
Erlotinib Treatment in Colorectal Cancer Suppresses Autophagy Based on KRAS Mutation
 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Culture
2.3. Erlotinib Preparation
2.4. Cell Viability Assay
2.5. Gene Expression Profiling Interactive Analysis 2 (GEPIA2)
2.6. TCGA Gene Expression
2.7. Protein Extraction
2.8. Protein Quantification
2.9. Western Blot Analysis
2.10. RNA Extraction and Quantification
2.11. cDNA Synthesis
2.12. Quantitative Polymerase Chain Reaction (qPCR)
2.13. Protein–Drug Docking
2.14. Protein–Protein Molecular Dynamics Simulation and Kinetic and Structural Analysis
2.15. Statistical Analysis
3. Results
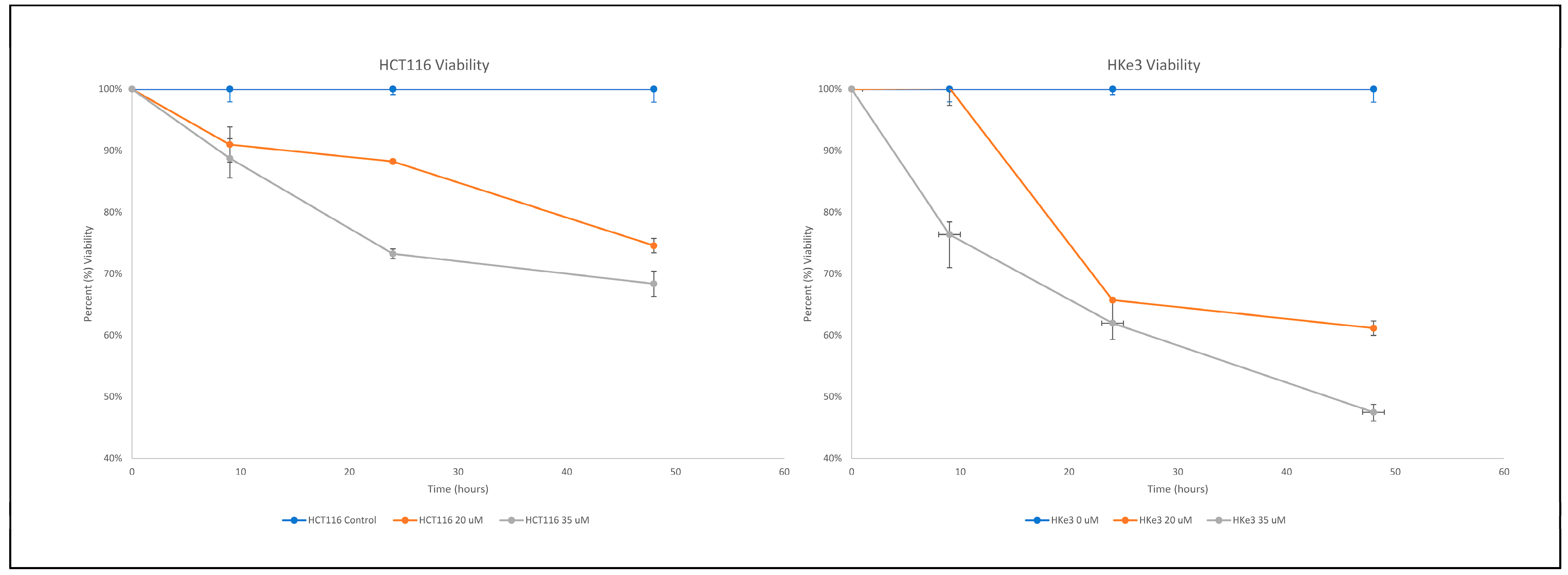
3.1. Erlotinib Induces Greater Cell Killing in KRAS Wildtype Hke3 Compared to KRAS Mutant HCT116 Cells
3.2. The Expression of Key Autophagy Proteins Decreases in KRAS-Mut upon Treatment with Erlotinib When Compared to KRAS-WT
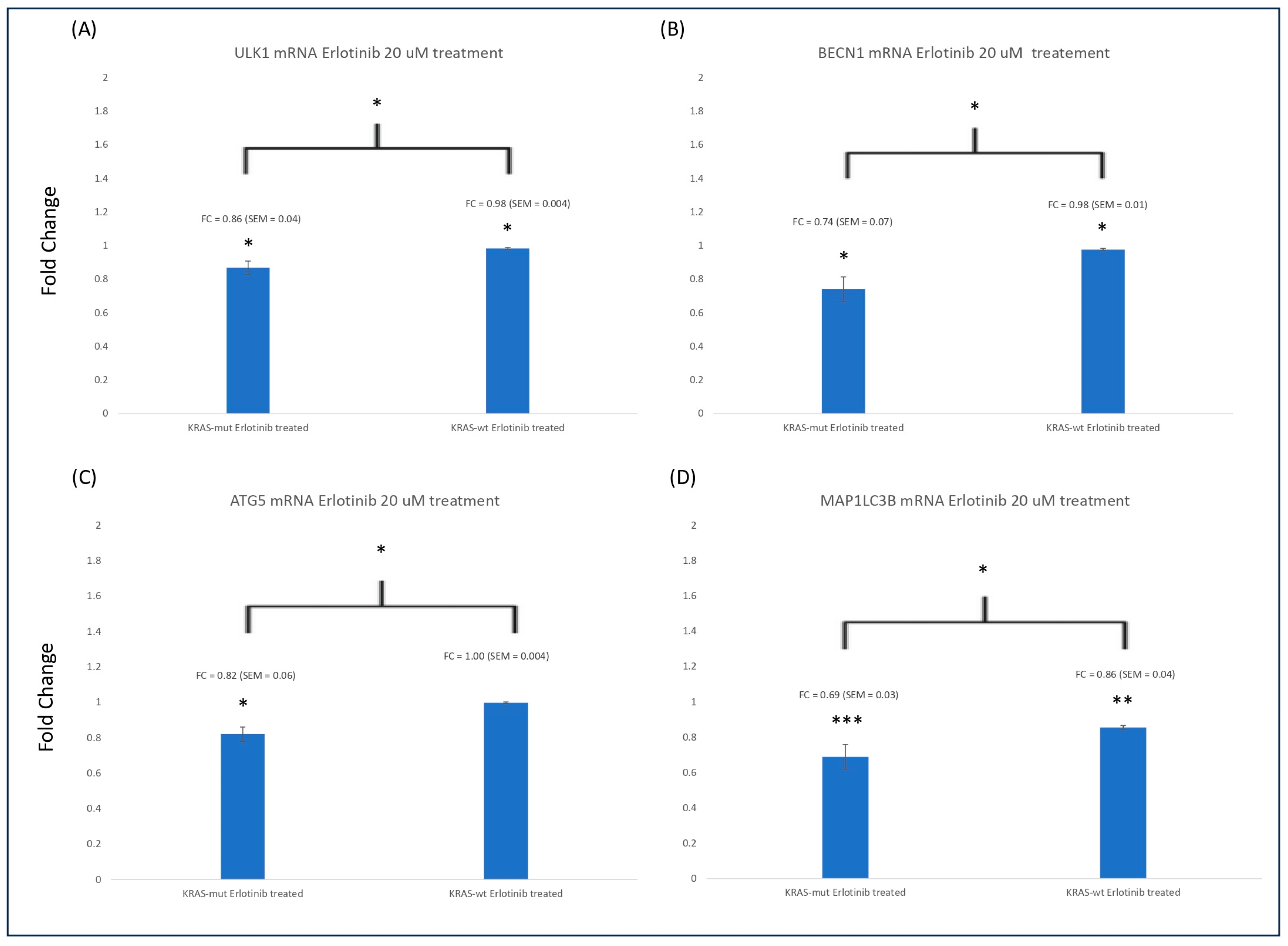
3.3. ULK1, BECN1, ATG5, and MAP1LC3B mRNA Expression Is Reduced in KRAS-mt CRC Compared to KRAS-WT CRC Cells
3.4. KRAS Expression Positively Correlates with the Expression of the Autophagy Genes MAP1LC3B, ATG5, ATG10, ATG13, and ATG14 in CRC Patients
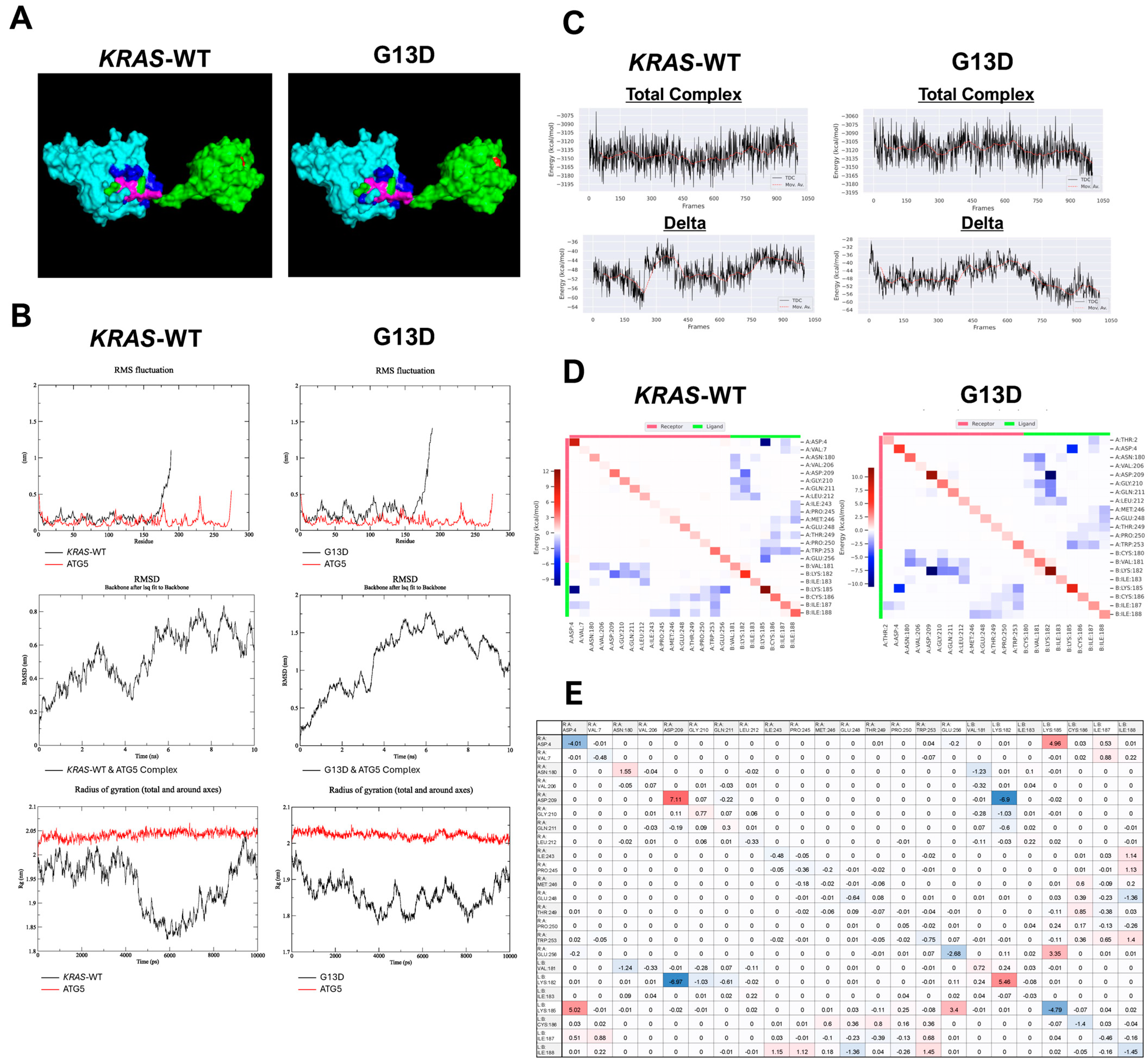
3.5. Destabilization of KRAS-ATG5 Complex when KRAS Is Mutated (G13D)
3.6. Determining Erlotinib Docking Site to EGFR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef]
- Zhu, G.; Pei, L.; Hia, H.; Tang, Q.; Bi, F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol. Cancer 2021, 20, 143. [Google Scholar] [CrossRef]
- Jass, J.R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2006, 50, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Taketo, M.M. Adenomatous polyposis coli (APC): A multi-functional tumor suppressor gene. J. Cell Sci. 2007, 120 Pt 19, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef]
- Gerber, M.; Goel, S.; Maitra, R. In silico comparative analysis of KRAS mutations at codons 12 and 13: Structural modifications of P-Loop, switch I & II regions preventing GTP hydrolysis. Comput. Biol. Med. 2022, 141, 105110. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Silverman, I.; Gerber, M.; Shaykevich, A.; Stein, Y.; Siegman, A.; Goel, S.; Maitra, R. Structural Modifications and Kinetic Effects of KRAS Interactions with HRAS and NRAS: An in-silico Comparative Analysis of KRAS Mutants. Front. Mol. Biosci. 2024, 11, 1436976. [Google Scholar] [CrossRef]
- Shaykevich, A.; Silverman, I.; Bandyopadhyaya, G.; Maitra, R. BRG1: Promoter or Suppressor of Cancer? The Outcome of BRG1’s Interaction with Specific Cellular Pathways. Int. J. Mol. Sci. 2023, 24, 2869. [Google Scholar] [CrossRef]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Lam, S.-K.; Mak, J.C.-W.; Zheng, C.-Y.; Ho, J.C.-M. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer 2013, 81, 354–361. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2016, 36, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Abdelgalil, A.A.; Al-Kahtani, H.M.; Al-Jenoobi, F.I. Erlotinib. In Profiles of Drug Substances, Excipients, and Related Methodology; Academic Press: Cambridge, MA, USA, 2020; pp. 93–117. [Google Scholar]
- Xu, C.; Jiang, Z.-B.; Shao, L.; Zhao, Z.-M.; Fan, X.-X.; Sui, X.; Yu, L.-L.; Wang, X.-R.; Zhang, R.-N.; Wang, W.-J.; et al. β-Elemene enhances erlotinib sensitivity through induction of ferroptosis by upregulating lncRNA H19 in EGFR-mutant non-small cell lung cancer. Pharmacol. Res. 2023, 191, 106739. [Google Scholar] [CrossRef] [PubMed]
- Shifteh, D.; Sapir, T.; Pahmer, M.; Haimowitz, A.; Goel, S.; Maitra, R. Protein Arginine Methyltransferase 5 as a Therapeutic Target for KRAS Mutated Colorectal Cancer. Cancers 2020, 12, 2091. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, R.H.; Swier, N.; van Beijnum, J.R.; Nowak-Sliwinska, P. Colorectal Cancer Growth Retardation through Induction of Apoptosis, Using an Optimized Synergistic Cocktail of Axitinib, Erlotinib, and Dasatinib. Cancers 2019, 11, 1878. [Google Scholar] [CrossRef] [PubMed]
- Chhouri, H.; Alexandre, D.; Grumolato, L. Mechanisms of Acquired Resistance and Tolerance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer. Cancers 2023, 15, 504. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.-K.; Lei, H.-M.; Liang, Q.; Tang, Y.-B.; Zhou, Y.; Wang, Y.; Zhang, S.; Li, W.-B.; Tong, Y.; Zhuang, G.; et al. Overcoming erlotinib resistance in EGFR mutation-positive lung adenocarcinomas through repression of phosphoglycerate dehydrogenase. Theranostics 2018, 8, 1808–1823. [Google Scholar] [CrossRef] [PubMed]
- Chellappan, S.P.; Lee, J.G.; Wu, R. Combination Erlotinib-Cisplatin and Atg3-Mediated Autophagy in Erlotinib Resistant Lung Cancer. PLoS ONE 2012, 7, e48532. [Google Scholar]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Lu, W.; Shen, X.; Wang, Q.; Lv, J.; Liu, M.; Cheng, F.; Zhao, Z.; Pang, X. Repurposing sertraline sensitizes non-small cell lung cancer cells to erlotinib by inducing autophagy. J. Clin. Investig. 2018, 3, e98921. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.-X.; Cao, Y. CB-Dock2: Improved protein–ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, Y.; Gan, J.; Xiao, Z.-X.; Cao, Y. FitDock: Protein–ligand docking by template fitting. Brief. Bioinform. 2022, 23, bbac087. [Google Scholar] [CrossRef]
- Desta, I.T.; Porter, K.A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and Its Limits in Rigid Body Protein-Protein Docking. Structure 2020, 28, 1071–1081.e3. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins Struct. Funct. Bioinform. 2013, 81, 2159–2166. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; A Porter, K.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the ClusPro server motivated by CAPRI. Proteins Struct. Funct. Bioinform. 2017, 85, 435–444. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Frias, E.M. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Zhang, W.; Wei, Y.; Yu, D.; Xu, J.; Peng, J. Gefitinib provides similar effectiveness and improved safety than erlotinib for advanced non-small cell lung cancer: A meta-analysis. Medicine 2018, 97, e0460. [Google Scholar] [CrossRef]
- Marsh, T.; Debnath, J. Autophagy suppresses breast cancer metastasis by degrading NBR1. Autophagy 2020, 16, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Pei, F.; Yang, F.; Li, L.; Amin, A.D.; Liu, S.; Buchan, J.R.; Cho, W.C. Role of Autophagy and Apoptosis in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2017, 18, 367. [Google Scholar] [CrossRef] [PubMed]
- Otahal, A.; Aydemir, D.; Tomasich, E.; Minichsdorfer, C. Delineation of cell death mechanisms induced by synergistic effects of statins and erlotinib in non-small cell lung cancer cell (NSCLC) lines. Sci. Rep. 2020, 10, 959. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Feng, H.; Sang, Q.; Li, F.; Chen, M.; Yu, B.; Xu, Z.; Pan, T.; Wu, X.; Hou, J.; et al. VPS35 promotes cell proliferation via EGFR recycling and enhances EGFR inhibitors response in gastric cancer. eBioMedicine 2023, 89, 104451. [Google Scholar] [CrossRef]
- Koulouridi, A.; Karagianni, M.; Messaritakis, I.; Sfakianaki, M.; Voutsina, A.; Trypaki, M.; Bachlitzanaki, M.; Koustas, E.; Karamouzis, M.V.; Ntavatzikos, A.; et al. Prognostic Value of KRAS Mutations in Colorectal Cancer Patients. Cancers 2022, 14, 3320. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef] [PubMed]
- Lypova, N.; Dougherty, S.M.; Lanceta, L.; Chesney, J.; Imbert-Fernandez, Y. PFKFB3 Inhibition Impairs Erlotinib-Induced Autophagy in NSCLCs. Cells 2021, 10, 1679. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Z.; Du, T.; Dong, X.; Li, Z.; Wu, G.; Zhang, R. Autophagy inhibition facilitates erlotinib cytotoxicity in lung cancer cells through modulation of endoplasmic reticulum stress. Int. J. Oncol. 2016, 48, 2558–2566. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dong, L.; Zheng, Z.; Zhu, Z.; Xie, B.; Xie, Y.; Li, X.; Chen, B.; Li, P. Effects of different KRAS mutants and Ki67 expression on diagnosis and prognosis in lung adenocarcinoma. Sci. Rep. 2024, 14, 4085. [Google Scholar] [CrossRef]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target Ther. 2023, 8, 9. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell. 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Ling, Y.-H.; Sironi, J.; Schwartz, E.L.; Perez-Soler, R.; Piperdi, B. The Autophagy Inhibitor Chloroquine Overcomes the Innate Resistance of Wild-Type EGFR Non-Small-Cell Lung Cancer Cells to Erlotinib. J. Thorac. Oncol. 2013, 8, 693–702. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Forward | Reverse |
|---|---|---|
| ULK1 | CACGCCACATAACAGACAAAAATAC | ACAAGGTGAGAATAAAGCCATCAAG |
| ATG5 | TTGGACGAATTCCAACTTGTTTC | ATTTCAGTGGTGTGCCTTCATATTC |
| MAP1LC3B | AGCAGCATCCAACCAAAATC | CTGTGTCCGTTCACCAACAG |
| BECN1 | ACAACAAGTTTGACCATGCAA | TTCAATCTTGCCTTTCTCCACAT |
| GAPDH | CTTTTGCGTCGCCAG | TTGATGGCAACAATATCC |
| Hours of Treatment | Erlotinib Treatment | HCT116 | HKe3 | p-Value |
|---|---|---|---|---|
| 9 | 20 μM | 0.91 | 1.00 | 0.026 |
| 35 μM | 0.89 | 0.76 | 0.013 | |
| 24 | 20 μM | 0.88 | 0.66 | 0.017 |
| 35 μM | 0.73 | 0.62 | 0.200 | |
| 48 | 20 μM | 0.75 | 0.61 | 0.049 |
| 35 μM | 0.68 | 0.47 | 0.010 |
| 20 μM of Erlotinib Treatment Protein Fold Change | |||
|---|---|---|---|
| Autophagy Protein | HCT116 | HKe3 | p-Value (HCT116 vs. HKe3) |
| ATG5 | 0.60 | 0.99 | 0.023 |
| ULK1 | 0.73 | 0.92 | 0.003 |
| Beclin-1 | 0.43 | 0.91 | 0.039 |
| LC3A | 0.40 | 0.80 | 0.019 |
| 20 μM of Erlotinib Treatment | |||
|---|---|---|---|
| Autophagy mRNA | HCT116 | HKe3 | p-Value (HCT116 vs. HKe3) |
| ATG5 | 0.82 | 1.00 | 0.019 |
| ULK1 | 0.87 | 0.98 | 0.029 |
| BECN1 | 0.74 | 0.98 | 0.019 |
| MAP1LC3B | 0.63 | 0.87 | 0.010 |
| mRNA Correlation Analysis (R) | ||
|---|---|---|
| mRNA | COADREAD | Normal Tissue |
| MAP1LC3B | 0.84 | 0.96 |
| ATG5 | 0.87 | 0.94 |
| ATG10 | 0.85 | 0.93 |
| ATG13 | 0.86 | 0.94 |
| ATG14 | 0.85 | 0.94 |
| mRNA Expression Change in CRC | |
|---|---|
| mRNA | Cancer Patients |
| MAP1LC3B | 76% |
| ATG5 | 81% |
| ATG10 | ---- |
| ATG13 | 85% |
| ATG14 | 86% |
| WT-ATG5 | G13D-ATG5 | |
|---|---|---|
| Total Complex Mean | −3142.8 | −3119.2 |
| Total Complex STD | 20.9 | 21.5 |
| Delta Mean | −48.4 | −47.7 |
| Delta STD | 5.3 | 6.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siegman, A.; Shaykevich, A.; Chae, D.; Silverman, I.; Goel, S.; Maitra, R. Erlotinib Treatment in Colorectal Cancer Suppresses Autophagy Based on KRAS Mutation. Curr. Issues Mol. Biol. 2024, 46, 7530-7547. https://doi.org/10.3390/cimb46070447
Siegman A, Shaykevich A, Chae D, Silverman I, Goel S, Maitra R. Erlotinib Treatment in Colorectal Cancer Suppresses Autophagy Based on KRAS Mutation. Current Issues in Molecular Biology. 2024; 46(7):7530-7547. https://doi.org/10.3390/cimb46070447
Chicago/Turabian StyleSiegman, Alexander, Aaron Shaykevich, Danbee Chae, Isaac Silverman, Sanjay Goel, and Radhashree Maitra. 2024. "Erlotinib Treatment in Colorectal Cancer Suppresses Autophagy Based on KRAS Mutation" Current Issues in Molecular Biology 46, no. 7: 7530-7547. https://doi.org/10.3390/cimb46070447
APA StyleSiegman, A., Shaykevich, A., Chae, D., Silverman, I., Goel, S., & Maitra, R. (2024). Erlotinib Treatment in Colorectal Cancer Suppresses Autophagy Based on KRAS Mutation. Current Issues in Molecular Biology, 46(7), 7530-7547. https://doi.org/10.3390/cimb46070447