
Rising Prevalence of Low-Frequency PPM1D Gene Mutations after Second HDCT in Multiple Myeloma
 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. NGS Amplicon Sequencing
2.3. Clinical Data Analysis
3. Results
3.1. Prevalence of PPM1D Mutations in Multiple Myeloma Patients after ASCT
3.2. Clinical Characteristics of Myeloma Patients
3.3. Therapeutic Interventions and Clinical Responses in Myeloma Patients
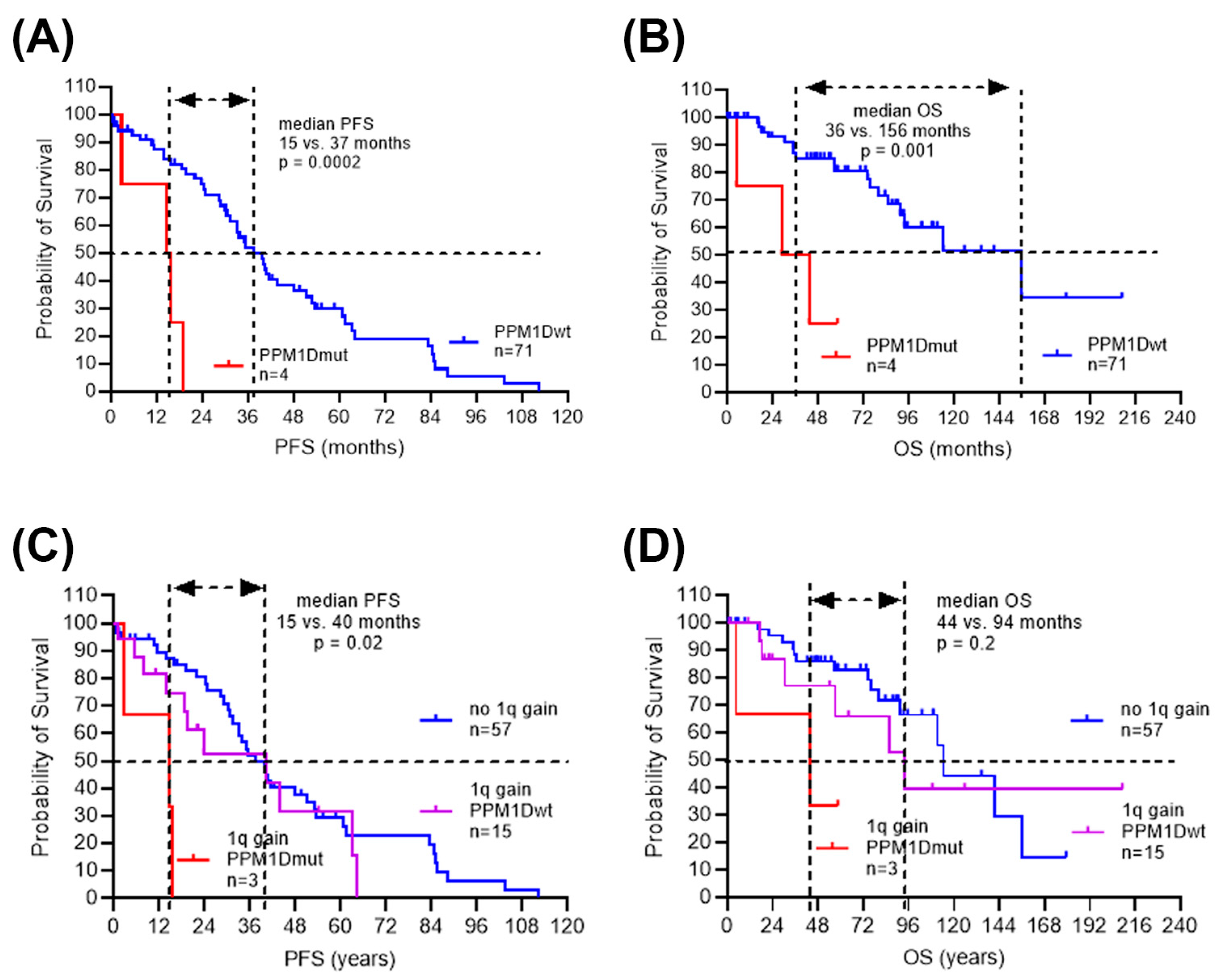
3.4. Inferior Clinical Outcome in Myeloma Patients with Low-Frequency PPM1D Mutations
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kyle, R.A.; Rajkumar, S.V. Multiple Myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef]
- Palumbo, A.; Anderson, K. Multiple Myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef]
- Willenbacher, E.; Balog, A.; Willenbacher, W. Short Overview on the Current Standard of Treatment in Newly Diagnosed Multiple Myeloma. Memo-Mag. Eur. Med. Oncol. 2018, 11, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V. Treatment of Multiple Myeloma. Nat. Rev. Clin. Oncol. 2011, 8, 479–491. [Google Scholar] [CrossRef]
- Callander, N.S.; Silbermann, R.; Kaufman, J.L.; Godby, K.N.; Laubach, J.; Schmidt, T.M.; Sborov, D.W.; Medvedova, E.; Reeves, B.; Dhakal, B.; et al. Daratumumab-Based Quadruplet Therapy for Transplant-Eligible Newly Diagnosed Multiple Myeloma with High Cytogenetic Risk. Blood Cancer J. 2024, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, H.; Ashcroft, J.; Szabo, Z.; Garderet, L. Navigating the Treatment Landscape in Multiple Myeloma: Which Combinations to Use and When? Ann. Hematol. 2019, 98, 1–18. [Google Scholar] [CrossRef]
- Farag, S.; Bacher, U.; Jeker, B.; Legros, M.; Rhyner, G.; Lüthi, J.-M.; Schardt, J.; Zander, T.; Daskalakis, M.; Mansouri, B.; et al. Adding Bendamustine to Melphalan before ASCT Improves CR Rate in Myeloma vs. Melphalan Alone: A Randomized Phase-2 Trial. Bone Marrow Transplant. 2022, 57, 990–997. [Google Scholar] [CrossRef]
- Gillich, C.; Akhoundova, D.; Hayoz, M.; Aebi, Y.; Largiadèr, C.R.; Seipel, K.; Daskalakis, M.; Bacher, U.; Pabst, T. Efficacy and Safety of High-Dose Chemotherapy with Treosulfan and Melphalan in Multiple Myeloma. Cancers 2023, 15, 2699. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Weinhold, N.; Diamond, B.; Kazandjian, D.; Rasche, L.; Morgan, G.; Landgren, O. The Mutagenic Impact of Melphalan in Multiple Myeloma. Leukemia 2021, 35, 2145–2150. [Google Scholar] [CrossRef]
- Pich, O.; Reyes-Salazar, I.; Gonzalez-Perez, A.; Lopez-Bigas, N. Discovering the Drivers of Clonal Hematopoiesis. Nat. Commun. 2022, 13, 4267. [Google Scholar] [CrossRef]
- Bernstein, N.; Spencer Chapman, M.; Nyamondo, K.; Chen, Z.; Williams, N.; Mitchell, E.; Campbell, P.J.; Cohen, R.L.; Nangalia, J. Analysis of Somatic Mutations in Whole Blood from 200,618 Individuals Identifies Pervasive Positive Selection and Novel Drivers of Clonal Hematopoiesis. Nat. Genet. 2024, 56, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Husby, S.; Hjermind Justesen, E.; Grønbæk, K. Protein Phosphatase, Mg2+/Mn2+-Dependent 1D (PPM1D) Mutations in Haematological Cancer. Br. J. Haematol. 2021, 192, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D-Truncating Mutations Confer Resistance to Chemotherapy and Sensitivity to PPM1D Inhibition in Hematopoietic Cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Kleiblova, P.; Shaltiel, I.A.; Benada, J.; Ševčík, J.; Pecháčková, S.; Pohlreich, P.; Voest, E.E.; Dundr, P.; Bartek, J.; Kleibl, Z.; et al. Gain-of-Function Mutations of PPM1D/Wip1 Impair the P53-Dependent G1 Checkpoint. J. Cell Biol. 2013, 201, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.J.; Lindsley, R.C.; Tchekmedyian, V.; Mar, B.G.; Shi, J.; Jaiswal, S.; Bosworth, A.; Francisco, L.; He, J.; Bansal, A.; et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J. Clin. Oncol. 2017, 35, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Lackraj, T.; Ben Barouch, S.; Medeiros, J.J.F.; Pedersen, S.; Danesh, A.; Bakhtiari, M.; Hong, M.; Tong, K.; Joynt, J.; Arruda, A.; et al. Clinical Significance of Clonal Hematopoiesis in the Setting of Autologous Stem Cell Transplantation for Lymphoma. Am. J. Hematol. 2022, 97, 1538–1547. [Google Scholar] [CrossRef] [PubMed]
- Mouhieddine, T.H.; Sperling, A.S.; Redd, R.; Park, J.; Leventhal, M.; Gibson, C.J.; Manier, S.; Nassar, A.H.; Capelletti, M.; Huynh, D.; et al. Clonal Hematopoiesis Is Associated with Adverse Outcomes in Multiple Myeloma Patients Undergoing Transplant. Nat. Commun. 2020, 11, 2996. [Google Scholar] [CrossRef] [PubMed]
- Stelmach, P.; Richter, S.; Sauer, S.; Fabre, M.A.; Gu, M.; Rohde, C.; Janssen, M.; Liebers, N.; Proynova, R.; Weinhold, N.; et al. Clonal Hematopoiesis with DNMT3A and PPM1D Mutations Impairs Regeneration in Autologous Stem Cell Transplant Recipients. Haematologica 2023, 108, 3308–3320. [Google Scholar] [CrossRef]
- Maechler, M.; Bacher, U.; Daskalakis, M.; Nilius, H.; Nagler, M.; Taleghani, B.M.; Jeker, B.; Pabst, T. Long-Term Safety of the Stem Cell Releasing Compound Plerixafor for Peripheral Stem Cell Collection in Myeloma Patients. Hematol. Oncol. 2023, 41, 583–586. [Google Scholar] [CrossRef]
- Mehl, J.; Akhoundova, D.; Bacher, U.; Jeker, B.; Rhyner Agocs, G.; Ruefer, A.; Soltermann, S.; Soekler, M.; Winkler, A.; Daskalakis, M.; et al. Daratumumab during Myeloma Induction Therapy Is Associated with Impaired Stem Cell Mobilization and Prolonged Post-Transplant Hematologic Recovery. Cancers 2024, 16, 1854. [Google Scholar] [CrossRef]
- Duggan, P.; Reece, D.E.; Song, K.; Jimenez-Zepeda, V.; McCurdy, A.; Louzada, M.L.; Mian, H.S.; Sebag, M.; White, D.J.; Stakiw, J.; et al. Is Tandem ASCT Needed in MM Patients with High Risk Cytogenetics in the Era of Maintenance Therapy? Results from the Canadian Myeloma Research Group (CMRG) Database. Blood 2020, 136, 18–19. [Google Scholar] [CrossRef]
- Talamo, G.; Dimaio, C.; Abbi, K.K.S.; Pandey, M.K.; Malysz, J.; Creer, M.H.; Zhu, J.; Mir, M.A.; Varlotto, J.M. Current Role of Radiation Therapy for Multiple Myeloma. Front. Oncol. 2015, 5, 40. [Google Scholar] [CrossRef]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal Hematopoiesis, with and without Candidate Driver Mutations, Is Common in the Elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef]
- Seipel, K.; Frey, M.; Nilius, H.; Akhoundova, D.; Banz, Y.; Bacher, U.; Pabst, T. Low-Frequency PPM1D Gene Mutations Affect Treatment Response to CD19-Targeted CAR T-Cell Therapy in Large B-Cell Lymphoma. Curr. Oncol. 2023, 30, 10463–10476. [Google Scholar] [CrossRef] [PubMed]
- Poczta, A.; Rogalska, A.; Marczak, A. Treatment of Multiple Myeloma and the Role of Melphalan in the Era of Modern Therapies—Current Research and Clinical Approaches. J. Clin. Med. 2021, 10, 1841. [Google Scholar] [CrossRef] [PubMed]
- Samur, M.K.; Roncador, M.; Aktas Samur, A.; Fulciniti, M.; Bazarbachi, A.H.; Szalat, R.; Shammas, M.A.; Sperling, A.S.; Richardson, P.G.; Magrangeas, F.; et al. High-Dose Melphalan Treatment Significantly Increases Mutational Burden at Relapse in Multiple Myeloma. Blood 2023, 141, 1724–1736. [Google Scholar] [CrossRef]
- Lim, H.; Im, M.; Seo, E.S.; Cho, H.W.; Ju, H.Y.; Yoo, K.H.; Cho, S.Y.; Kim, J.-W.; Lim, D.H.; Sung, K.W.; et al. Tandem High-Dose Chemotherapy Increases the Risk of Secondary Malignant Neoplasm in Pediatric Solid Tumors. Cancer Res. Treat. 2024, 56, 642–651. [Google Scholar] [CrossRef]
- Neupane, K.; Fortuna, G.G.; Dahal, R.; Schmidt, T.; Fonseca, R.; Chakraborty, R.; Koehn, K.A.; Mohan, M.; Mian, H.; Costa, L.J.; et al. Alterations in Chromosome 1q in Multiple Myeloma Randomized Clinical Trials: A Systematic Review. Blood Cancer J. 2024, 14, 20. [Google Scholar] [CrossRef]
- Abdallah, N.; Greipp, P.; Kapoor, P.; Gertz, M.A.; Dispenzieri, A.; Baughn, L.B.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; et al. Clinical Characteristics and Treatment Outcomes of Newly Diagnosed Multiple Myeloma with Chromosome 1q Abnormalities. Blood Adv. 2020, 4, 3509–3519. [Google Scholar] [CrossRef]
- Tirier, S.M.; Mallm, J.-P.; Steiger, S.; Poos, A.M.; Awwad, M.H.S.; Giesen, N.; Casiraghi, N.; Susak, H.; Bauer, K.; Baumann, A.; et al. Subclone-Specific Microenvironmental Impact and Drug Response in Refractory Multiple Myeloma Revealed by Single-Cell Transcriptomics. Nat. Commun. 2021, 12, 6960. [Google Scholar] [CrossRef]
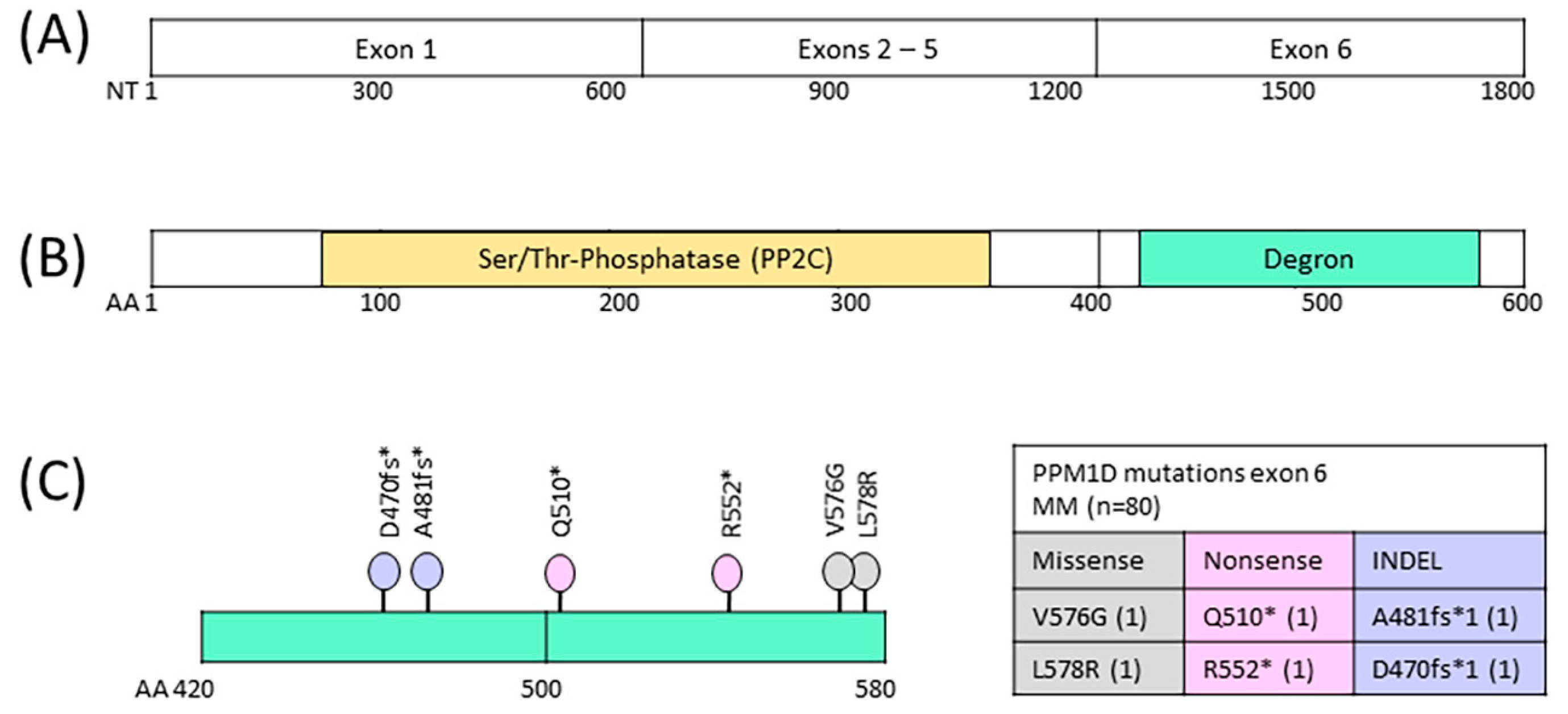

{kind=link}
{kind=link}
{kind=link}
| Classification | Locus Chr7 | VAF | NT Change | AA Change |
|---|---|---|---|---|
| indel | 60,663,137 | 0.014 | CA/C | D470fs * |
| indel | 60,663,171 | 0.011 | TA/T | A481fs * |
| nonsense | 60,663,262 | 0.011 | C/T | Q510 * |
| nonsense | 60,663,388 | 0.016 | C/T | R552 * |
| missense | 60,663,461 | 0.051 | T/G | V576G |
| missense | 60,663,467 | 0.025 | T/G | L578R |
| All Patients (n = 75) | PPM1Dwt (n = 71) | PPM1Dmut (n = 4) | p-Value | |
|---|---|---|---|---|
| Sex | 0.58 | |||
| Female, n (%) | 23 (31%) | 21 (30%) | 2 (50%) | |
| Male, n (%) | 52 (69%) | 50 (70%) | 2 (50%) | |
| Age at ID, median (range) | 59 (39–74) | 60 (39–74) | 55 (46–65) | 0.40 |
| Initial Disease Stage (ISS) | 0.67 | |||
| I | 25 (33%) | 24 (34%) | 1 (25%) | |
| II | 29 (39%) | 28 (39%) | 1 (25%) | |
| III | 21 (28%) | 19 (27%) | 2 (50%) | |
| Cytogenetic risk category | n = 66 | n = 62 | n = 4 | 0.99 |
| high risk * | 18 (27%) | 17 (27%) | 1 (25%) | |
| standard risk | 48 (73%) | 45 (73%) | 3 (75%) | |
| Cytogenetic aberrations | n = 66 | n = 62 | n = 4 | |
| −13 | 12 (21%) | 10 (19%) | 2 (50%) | 0.19 |
| −14 | 5 (9%) | 4 (8%) | 1 (25%) | 0.31 |
| −16 | 4 (7%) | 3 (6%) | 1 (25%) | 0.26 |
| +1q | 18 (32%) | 15 (29%) | 3 (75%) | 0.09 |
| del17p * | 6 (11%) | 6 (12%) | 0 | 0.99 |
| t(4;14) * | 9 (16%) | 8 (15%) | 1 (25%) | 0.51 |
| t(14;16) * | 3 (5%) | 3 (6%) | 0 | 0.99 |
| t(11;14) | 9 (16%) | 8 (15%) | 1 (25%) | 0.51 |
| Paraprotein-type | ||||
| Heavy chain IgG | 41 (55%) | 40 (57%) | 1 (25%) | 0.32 |
| Heavy chain IgA | 16 (22%) | 13 (19%) | 3 (75%) | 0.03 |
| Light chain only | 17 (23%) | 17 (24%) | 0 | 0.57 |
| Light chain kappa | 54 (73%) | 52 (74%) | 2 (50%) | 0.29 |
| Light chain lambda | 20 (27%) | 18 (26%) | 2 (50%) | 0.29 |
| Anemia | ||||
| Hb (g/L), average (range) | 109 (46–167) | 109 (46–167) | 113 (92–128) | 0.76 |
| Hb < 110 g/L | 34 (49%) | 32 (48%) | 2 (50%) | |
| Hb > 110 g/L | 36 (51%) | 34 (51%) | 2 (50%) | |
| Hypercalcemia | ||||
| Ca (mmol/L), median (range) | 2.4 (2–4.2) | 2.4 (2–4.2) | 2.5 (2.3–4.1) | 0.55 |
| >2.6 mmol/L | 16 (23%) | 16 (24%) | 0 | |
| <2.6 mmol/L | 55 (77%) | 51 (76%) | 4 (100%) | |
| Beta-2-microglobulin | ||||
| B2M (mg/L), median (range) | 3.4 (1–38) | 3.4 (1–38) | 3.9 (2–15) | 0.58 |
| >3.5 mg/L | 35 (47%) | 33 (46%) | 2 (50%) | |
| <3.5 mg/L | 40 (53%) | 38 (54%) | 2 (50%) | |
| Lactate-dehydrogenase | ||||
| LDH (U/L), median (range) | 336 (110–3277) | 342 (110–3277) | 251 (154–428) | 0.41 |
| >480 | 9 (13%) | 9 (14%) | 0 | |
| <480 | 59 (87%) | 55 (86%) | 4 (100%) | |
| Serum albumin | ||||
| g/dL, median (range) | 3.5 (1.8–5.1) | 3.5 (1.8–5.1) | 3.6 (3.3–4.0) | 0.72 |
| >3.5 g/dL | 32 (47%) | 30 (47%) | 2 (50%) | |
| <3.5 g/dL | 36 (53%) | 34 (53%) | 2 (50%) | |
| Bone marrow infiltration | ||||
| Percent, median (range) | 70 (3–100) | 70 (3–100) | 55 (40–100) | 0.94 |
| 3–39% | 16 (23%) | 16 (24%) | 0 | |
| 40–100% | 55 (77%) | 51 (76%) | 4 (100%) | |
| Osteolytic lesions | 57 (76%) | 53 (75%) | 4 (100%) | 0.57 |
| Renal dysfunction | 15 (21%) | 14 (21%) | 1 (25%) | 0.99 |
| All Patients (n = 75) | PPM1Dwt (n = 71) | PPM1Dmut (n = 4) | p-Value | |
|---|---|---|---|---|
| 1st line therapy (ICT) | 0.99 | |||
| VD | 13 (16%) | 13 (17%) | 0 | |
| VRD | 35 (47%) | 33 (47%) | 2 (50%) | |
| VCD | 28 (37%) | 26 (36%) | 2 (50%) | |
| Response to 1st line ICT | 0.99 | |||
| CR | 12 (16%) | 11 (16%) | 1 (25%) | |
| VGPR/PR | 56 (75%) | 53 (75%) | 3 (75%) | |
| SD/PD | 5 (7%) | 5 (7%) | 0 | |
| not reported | 2 (3%) | 2 (3%) | 0 | |
| Response to 2nd line ICT | n = 52 | n = 48 | n = 4 | 0.99 |
| CR | 8 (15%) | 8 (17%) | 0 | |
| VGPR/PR | 27 (52%) | 24 (50%) | 3 (75%) | |
| SD/PD | 9 (17%) | 8 (17%) | 1 (25%) | |
| not reported | 8 (15%) | 8 (17%) | 0 | |
| ICT lines | ||||
| n, median (range) | 2 (1–7) | 2 (1–7) | 3 (2–6) | 0.31 |
| 1–2 | 47 (63%) | 46 (65%) | 2 (50%) | |
| 3–7 | 28 (37%) | 26 (35%) | 2 (50%) | |
| Relapse/Progression | ||||
| n, median (range) | 1 (0–7) | 1 (0–7) | 2 (1–6) | 0.26 |
| 0 | 21 (28%) | 21 (30%) | 0 | |
| 1 | 26 (35%) | 25 (35%) | 1 (25%) | |
| 2–7 | 28 (37%) | 26 (35%) | 3 (75%) | |
| 1st PBSC mobilization | ||||
| CD34+ cells (×106/kg) (range) | 10.7 (1–28) | 10.7 (1–28) | 10.6 (10–11) | 0.96 |
| <4 × 106/kg | 3 (4%) | 3 (4%) | 0 | |
| 1st HDCT | 0.08 | |||
| M | 51 (68%) | 49 (68%) | 2 (50%) | |
| MB | 8 (11%) | 6 (9%) | 2 (50%) | |
| MT | 16 (21%) | 16 (23%) | 0 | |
| 1st ASCT | ||||
| CD34+ cells (×106/kg) (range) | 3.8 (1.3–9.9) | 3.8 (1.3–9.9) | 3.4 (2.9–3.9) | 0.53 |
| <2.5 × 106/kg | 4 (5%) | 4 (5%) | 0 | |
| Platelet transfusions n, median (range) | 3 (0–45) | 3 (0–45) | 9 (2–27) | 0.15 |
| Response to 1st ASCT | 0.99 | |||
| CR/sCR | 48 (64%) | 45 (63%) | 3 (75%) | |
| PR/VGPR | 22 (29%) | 21 (29%) | 1 (25%) | |
| SD/PD | 1 (1%) | 1 (1%) | 0 | |
| not reported | 4 (5%) | 4 (6%) | 0 | |
| 2nd ASCT | n = 41 | n = 37 | n = 4 | 0.93 |
| CD34+ cells (×106/kg) (range) | 3.8 (0.1–10) | 3.8 (0.1–10) | 3.7 (3.4–3.9) | |
| <2.5 × 106/kg | 3 (7%) | 3 (8%) | 0 | |
| Response to 2nd ASCT | n = 41 | n = 37 | n = 4 | 0.17 |
| CR/sCR | 33 (75%) | 31 (84%) | 2 (50%) | |
| VGPR/PR | 7 (17%) | 5 (14%) | 2 (50%) | |
| SD/PD | 1 (3%) | 1 (2%) | 0 | |
| HDCT/ASCT lines | ||||
| n, median (range) | 2 (1–4) | 2 (1–4) | 2.5 (2–3) | 0.01 |
| 1 | 34 (45%) | 34 (47%) | 0 | |
| 2 | 34 (45%) | 32 (46%) | 2 (50%) | |
| 3 | 6 (8%) | 4 (6%) | 2 (50%) | |
| 4 | 1 (1%) | 1 (1%) | 0 | |
| Radiotherapy | 28 (37%) | 26 (37%) | 2 (50%) | 0.63 |
| PFS | OS | |||
|---|---|---|---|---|
| Predictors | HR (95% CI) | p-Value | HR (95% CI) | p-Value |
| PPM1Dmut | 6.42 (1.0, 20.7) | 0.002 | 8.05 (2.0, 33.3) | 0.004 |
| Age > 58 | 1.30 (0.7, 2.4) | 0.2 | 1.79 (0.7, 4.5) | 0.2 |
| ISS2/3 vs. ISS1 | 1.72 (0.9, 3.4) | 0.12 | 1.32 (0.5, 3.5) | 0.6 |
| Cytogenetic high risk | 0.98 (0.5, 2.1) | >0.9 | 0.93 (0.3, 3.2) | >0.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seipel, K.; Veglio, N.Z.; Nilius, H.; Jeker, B.; Bacher, U.; Pabst, T. Rising Prevalence of Low-Frequency PPM1D Gene Mutations after Second HDCT in Multiple Myeloma. Curr. Issues Mol. Biol. 2024, 46, 8197-8208. https://doi.org/10.3390/cimb46080484
Seipel K, Veglio NZ, Nilius H, Jeker B, Bacher U, Pabst T. Rising Prevalence of Low-Frequency PPM1D Gene Mutations after Second HDCT in Multiple Myeloma. Current Issues in Molecular Biology. 2024; 46(8):8197-8208. https://doi.org/10.3390/cimb46080484
Chicago/Turabian StyleSeipel, Katja, Nuria Z. Veglio, Henning Nilius, Barbara Jeker, Ulrike Bacher, and Thomas Pabst. 2024. "Rising Prevalence of Low-Frequency PPM1D Gene Mutations after Second HDCT in Multiple Myeloma" Current Issues in Molecular Biology 46, no. 8: 8197-8208. https://doi.org/10.3390/cimb46080484