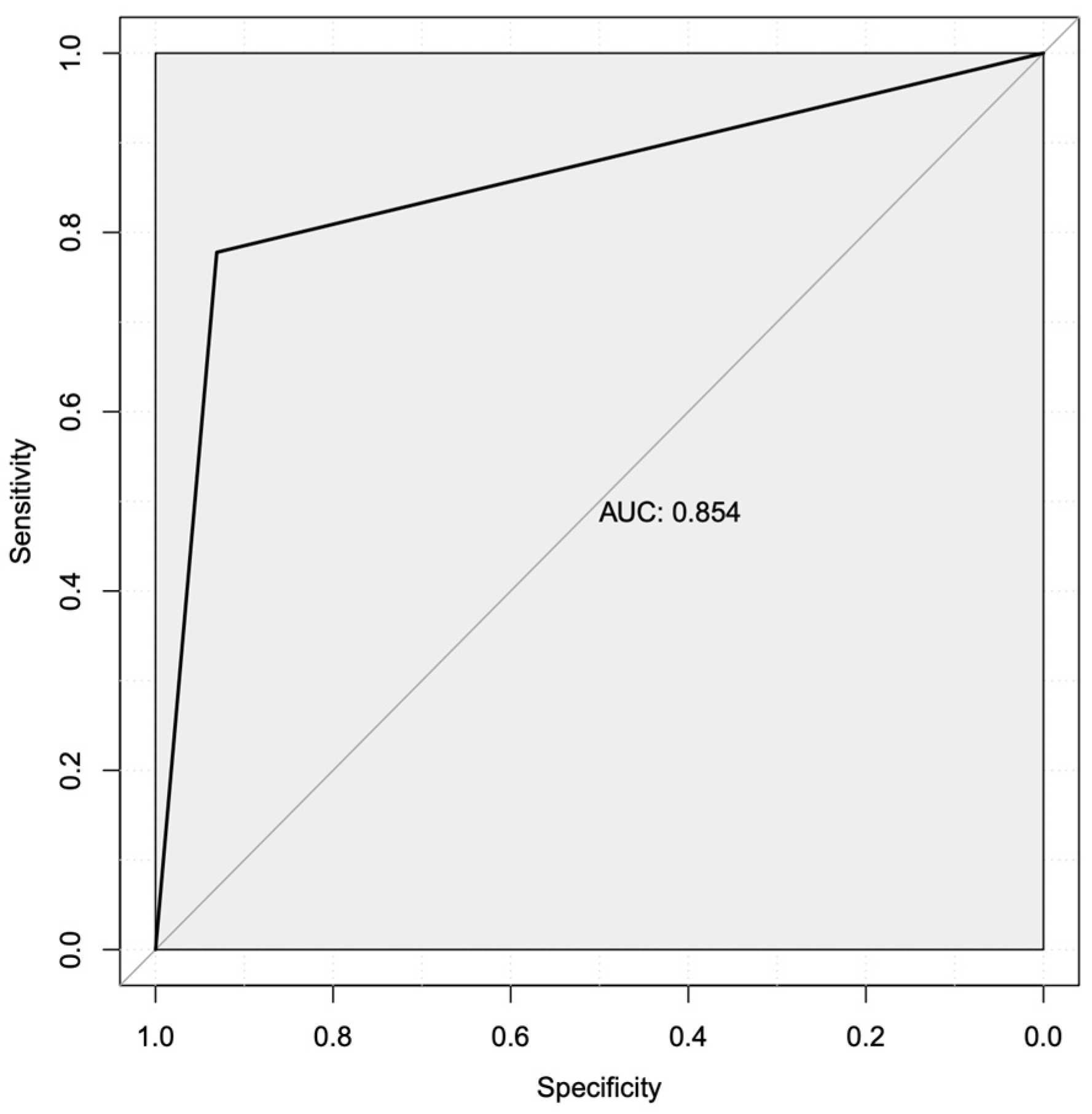
3.4. Machine Learning Predicted Two Robust Mechanistic Transcriptional Markers
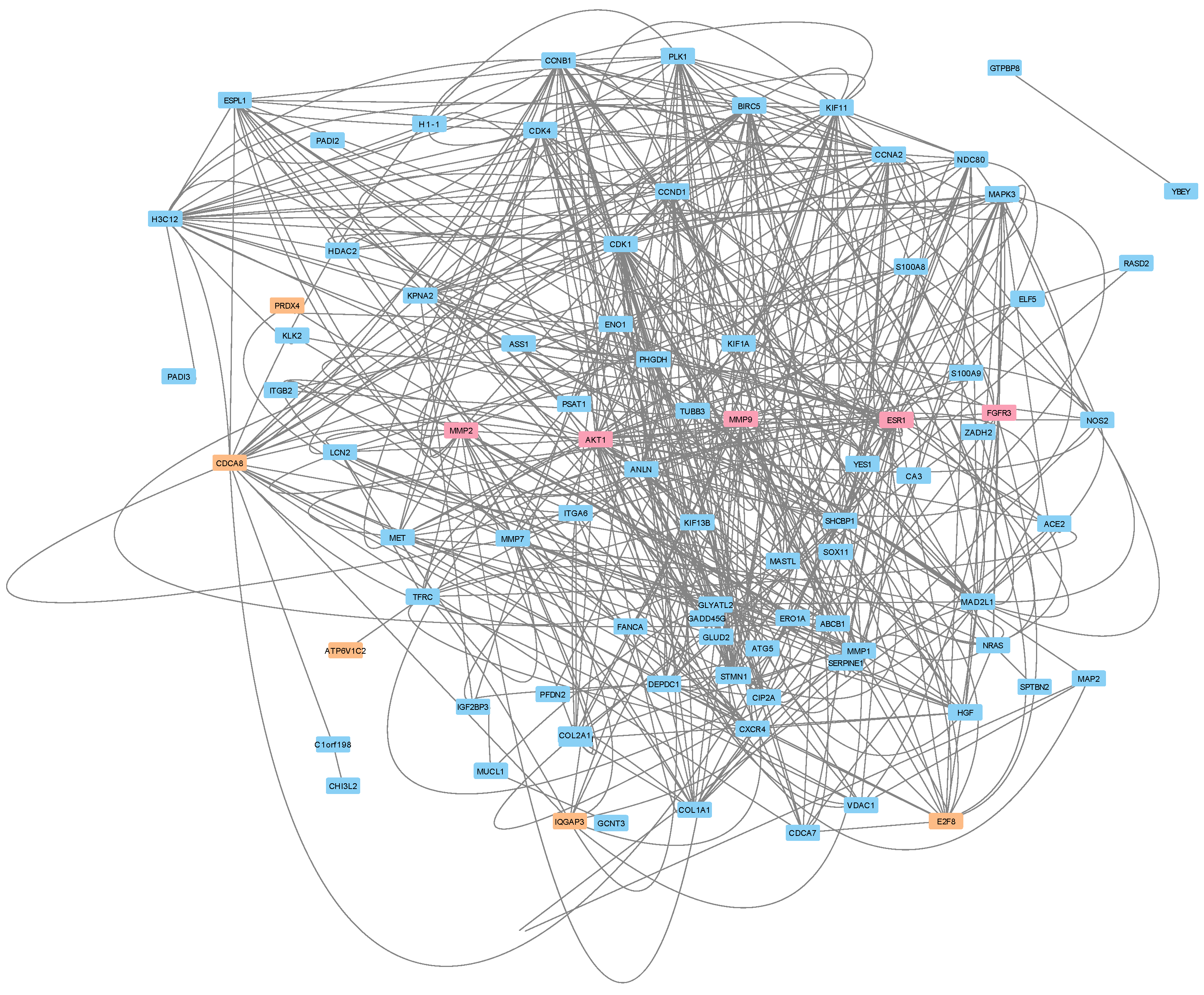
3.5. Protein–Protein Interactions Reveal a Potential Treatment Resistance Network
4. Discussion
4.1. Differentially Expressed Genes
4.2. Intracellular Signaling Pathways
4.3. Target Prioritization and Repurposing
4.4. Mechanistic Transcriptional Marker Analysis
4.5. Potential Treatment Resistance Mechanisms
4.6. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA A Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef]
- Postmenopause. Cleveland Clinic. Available online: https://my.clevelandclinic.org/health/diseases/21837-postmenopause (accessed on 8 June 2024).
- US Census Bureau. The Older Population: 2020; US Census Bureau: Washington, DC, USA, 2023.
- Breast Cancer Statistics. Available online: https://www.cancer.org/cancer/types/breast-cancer/about/how-common-is-breast-cancer.html (accessed on 8 June 2024).
- Rosenberg, P.S.; Barker, K.A.; Anderson, W.F. Estrogen Receptor Status and the Future Burden of Invasive and In Situ Breast Cancers in the United States. JNCI J. Natl. Cancer Inst. 2015, 107, djv159. [Google Scholar] [CrossRef]
- Breast Cancer Types: What your Type Means. Mayo Clinic. 2022. Available online: https://www.mayoclinic.org/diseases-conditions/breast-cancer/in-depth/breast-cancer/art-20045654 (accessed on 8 June 2024).
- Korourian, S.; Kumarapeli, A.R.; Klimberg, V.S. Breast Biomarker Immunocytochemistry. In The Breast; Elsevier: Amsterdam, The Netherlands, 2018; pp. 197–206.e6. [Google Scholar]
- Siddiqa, A.; Long, L.M.; Li, L.; Marciniak, R.A.; Kazhdan, I. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins Survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer 2008, 8, 129. [Google Scholar] [CrossRef]
- Zambrano, J.; Yeh, E.S. Autophagy and Apoptotic Crosstalk: Mechanism of Therapeutic Resistance in HER2-Positive Breast Cancer. Breast Cancer 2016, 10, 13–23. [Google Scholar] [CrossRef]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast cancer subtypes based on ER/PR and Her2 expression: Comparison of clinicopathologic features and survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef]
- Calhoun, B.C.; Collins, L.C. Predictive markers in breast cancer: An update on ER and HER2 testing and reporting. Semin. Diagn. Pathol. 2015, 32, 362–369. [Google Scholar] [CrossRef]
- Caldarella, A.; Crocetti, E.; Bianchi, S.; Vezzosi, V.; Urso, C.; Biancalani, M.; Zappa, M. Female breast cancer status according to ER, PR and HER2 expression: A population based analysis. Pathol. Oncol. Res. 2011, 17, 753–758. [Google Scholar] [CrossRef]
- Bagaria, S.P.; Ray, P.S.; Sim, M.-S.; Ye, X.; Shamonki, J.M.; Cui, X.; Giuliano, A.E. Personalizing breast cancer staging by the inclusion of ER, PR, and HER2. JAMA Surg. 2014, 149, 125–129. [Google Scholar] [CrossRef]
- Bhatnagar, A.S. The discovery and mechanism of action of letrozole. Breast Cancer Res. Treat. 2007, 105 (Suppl. 1), 7–17. [Google Scholar] [CrossRef]
- Sohl, C.D.; Guengerich, F.P. Kinetic analysis of the three-step steroid aromatase reaction of human cytochrome P450 19A1. J. Biol. Chem. 2010, 285, 17734–17743. [Google Scholar] [CrossRef]
- Mukherjee, A.G.; Wanjari, U.R.; Nagarajan, D.; Vibhaa, K.K.; Anagha, V.; Chakraborty, R.; Renu, K.; Dey, A.; Vellingiri, B.; Gopalakrishnan, A.V. Letrozole: Pharmacology, toxicity and potential therapeutic effects. Life Sci. 2022, 310, 121074. [Google Scholar] [CrossRef]
- Ellis, M.J.; Ma, C. Letrozole in the neoadjuvant setting the P024 trial. Breast Cancer Res. Treat. 2008, 112, 371. [Google Scholar] [CrossRef]
- Guarneri, V.; Generali, D.G.; Frassoldati, A.; Artioli, F.; Boni, C.; Cavanna, L.; Tagliafico, E.; Maiorana, A.; Bottini, A.; Cagossi, K.; et al. Double-blind, placebo-controlled, multicenter, randomized, phase IIb neoadjuvant study of letrozole-lapatinib in postmenopausal hormone receptor-positive, human epidermal growth factor receptor 2-negative, operable breast cancer. J. Clin. Oncol. 2014, 32, 1050–1057. [Google Scholar] [CrossRef]
- Sachdev, A.; Sharpe, I.; Bowman, M.; Booth, C.M.; Gyawali, B. Objective response rate of placebo in randomized controlled trials of anticancer medicines. eClinicalMedicine 2023, 55, 101753. [Google Scholar] [CrossRef]
- Lee, K.M.; Guerrero-Zotano, A.L.; Servetto, A.; Sudhan, D.R.; Lin, C.C.; Formisano, L.; Jansen, V.M.; González-Ericsson, P.; Sanders, M.E.; Stricker, T.P.; et al. Proline rich 11 (PRR11) overexpression amplifies PI3K signaling and promotes antiestrogen resistance in breast cancer. Nat. Commun. 2020, 11, 5488. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, J.; Choi, H.H.; Han, C.; Fang, Y.; Li, Y.; Van der Jeught, K.; Xu, H.; Zhang, L.; Frieden, M.; et al. Targeting 17q23 amplicon to overcome the resistance to anti-HER2 therapy in HER2+ breast cancer. Nat. Commun. 2018, 9, 4718. [Google Scholar] [CrossRef]
- Kodama, Y.; Shumway, M.; Leinonen, R.; International Nucleotide Sequence Database Collaboration. The Sequence Read Archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef]
- Orjuela, S.; Huang, R.; Hembach, K.M.; Robinson, M.D.; Soneson, C. ARMOR: An utomated eproducible dular Workflow for Preprocessing and Differential Analysis of NA-seq Data. G3 2019, 9, 2089–2096. [Google Scholar] [CrossRef]
- Köster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2018, 34, 3600. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 June 2024).
- Babraham Bioinformatics—Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 8 June 2024).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Wu, D.; Smyth, G.K. Camera: A competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012, 40, e133. [Google Scholar] [CrossRef]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.-S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef]
- Gifford, K.T.; Pickett, B.E. Comparative meta-analysis of host transcriptional response during Streptococcus pneumoniae carriage or infection. Microb. Pathog. 2022, 173, 105816. [Google Scholar] [CrossRef]
- Ferrarini, M.G.; Lal, A.; Rebollo, R.; Gruber, A.J.; Guarracino, A.; Gonzalez, I.M.; Floyd, T.; de Oliveira, D.S.; Shanklin, J.; Beausoleil, E.; et al. Genome-wide bioinformatic analyses predict key host and viral factors in SARS-CoV-2 pathogenesis. Commun. Biol. 2021, 4, 590. [Google Scholar] [CrossRef]
- Viedma, M.d.P.M.; Pickett, B.E. Characterizing the Different Effects of Zika Virus Infection in Placenta and Microglia Cells. Viruses 2018, 10, 649. [Google Scholar] [CrossRef]
- Spendlove, M.D.; Gibson, T.M.; McCain, S.; Stone, B.C.; Gill, T.; Pickett, B.E. Pathway2Targets: An open-source pathway-based approach to repurpose therapeutic drugs and prioritize human targets. PeerJ 2023, 11, e16088. [Google Scholar] [CrossRef]
- Scott, T.M.; Solis-Leal, A.; Lopez, J.B.; Robison, R.A.; Berges, B.K.; Pickett, B.E. Comparison of Intracellular Transcriptional Response of NHBE Cells to Infection with SARS-CoV-2 Washington and New York Strains. Front. Cell. Infect. Microbiol. 2022, 12, 1009328. [Google Scholar] [CrossRef]
- Gray, M.; Guerrero-Arguero, I.; Solis-Leal, A.; Robison, R.A.; Berges, B.K.; Pickett, B.E. Chikungunya virus time course infection of human macrophages reveals intracellular signaling pathways relevant to repurposed therapeutics. PeerJ 2022, 10, e13090. [Google Scholar] [CrossRef]
- Scott, T.M.; Jensen, S.; Pickett, B.E. A signaling pathway-driven bioinformatics pipeline for predicting therapeutics against emerging infectious diseases. F1000Research 2021, 10, 330. [Google Scholar] [CrossRef]
- Ochoa, D.; Hercules, A.; Carmona, M.; Suveges, D.; Gonzalez-Uriarte, A.; Malangone, C.; Miranda, A.; Fumis, L.; Carvalho-Silva, D.; Spitzer, M.; et al. Open Targets Platform: Supporting systematic drug–target identification and prioritisation. Nucleic Acids Res. 2021, 49, D1302–D1310. [Google Scholar] [CrossRef] [PubMed]
- GitHub—dmlc/xgboost: Scalable, Portable and Distributed Gradient Boosting (GBDT, GBRT or GBM) Library, for Python, R, Java, Scala, C++ and More. Runs on Single Machine, Hadoop, Spark, Dask, Flink and DataFlow. GitHub. Available online: https://github.com/dmlc/xgboost (accessed on 8 June 2024).
- Rapier-Sharman, N.; Clancy, J.; E Pickett, B. Joint Secondary Transcriptomic Analysis of Non-Hodgkin’s B-Cell Lymphomas Predicts Reliance on Pathways Associated with the Extracellular Matrix and Robust Diagnostic Biomarkers. J. Bioinform. Syst. Biol. 2022, 5, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Bybee, E.; Tellez Freitas, C.M.; Pickett, B.E.; Weber, K.S. Meta-Analysis of Two Human RNA-seq Datasets to Determine Periodontitis Diagnostic Biomarkers and Drug Target Candidates. Int. J. Mol. Sci. 2022, 23, 5580. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.; Hoffmann, C.S.; Pickett, B.E. Transcriptomics secondary analysis of severe human infection with SARS-CoV-2 identifies gene expression changes and predicts three transcriptional biomarkers in leukocytes. Comput. Struct. Biotechnol. J. 2023, 21, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.R.; Mecham, A.; Golightly, N.P.; Johnson, J.L.; Miller, D.B. The ability to classify patients based on gene-expression data varies by algorithm and performance metric. PLoS Comput. Biol. 2022, 18, e1009926. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Srivastava, G.; Hsieh, W.-S.; Gao, Z.; Murray, P.; Liao, S.-K.; Ambinder, R.; Tao, Q. The Stress-Responsive Gene GADD45G Is a Functional Tumor Suppressor, with Its Response to Environmental Stresses Frequently Disrupted Epigenetically in Multiple Tumors. Clin. Cancer Res. 2005, 11, 6442–6449. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Zotano, A.L.; Stricker, T.P.; Formisano, L.; Hutchinson, K.E.; Stover, D.G.; Lee, K.M.; Schwarz, L.J.; Giltnane, J.M.; Estrada, M.V.; Jansen, V.M.; et al. ER Breast Cancers Resistant to Prolonged Neoadjuvant Letrozole Exhibit an E2F4 Transcriptional Program Sensitive to CDK4/6 Inhibitors. Clin. Cancer Res. 2018, 24, 2517–2529. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Kundu, M.; Das, S.; Jena, B.C.; Mandal, M. Understanding the function and regulation of Sox2 for its therapeutic potential in breast cancer. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2022, 1877, 188692. [Google Scholar] [CrossRef] [PubMed]
- She, Z.-Y.; Yang, W.-X. SOX family transcription factors involved in diverse cellular events during development. Eur. J. Cell Biol. 2015, 94, 547–563. [Google Scholar] [CrossRef]
- Potzner, M.R.; Tsarovina, K.; Binder, E.; Penzo-Méndez, A.; Lefebvre, V.; Rohrer, H.; Wegner, M.; Sock, E. Sequential requirement of Sox4 and Sox11 during development of the sympathetic nervous system. Development 2010, 137, 775–784. [Google Scholar] [CrossRef]
- Shepherd, J.H.; Uray, I.P.; Mazumdar, A.; Tsimelzon, A.; Savage, M.; Hilsenbeck, S.G.; Brown, P.H. The SOX11 transcription factor is a critical regulator of basal-like breast cancer growth, invasion, and basal-like gene expression. Oncotarget 2016, 7, 13106–13121. [Google Scholar] [CrossRef]
- Wang, H.; Liu, B.; Long, J.; Yu, J.; Ji, X.; Li, J.; Zhu, N.; Zhuang, X.; Li, L.; Chen, Y.; et al. Integrative analysis identifies two molecular and clinical subsets in Luminal B breast cancer. iScience 2023, 26, 107466. [Google Scholar] [CrossRef]
- Zvelebil, M.; Oliemuller, E.; Gao, Q.; Wansbury, O.; Mackay, A.; Kendrick, H.; Smalley, M.J.; Reis-Filho, J.S.; Howard, B.A. Embryonic mammary signature subsets are activated in Brca1-/- and basal-like breast cancers. Breast Cancer Res. 2013, 15, R25. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Németh, J.; Angel, P.; Hess, J. S100A8 and S100A9 in inflammation and cancer. Biochem. Pharmacol. 2006, 72, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shu, X.; Xu, J.; Su, S.M.; Chan, U.I.; Mo, L.; Liu, J.; Zhang, X.; Adhav, R.; Chen, Q.; et al. S100A9-CXCL12 activation in BRCA1-mutant breast cancer promotes an immunosuppressive microenvironment associated with resistance to immunotherapy. Nat. Commun. 2022, 13, 1481. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ouyang, Y.; Li, Z.; Wang, X.; Ma, J. S100A8 and S100A9 in Cancer. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2023, 1878, 188891. [Google Scholar] [CrossRef]
- Rammes, A.; Roth, J.; Goebeler, M.; Klempt, M.; Hartmann, M.; Sorg, C. Myeloid-related Protein (MRP) 8 and MRP14, Calcium-binding Proteins of the S100 Family, Are Secreted by Activated Monocytes via a Novel, Tubulin-dependent Pathway. J. Biol. Chem. 1997, 272, 9496–9502. [Google Scholar] [CrossRef]
- Wang, L.; Di, L.-J. BRCA1 and estrogen/estrogen receptor in breast cancer: Where they interact? Int. J. Biol. Sci. 2014, 10, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Karpisheh, V.; Ahmadi, M.; Abbaszadeh-Goudarzi, K.; Saray, M.M.; Barshidi, A.; Mohammadi, H.; Yousefi, M.; Jadidi-Niaragh, F. The role of Th17 cells in the pathogenesis and treatment of breast cancer. Cancer Cell Int. 2022, 22, 108. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Takano, S.; Teratani, T.; Ito, Y.; Yamada, T.; Nozawa, R. S100A8 and S100A9 overexpression is associated with poor pathological parameters in invasive ductal carcinoma of the breast. Curr. Cancer Drug Targets 2008, 8, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Boutas, I.; Kontogeorgi, A.; Koufopoulos, N.; Dimitrakakis, C.; Dimas, D.; Kalantaridou, S.N.; Korou, L.-M.; Perrea, D. Effect of Aromatase Inhibitors on Serum Calprotectin Levels in an Animal Experimental Model: Trial. Cancer Diagn. Progn. 2022, 2, 720–730. [Google Scholar] [CrossRef]
- Ralli, S.; Jones, S.J.; Leach, S.; Lynch, H.T.; Brooks-Wilson, A.R. Gene and pathway based burden analyses in familial lymphoid cancer cases: Rare variants in immune pathway genes. PLoS ONE 2023, 18, e0287602. [Google Scholar] [CrossRef]
- Patil, S.; Linge, A.; Grosser, M.; Lohaus, F.; Gudziol, V.; Kemper, M.; Nowak, A.; Haim, D.; Tinhofer, I.; Budach, V.; et al. Development and validation of a 6-gene signature for the prognosis of loco-regional control in patients with HPV-negative locally advanced HNSCC treated by postoperative radio(chemo)therapy. Radiother. Oncol. 2022, 171, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Xie, L.; Shui, Y.; Li, J.; Wei, Q. Identification of Tumor Microenvironment-Related Prognostic Genes in Sarcoma. Front. Genet. 2021, 12, 620705. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liu, X.; Zhou, X.J.; Chen, X.; Li, J.P.; Yin, Y.H.; Qu, Y.Q. Identification and validation of the prognostic value of immune-related genes in non-small cell lung cancer. Am. J. Transl. Res. 2020, 12, 5844–5865. [Google Scholar] [PubMed]
- Liao, H.Y.; Da, C.M.; Liao, B. Roles of matrix metalloproteinase-7 (MMP-7) in cancer. Clin. Biochem. 2021, 92, 9–18. [Google Scholar] [CrossRef]
- Köhrmann, A.; Kammerer, U.; Kapp, M.; Dietl, J.; Anacker, J. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer and breast cancer cell lines: New findings and review of the literature. BMC Cancer 2009, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Bouris, P.; Skandalis, S.S.; Piperigkou, Z.; Afratis, N.; Karamanou, K.; Aletras, A.J.; Moustakas, A.; Theocharis, A.D.; Karamanos, N.K. Estrogen receptor alpha mediates epithelial to mesenchymal transition, expression of specific matrix effectors and functional properties of breast cancer cells. Matrix Biol. 2015, 43, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, S.T.; Keri, R.A. The Forkhead Box Transcription Factor FOXC1 Promotes Breast Cancer Invasion by Inducing Matrix Metalloprotease 7 (MMP7) Expression. J. Biol. Chem. 2012, 287, 24631–24640. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhan, Y.; Yan, L.; Hao, D. How zoledronic acid improves osteoporosis by acting on osteoclasts. Front. Pharmacol. 2022, 13, 961941. [Google Scholar] [CrossRef]
- Reid, I.R.; Green, J.R.; Lyles, K.W.; Reid, D.M.; Trechsel, U.; Hosking, D.J.; Black, D.M.; Cummings, S.R.; Russell, R.G.G.; Eriksen, E.F. Zoledronate. Bone 2020, 137, 115390. [Google Scholar] [CrossRef]
- Black, D.M.; Delmas, P.D.; Eastell, R.; Reid, I.R.; Boonen, S.; Cauley, J.A.; Cosman, F.; Lakatos, P.; Leung, P.C.; Man, Z.; et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N. Engl. J. Med. 2007, 356, 1809–1822. [Google Scholar] [CrossRef] [PubMed]
- Dedes, P.; Gialeli, C.; Tsonis, A.; Kanakis, I.; Theocharis, A.; Kletsas, D.; Tzanakakis, G.; Karamanos, N. Expression of matrix macromolecules and functional properties of breast cancer cells are modulated by the bisphosphonate zoledronic acid. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2012, 1820, 1926–1939. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Guo, C.; Li, J.; Liang, Z.; Wang, Y.; Yu, A.; Liu, R.; Guo, Y.; Chen, J.; Huang, S. EN1 Regulates Cell Growth and Proliferation in Human Glioma Cells via Hedgehog Signaling. Int. J. Mol. Sci. 2022, 23, 1123. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Singh, M.; Tyagi, S.C. Post-menopausal breast cancer: From estrogen to androgen receptor. Oncotarget 2017, 8, 102739–102758. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Sung, M.; Oh, E.; Van Vrancken, M.; Song, J.-Y.; Jung, K.; Choi, Y.-L. Engrailed 1 overexpression as a potential prognostic marker in quintuple-negative breast cancer. Cancer Biol. Ther. 2018, 19, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Beltran, A.S.; Graves, L.M.; Blancafort, P. Novel role of Engrailed 1 as a prosurvival transcription factor in basal-like breast cancer and engineering of interference peptides block its oncogenic function. Oncogene 2013, 33, 4767–4777. [Google Scholar] [CrossRef]
- Yang, L.; Li, X.; Luo, Y.; Yang, T.; Wang, H.; Shi, L.; Feng, M.; Xie, W. Weighted gene co expression network analysis of the association between upregulated AMD1, EN1 and VGLL1 and the progression and poor prognosis of breast cancer. Exp. Ther. Med. 2021, 22, 1030. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef]
- Chiappa, M.; Petrella, S.; Damia, G.; Broggini, M.; Guffanti, F.; Ricci, F. Present and Future Perspective on PLK1 Inhibition in Cancer Treatment. Front. Oncol. 2022, 12, 903016. [Google Scholar] [CrossRef]
- Wierer, M.; Verde, G.; Pisano, P.; Molina, H.; Font-Mateu, J.; Di Croce, L.; Beato, M. PLK1 Signaling in Breast Cancer Cells Cooperates with Estrogen Receptor-Dependent Gene Transcription. Cell Rep. 2013, 3, 2021–2032. [Google Scholar] [CrossRef]
- Kahl, I.; Mense, J.; Finke, C.; Boller, A.; Lorber, C.; Győrffy, B.; Greve, B.; Götte, M.; A Espinoza-Sánchez, N. The cell cycle-related genes RHAMM, AURKA, TPX2, PLK1, and PLK4 are associated with the poor prognosis of breast cancer patients. J. Cell. Biochem. 2022, 123, 581–600. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, R.E.A.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Jansen, V.M.; Bafna, S.; Giltnane, J.M.; Balko, J.M.; Estrada, M.V.; Meszoely, I.; Mayer, I.; Abramson, V.; Ye, F.; et al. Kinome-wide Functional Screen Identifies Role of PLK1 in Hormone-Independent, ER-Positive Breast Cancer. Cancer Res. 2015, 75, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Couchman, J.R. Syndecan-1 (CD138), Carcinomas and EMT. Int. J. Mol. Sci. 2021, 22, 4227. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chen, S.; Ying, H.; Yao, W. Targeting syndecan-1: New opportunities in cancer therapy. Am. J. Physiol. Physiol. 2022, 323, C29–C45. [Google Scholar] [CrossRef] [PubMed]
- Fleurot, E.; Goudin, C.; Hanoux, V.; Bonnamy, P.-J.; Levallet, J. Estrogen receptor α regulates the expression of syndecan-1 in human breast carcinoma cells. Endocr.-Relat. Cancer 2019, 26, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Barbareschi, M.; Maisonneuve, P.; Aldovini, D.; Cangi, M.G.; Pecciarini, L.; Mauri, F.A.; Veronese, S.; Caffo, O.; Lucenti, A.; Palma, P.D.; et al. High syndecan-1 expression in breast carcinoma is related to an aggressive phenotype and to poorer prognosis. Cancer 2003, 98, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Al Kawas, H.; Saaid, I.; Jank, P.; Westhoff, C.C.; Denkert, C.; Pross, T.; Weiler, K.B.S.; Karsten, M.M. How VEGF-A and its splice variants affect breast cancer development—Clinical implications. Cell. Oncol. 2022, 45, 227–239. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations—A mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef]
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 mutations in breast cancer. Cancer 2019, 125, 3714–3728. [Google Scholar] [CrossRef]
- Brett, J.O.; Spring, L.M.; Bardia, A.; Wander, S.A. ESR1 mutation as an emerging clinical biomarker in metastatic hormone receptor-positive breast cancer. Breast Cancer Res. 2021, 23, 85. [Google Scholar] [CrossRef] [PubMed]
- Joseph, C.; Alsaleem, M.; Orah, N.; Narasimha, P.L.; Miligy, I.M.; Kurozumi, S.; Ellis, I.O.; Mongan, N.P.; Green, A.R.; Rakha, E.A. Elevated MMP9 expression in breast cancer is a predictor of shorter patient survival. Breast Cancer Res. Treat. 2020, 182, 267–282. [Google Scholar] [CrossRef]
- Cancemi, P.; Aiello, A.; Accardi, G.; Caldarella, R.; Candore, G.; Caruso, C.; Ciaccio, M.; Cristaldi, L.; Di Gaudio, F.; Siino, V.; et al. The Role of Matrix Metalloproteinases (MMP-2 and MMP-9) in Ageing and Longevity: Focus on Sicilian Long-Living Individuals (LLIs). Mediat. Inflamm. 2020, 2020, 8635158. [Google Scholar] [CrossRef] [PubMed]
- Nikolov, A.; Popovski, N. Role of Gelatinases MMP-2 and MMP-9 in Healthy and Complicated Pregnancy and Their Future Potential as Preeclampsia Biomarkers. Diagnostics 2021, 11, 480. [Google Scholar] [CrossRef]
- Owyong, M.; Chou, J.; Bijgaart, R.J.v.D.; Kong, N.; Efe, G.; Maynard, C.; Talmi-Frank, D.; Solomonov, I.; Koopman, C.; Hadler-Olsen, E.; et al. MMP9 modulates the metastatic cascade and immune landscape for breast cancer anti-metastatic therapy. Life Sci. Alliance 2019, 2, e201800226. [Google Scholar] [CrossRef]
- Mendes, O.; Kim, H.-T.; Lungu, G.; Stoica, G. MMP2 role in breast cancer brain metastasis development and its regulation by TIMP2 and ERK1/2. Clin. Exp. Metastasis 2007, 24, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Mendes, O.; Kim, H.-T.; Stoica, G. Expression of MMP2, MMP9 and MMP3 in Breast Cancer Brain Metastasis in a Rat Model. Clin. Exp. Metastasis 2005, 22, 237–246. [Google Scholar] [CrossRef]
- Jiang, H.; Li, H. Prognostic values of tumoral MMP2 and MMP9 overexpression in breast cancer: A systematic review and meta-analysis. BMC Cancer 2021, 21, 149. [Google Scholar] [CrossRef]
- Somiari, S.B.; Somiari, R.I.; Heckman, C.M.; Olsen, C.H.; Jordan, R.M.; Russell, S.J.; Shriver, C.D. Circulating MMP2 and MMP9 in breast cancer—Potential role in classification of patients into low risk, high risk, benign disease and breast cancer categories. Int. J. Cancer 2006, 119, 1403–1411. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/study/NCT00003010 (accessed on 8 June 2024).
- Chew, N.J.; Nguyen, E.V.; Su, S.-P.; Novy, K.; Chan, H.C.; Nguyen, L.K.; Luu, J.; Simpson, K.J.; Lee, R.S.; Daly, R.J. FGFR3 signaling and function in triple negative breast cancer. Cell Commun. Signal. 2020, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Shi, Y.; Ye, P.; Guo, J.; Zhou, Q.; Tang, Y. MicroRNA-99a Suppresses Breast Cancer Progression by Targeting FGFR3. Front. Oncol. 2020, 9, 1473. [Google Scholar] [CrossRef]
- Xie, J.; Wan, Y.; Zhang, M.; Jin, Z.; Yao, Y. Circ_0061825 Acts as a miR-593-3p Sponge to Promote Breast Cancer Progression by Regulating FGFR3 Expression. Cancer Manag. Res. 2020, 12, 11243–11255. [Google Scholar] [CrossRef] [PubMed]
- Hinz, N.; Jücker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef] [PubMed]
- Riggio, M.; Perrone, M.C.; Polo, M.L.; Rodriguez, M.J.; May, M.; Abba, M.; Lanari, C.; Novaro, V. AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins. Sci. Rep. 2017, 7, srep44244. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.G.; Ma, C.X.; Crowder, R.J.; Guintoli, T.; Phommaly, C.; Gao, F.; Lin, L.; Ellis, M.J. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011, 13, R21. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Chen, P.; Cheng, Y. PRDX4 and Its Roles in Various Cancers. Technol. Cancer Res. Treat. 2019, 18, 1533033819864313. [Google Scholar] [CrossRef]
- Elko, E.A.; Manuel, A.M.; White, S.; Zito, E.; van der Vliet, A.; Anathy, V.; Janssen-Heininger, Y.M. Oxidation of peroxiredoxin-4 induces oligomerization and promotes interaction with proteins governing protein folding and endoplasmic reticulum stress. J. Biol. Chem. 2021, 296, 100665. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Jo, M.; Kim, Y.R.; Lee, C.K.; Hong, J.T. Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 2016, 163, 1–23. [Google Scholar] [CrossRef]
- Kocatürk, B. In silico analysis reveals PRDX4 as a prognostic and oncogenic marker in renal papillary cell carcinoma. Gene 2023, 859, 147201. [Google Scholar] [CrossRef]
- Mei, J.; Hao, L.; Liu, X.; Sun, G.; Xu, R.; Wang, H.; Liu, C. Comprehensive analysis of peroxiredoxins expression profiles and prognostic values in breast cancer. Biomark. Res. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, S.; Tiedemann, K.; Tabariès, S.; Siegel, P.M.; Komarova, S.V. Peroxiredoxin 4: A novel secreted mediator of cancer induced osteoclastogenesis. Cancer Lett. 2015, 361, 262–270. [Google Scholar] [CrossRef]
- Wang, G.; Zhong, W.-C.; Bi, Y.-H.; Tao, S.-Y.; Zhu, H.-X.; Xu, A.-M. The Prognosis of Peroxiredoxin Family in Breast Cancer. Cancer Manag. Res. 2019, 11, 9685–9699. [Google Scholar] [CrossRef] [PubMed]
- Ummanni, R.; Mundt, F.; Pospisil, H.; Venz, S.; Scharf, C.; Barett, C.; Fälth, M.; Köllermann, J.; Walther, R.; Schlomm, T.; et al. Identification of clinically relevant protein targets in prostate cancer with 2D-DIGE coupled mass spectrometry and systems biology network platform. PLoS ONE 2011, 6, e16833. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.A.; Hou, M.-F.; Chen, C.-C.; Wang, C.-L.; Chang, H.-W. Androgen receptor and gene network: Micromechanics reassemble the signaling machinery of TMPRSS2-ERG positive prostate cancer cells. Cancer Cell Int. 2014, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Chun, J.N.; Cho, M.; So, I.; Jeon, J.H. Emerging role of E2F8 in human cancer. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2023, 1869, 166745. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef] [PubMed]
- Weijts, B.G.M.W.; Bakker, W.J.; A Cornelissen, P.W.; Liang, K.-H.; Schaftenaar, F.H.; Westendorp, B.; Wolf, C.A.C.M.T.d.; Paciejewska, M.; Scheele, C.L.G.J.; Kent, L.; et al. E2F7 and E2F8 promote angiogenesis through transcriptional activation of VEGFA in cooperation with HIF1. EMBO J. 2012, 31, 3871–3884. [Google Scholar] [CrossRef]
- Whittle, J.R.; Vaillant, F.; Surgenor, E.; Policheni, A.N.; Giner, G.; Capaldo, B.D.; Chen, H.R.; Liu, H.K.; Dekkers, J.F.; Sachs, N.; et al. Dual Targeting of CDK4/6 and BCL2 Pathways Augments Tumor Response in Estrogen Receptor–Positive Breast Cancer. Clin. Cancer Res. 2020, 26, 4120–4134. [Google Scholar] [CrossRef]
- Ye, L.; Guo, L.; He, Z.; Wang, X.; Lin, C.; Zhang, X.; Wu, S.; Bao, Y.; Yang, Q.; Song, L.; et al. Upregulation of E2F8 promotes cell proliferation and tumorigenicity in breast cancer by modulating G1/S phase transition. Oncotarget 2016, 7, 23757–23771. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; Yang, D.; Xiang, S.; Sun, J.; Li, H.; Ren, G. Expression patterns of E2F transcription factors and their potential prognostic roles in breast cancer. Oncol. Lett. 2018, 15, 9216–9230. [Google Scholar] [CrossRef] [PubMed]
- Oshi, M.; Takahashi, H.; Tokumaru, Y.; Yan, L.; Rashid, O.M.; Nagahashi, M.; Matsuyama, R.; Endo, I.; Takabe, K. The E2F Pathway Score as a Predictive Biomarker of Response to Neoadjuvant Therapy in ER+/HER2- Breast Cancer. Cells 2020, 9, 1643. [Google Scholar] [CrossRef] [PubMed]
- Hoogstraat, M.; Lips, E.H.; Mayayo-Peralta, I.; Mulder, L.; Kristel, P.; van der Heijden, I.; Annunziato, S.; van Seijen, M.; Nederlof, P.M.; Sonke, G.S.; et al. Comprehensive characterization of pre- and post-treatment samples of breast cancer reveal potential mechanisms of chemotherapy resistance. NPJ Breast Cancer 2022, 8, 60. [Google Scholar] [CrossRef]
- Selli, C.; Turnbull, A.K.; Pearce, D.A.; Li, A.; Fernando, A.; Wills, J.; Renshaw, L.; Thomas, J.S.; Dixon, J.M.; Sims, A.H. Molecular changes during extended neoadjuvant letrozole treatment of breast cancer: Distinguishing acquired resistance from dormant tumours. Breast Cancer Res. BCR 2019, 21, 2. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhou, J.; Chen, H.; Li, J.; Zhang, C.; Jiang, X.; Ni, C. The immunomodulatory effects of endocrine therapy in breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 19. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.B.; Panis, C.; Alves, V.D.; Herrera, A.C.S.A.; Binato, R.; Pizzatti, L.; Cecchini, R.; Abdelhay, E. Label-Free Proteomics Revealed Oxidative Stress and Inflammation as Factors That Enhance Chemoresistance in Luminal Breast Cancer. Oxidative Med. Cell. Longev. 2019, 2019, 5357649. [Google Scholar] [CrossRef] [PubMed]
- Dabydeen, S.A.; Kang, K.; Díaz-Cruz, E.S.; Alamri, A.; Axelrod, M.L.; Bouker, K.B.; Al-Kharboosh, R.; Clarke, R.; Hennighausen, L.; Furth, P.A. Comparison of tamoxifen and letrozole response in mammary preneoplasia of ER and aromatase overexpressing mice defines an immune-associated gene signature linked to tamoxifen resistance. Carcinog. 2015, 36, 122–132. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metrics | Study Information |
|---|---|
| Gene Expression Omnibus (GEO) Study Identifier | GSE145325 |
| Title | RNA sequencing of ER+ breast tumor treated with Letrozole |
| Platform | Illumina HiSeq 3000 |
| Diagnostic criteria | Resistance to estrogen suppression was defined by a preoperative endocrine prognostic index (PEPI) ≥ 4 and/or evidence of cancer relapse after a median follow-up of 5 years |
| Number of responders vs. non-responders | 36 vs. 22 |
| Gene Symbol | Log2 Fold-Change | Log Counts Per Million (CPM) | FDR p-Value | Gene Name |
|---|---|---|---|---|
| SOX11 | 3.87 | 3.74 | 8.07 × 10−6 | SRY-box transcription factor 11 |
| S100A9 | 4.53 | 5.06 | 1.77 × 10−5 | S100 calcium-binding protein A9 |
| S100A8 | 4.7 | 2.96 | 1.77 × 10−5 | S100 calcium-binding protein A8 |
| IGLV3–25 | 5.15 | 6.27 | 0.000184 | Immunoglobulin lambda variable 3–25 |
| MMP7 | 4.11 | 4.84 | 0.000665 | Matrix metallopeptidase 7 |
| Rank | Pathway Name | Total # of Pathway Members | # DEGs in Pathway | Bonferroni p-Value | Predicted Modulation |
|---|---|---|---|---|---|
| 1 | PLK1 signaling events/anti-tumoral activity | 44 | 23 | 5.84 × 10−10 | Activated |
| 2 | Syndecan-1-mediated signaling events/leukocyte adhesion | 30 | 8 | 0.00018429 | Activated |
| 3 | FOXM1 proliferation-associated transcription factor network | 36 | 14 | 0.0105863 | Activated |
| 4 | HIF-1-alpha transcription factor network/oxygen homeostasis | 60 | 17 | 0.01923722 | Inhibited |
| Target Symbol | Target Name | Weighted Score (Higher Is Better) |
|---|---|---|
| VEGFA | Vascular endothelial growth factor A | 2220.5 |
| ESR1 | Estrogen Receptor 1 | 1771 |
| MMP9 | Matrix Metallopeptidase 9 | 1699 |
| FGFR3 | Fibroblast Growth Factor Receptor 9 | 1589 |
| AKT1 | AKT serine/threonine kinase 1 | 1571 |
| Feature | Gain | Cover | Frequency | Antibody Available | Log2FC (FDR p-Value) | Location |
|---|---|---|---|---|---|---|
| PRDX4 | 0.032 | 0.027 | 0.023 | Monoclonal | 0.807 (0.0326) | Secretory granules; the ER; and exosomes |
| E2F8 | 0.031 | 0.025 | 0.021 | Monoclonal | 1.4 (0.266) | Cytosol and nucleus |
| IQGAP3 | 0.024 | 0.020 | 0.016 | Monoclonal | 1.2 (0.266) | Cytosol and plasma membrane |
| ATP6V1C2 | 0.021 | 0.018 | 0.015 | Monoclonal | 2.44 (0.0263) | Cytosol; lysosomes; and exosomes |
| CDCA8 | 0.017 | 0.014 | 0.011 | Monoclonal | 1.37 (0.101) | Cytosol and nucleus |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutherland, L.; Lang, J.; Gonzalez-Juarbe, N.; Pickett, B.E. Secondary Analysis of Human Bulk RNA-Seq Dataset Suggests Potential Mechanisms for Letrozole Resistance in Estrogen-Positive (ER+) Breast Cancer. Curr. Issues Mol. Biol. 2024, 46, 7114-7133. https://doi.org/10.3390/cimb46070424
Sutherland L, Lang J, Gonzalez-Juarbe N, Pickett BE. Secondary Analysis of Human Bulk RNA-Seq Dataset Suggests Potential Mechanisms for Letrozole Resistance in Estrogen-Positive (ER+) Breast Cancer. Current Issues in Molecular Biology. 2024; 46(7):7114-7133. https://doi.org/10.3390/cimb46070424
Chicago/Turabian StyleSutherland, Lincoln, Jacob Lang, Norberto Gonzalez-Juarbe, and Brett E. Pickett. 2024. "Secondary Analysis of Human Bulk RNA-Seq Dataset Suggests Potential Mechanisms for Letrozole Resistance in Estrogen-Positive (ER+) Breast Cancer" Current Issues in Molecular Biology 46, no. 7: 7114-7133. https://doi.org/10.3390/cimb46070424