
Dexamethasone Impairs ATP Production and Mitochondrial Performance in Human Trabecular Meshwork Cells


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Isolation and Culture
2.2. Quantitative Polymerase Chain Reaction (qPCR)
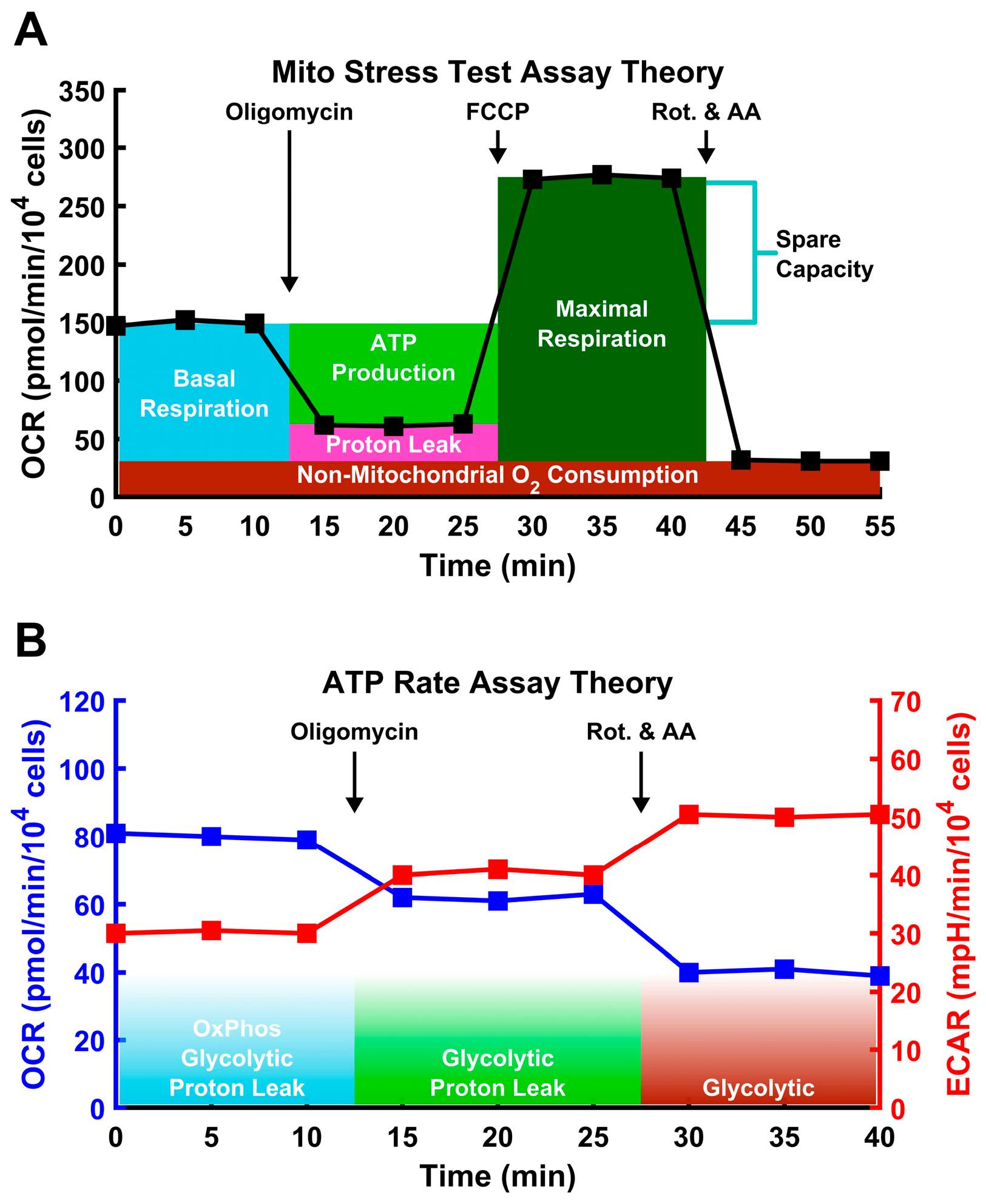
2.3. Metabolic Assays
2.4. Statistical Analysis
3. Results
3.1. Dexamethasone Treatment Decreases Relative Mitochondrial ATP Generation
3.2. TGF-β2 Does Not Significantly Alter ATP Production
3.3. Dexamethasone Impairs Mitochondrial Function
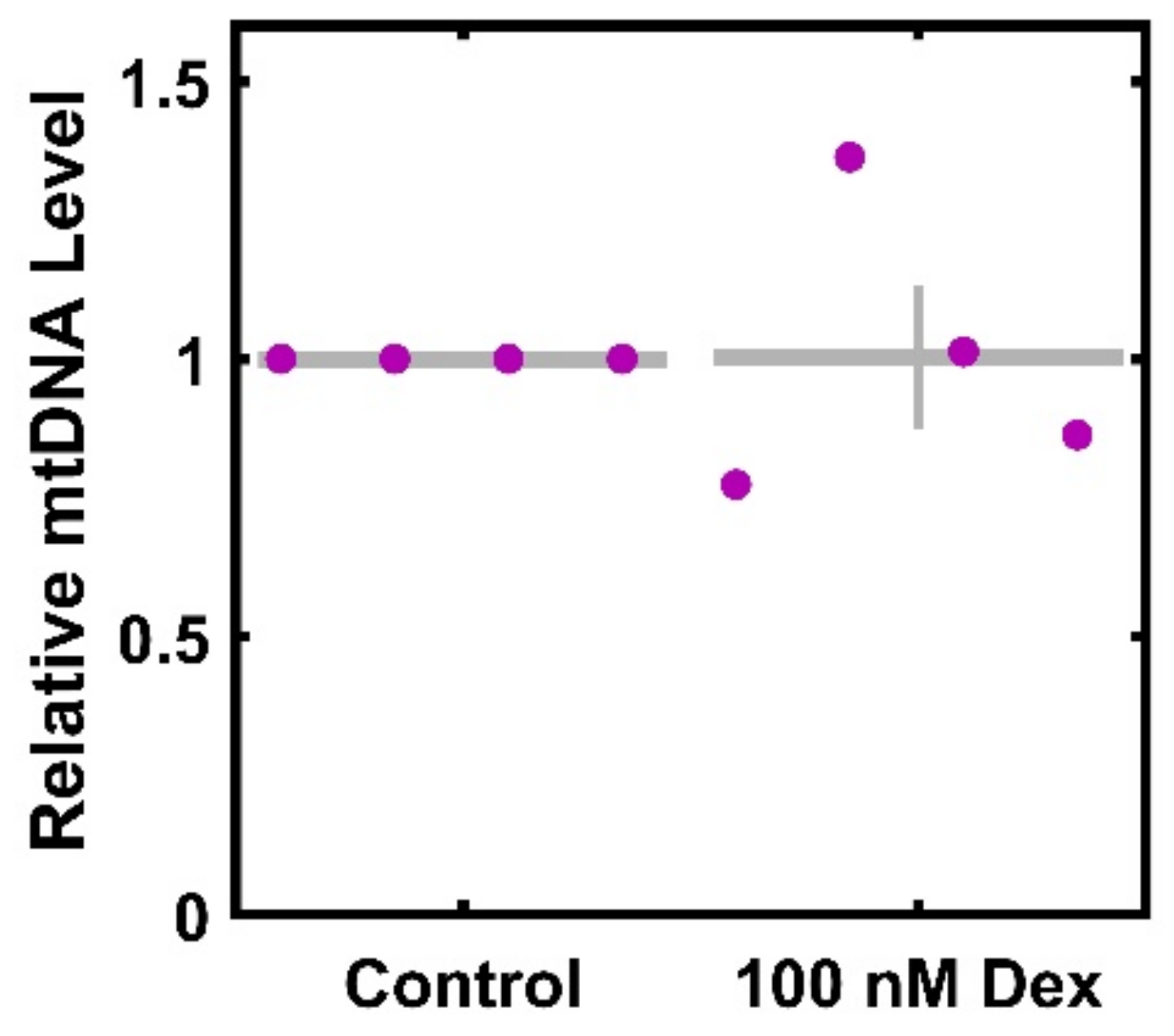
3.4. Dexamethasone Does Not Result in a Significant Decrease in mtDNA per Cell
3.5. Dexamethasone Does Not Result in a Significant Change in ROS-Related mRNA Transcripts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tham, Y.-C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.-Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.V.; Quigley, H.A. Risk factors and open-angle glaucoma: Classification and application. J. Glaucoma 2007, 16, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A. Glaucoma. Lancet 2011, 377, 1367–1377. [Google Scholar] [CrossRef]
- Stamer, W.D.; Acott, T.S. Current understanding of conventional outflow dysfunction in glaucoma. Curr. Opin. Ophthalmol. 2012, 23, 135–143. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Izumchenko, E.; Kanherkar, R.R.; Teka, M.; Cantor, C.; Manaye, K.; Sidransky, D.; West, M.D.; Makarev, E.; Csoka, A.B. Pro-fibrotic pathway activation in trabecular meshwork and lamina cribrosa is the main driving force of glaucoma. Cell Cycle 2016, 15, 1643–1652. [Google Scholar] [CrossRef]
- Liton, P.B.; Challa, P.; Stinnett, S.; Luna, C.; Epstein, D.L.; Gonzalez, P. Cellular senescence in the glaucomatous outflow pathway. Exp. Gerontol. 2005, 40, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Saccà, S.C.; Pulliero, A.; Izzotti, A. The dysfunction of the trabecular meshwork during glaucoma course. J. Cell. Physiol. 2015, 230, 510–525. [Google Scholar] [CrossRef]
- Aktas, Z.; Karaca, E.E.; Gonul, I.I.; Hasanreisoglu, M.; Onol, M. Apoptosis in the iris and trabecular meshwork of medically treated and untreated primary open angle glaucoma patients. Int. J. Ophthalmol. 2013, 6, 827–830. [Google Scholar] [CrossRef]
- Caballero, M.; Liton, P.B.; Challa, P.; Epstein, D.L.; Gonzalez, P. Effects of donor age on proteasome activity and senescence in trabecular meshwork cells. Biochem. Biophys. Res. Commun. 2004, 323, 1048–1054. [Google Scholar] [CrossRef]
- Schmidl, D.; Schmetterer, L.; Garhöfer, G.; Popa-Cherecheanu, A. Pharmacotherapy of glaucoma. J. Ocul. Pharmacol. Ther. Off. J. Assoc. Ocul. Pharmacol. Ther. 2015, 31, 63–77. [Google Scholar] [CrossRef]
- Mehran, N.A.; Sinha, S.; Razeghinejad, R. New glaucoma medications: Latanoprostene bunod, netarsudil, and fixed combination netarsudil-latanoprost. Eye 2020, 34, 72–88. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, S.; Zhong, W.; Yang, B.; Sun, L.; Zheng, Y. Oxidative stress in the trabecular meshwork (Review). Int. J. Mol. Med. 2016, 38, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Leung, K.W.; Zhang, Y.-H.; Duan, S.; Zhong, X.-F.; Jiang, R.-Z.; Peng, Z.; Tombran-Tink, J.; Ge, J. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: Protection by antioxidants. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1447–1458. [Google Scholar] [CrossRef]
- Izzotti, A.; Longobardi, M.; Cartiglia, C.; Saccà, S.C. Mitochondrial damage in the trabecular meshwork occurs only in primary open-angle glaucoma and in pseudoexfoliative glaucoma. PLoS ONE 2011, 6, e14567. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Saccà, S.C.; Longobardi, M.; Cartiglia, C. Mitochondrial damage in the trabecular meshwork of patients with glaucoma. Arch. Ophthalmol. 2010, 128, 724–730. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Breitenbach, M.; Rinnerthaler, M.; Hartl, J.; Stincone, A.; Vowinckel, J.; Breitenbach-Koller, H.; Ralser, M. Mitochondria in ageing: There is metabolism beyond the ROS. FEMS Yeast Res. 2014, 14, 198–212. [Google Scholar] [CrossRef]
- Epstein, D.L.; Anderson, P.J. In vitro biochemistry of trabecular meshwork. Vis. Res. 1981, 21, 161. [Google Scholar] [CrossRef]
- Anderson, P.J.; Wang, J.; Epstein, D.L. Metabolism of calf trabecular (reticular) meshwork. Investig. Ophthalmol. Vis. Sci. 1980, 19, 13–20. [Google Scholar]
- Graybeal, K.; Sanchez, L.; Zhang, C.; Stiles, L.; Zheng, J.J. Characterizing the metabolic profile of dexamethasone treated human trabecular meshwork cells. Exp. Eye Res. 2022, 214, 108888. [Google Scholar] [CrossRef]
- Marcoccia, R.; Nesci, S.; Merlo, B.; Ballotta, G.; Algieri, C.; Pagliarani, A.; Iacono, E. Biological characteristics and metabolic profile of canine mesenchymal stem cells isolated from adipose tissue and umbilical cord matrix. PLoS ONE 2021, 16, e0247567. [Google Scholar] [CrossRef] [PubMed]
- Duraj, T.; Carrión-Navarro, J.; Seyfried, T.N.; García-Romero, N.; Ayuso-Sacido, A. Metabolic therapy and bioenergetic analysis: The missing piece of the puzzle. Mol. Metab. 2021, 54, 101389. [Google Scholar] [CrossRef]
- Gropman, A.; Uittenbogaard, M.; Brantner, C.A.; Wang, Y.; Wong, L.-J.; Chiaramello, A. Molecular genetic and mitochondrial metabolic analyses confirm the suspected mitochondrial etiology in a pediatric patient with an atypical form of alternating hemiplegia of childhood. Mol. Genet. Metab. Rep. 2020, 24, 100609. [Google Scholar] [CrossRef]
- Chaphalkar, R.M.; Stankowska, D.L.; He, S.; Kodati, B.; Phillips, N.; Prah, J.; Yang, S.; Krishnamoorthy, R.R. Endothelin-1 Mediated Decrease in Mitochondrial Gene Expression and Bioenergetics Contribute to Neurodegeneration of Retinal Ganglion Cells. Sci. Rep. 2020, 10, 3571. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, G.F.; Galiano, S.; Canepa, L.; Ferraris, A.M.; Kirkman, H.N. Catalase and glutathione peroxidase are equally active in detoxification of hydrogen peroxide in human erythrocytes. Blood 1989, 73, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Prieto, M.; Biarnés, X.; Vidossich, P.; Rovira, C. The Molecular Mechanism of the Catalase Reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: Function or dysfunction? Oncogene 2024, 43, 295–303. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Hernandez-Saavedra, D.; Swain, K.; Tuder, R.; Petersen, S.V.; Nozik-Grayck, E. Redox Regulation of the Superoxide Dismutases SOD3 and SOD2 in the Pulmonary Circulation. Adv. Exp. Med. Biol. 2017, 967, 57–70. [Google Scholar] [CrossRef]
- Hernansanz-Agustín, P.; Enríquez, J.A. Generation of Reactive Oxygen Species by Mitochondria. Antioxidants 2021, 10, 415. [Google Scholar] [CrossRef]
- Becker, S.; L’Ecuyer, Z.; Jones, B.W.; Zouache, M.A.; McDonnell, F.S.; Vinberg, F. Modeling complex age-related eye disease. Prog. Retin. Eye Res. 2024, 100, 101247. [Google Scholar] [CrossRef]
- Puigserver, P.; Adelmant, G.; Wu, Z.; Fan, M.; Xu, J.; O’Malley, B.; Spiegelman, B.M. Activation of PPARgamma coactivator-1 through transcription factor docking. Science 1999, 286, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Redondo, V.; Pettersson, A.T.; Ruas, J.L. The hitchhiker’s guide to PGC-1α isoform structure and biological functions. Diabetologia 2015, 58, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. TEM 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, J.; Rowe, G.C.; Khadka, A.; Diaz-Aguilar, D.; Spencer, C.; Arany, Z.; Saint-Geniez, M. PGC-1α Induces Human RPE Oxidative Metabolism and Antioxidant Capacity. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1038–1051. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Choi, H.-I.; Kim, H.-J.; Park, J.-S.; Kim, I.-J.; Bae, E.H.; Ma, S.K.; Kim, S.W. PGC-1α attenuates hydrogen peroxide-induced apoptotic cell death by upregulating Nrf-2 via GSK3β inactivation mediated by activated p38 in HK-2 Cells. Sci. Rep. 2017, 7, 4319. [Google Scholar] [CrossRef]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef]
- Hock, M.B.; Kralli, A. Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef]
- Xu, W.; Boyd, R.M.; Tree, M.O.; Samkari, F.; Zhao, L. Mitochondrial transcription factor A promotes DNA strand cleavage at abasic sites. Proc. Natl. Acad. Sci. USA 2019, 116, 17792–17799. [Google Scholar] [CrossRef]
- Fuchshofer, R.; Tamm, E.R. The role of TGF-β in the pathogenesis of primary open-angle glaucoma. Cell Tissue Res. 2012, 347, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Daher, A.M.; Agarwal, R. Aqueous humor TGF-β2 levels in patients with open-angle glaucoma: A meta-analysis. Mol. Vis. 2015, 21, 612–620. [Google Scholar] [PubMed]
- Fleenor, D.L.; Shepard, A.R.; Hellberg, P.E.; Jacobson, N.; Pang, I.-H.; Clark, A.F. TGFbeta2-induced changes in human trabecular meshwork: Implications for intraocular pressure. Investig. Ophthalmol. Vis. Sci. 2006, 47, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Puca, F.; Yu, F.; Bartolacci, C.; Pettazzoni, P.; Carugo, A.; Huang-Hobbs, E.; Liu, J.; Zanca, C.; Carbone, F.; Del Poggetto, E.; et al. Medium-Chain Acyl-CoA Dehydrogenase Protects Mitochondria from Lipid Peroxidation in Glioblastoma. Cancer Discov. 2021, 11, 2904–2923. [Google Scholar] [CrossRef]
- Morgan, J.T.; Wood, J.A.; Walker, N.J.; Raghunathan, V.K.; Borjesson, D.L.; Murphy, C.J.; Russell, P. Human trabecular meshwork cells exhibit several characteristics of, but are distinct from, adipose-derived mesenchymal stem cells. J. Ocul. Pharmacol. Ther. Off. J. Assoc. Ocul. Pharmacol. Ther. 2014, 30, 254–266. [Google Scholar] [CrossRef]
- Keller, K.E.; Bhattacharya, S.K.; Borrás, T.; Brunner, T.M.; Chansangpetch, S.; Clark, A.F.; Dismuke, W.M.; Du, Y.; Elliott, M.H.; Ethier, C.R.; et al. Consensus recommendations for trabecular meshwork cell isolation, characterization and culture. Exp. Eye Res. 2018, 171, 164–173. [Google Scholar] [CrossRef]
- Clemente, M.G.; Patton, J.T.; Anders, R.A.; Yolken, R.H.; Schwarz, K.B. Rotavirus Infects Human Biliary Epithelial Cells and Stimulates Secretion of Cytokines IL-6 and IL-8 via MAPK Pathway. BioMed Res. Int. 2015, 2015, 697238. [Google Scholar] [CrossRef]
- Park, B.-C.; Tibudan, M.; Samaraweera, M.; Shen, X.; Yue, B.Y.J.T. Interaction between two glaucoma genes, optineurin and myocilin. Genes Cells Devoted Mol. Cell. Mech. 2007, 12, 969–979. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Goncalves, R.L.S.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta 2015, 1847, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, S.A.; Nicholls, D.G.; Brand, M.D. Determining Maximum Glycolytic Capacity Using Extracellular Flux Measurements. PLoS ONE 2016, 11, e0152016. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef] [PubMed]
- Preibisch, S.; Saalfeld, S.; Tomancak, P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 2009, 25, 1463–1465. [Google Scholar] [CrossRef]
- Romero, N.; Swain, P.M.; Kam, Y.; Rogers, G. Bioenergetic profiling of cancer cell lines: Quantifying the impact of glycolysis on cell proliferation. Cancer Res. 2018, 78, 3487. [Google Scholar] [CrossRef]
- Giddings, E.L.; Champagne, D.P.; Wu, M.-H.; Laffin, J.M.; Thornton, T.M.; Valenca-Pereira, F.; Culp-Hill, R.; Fortner, K.A.; Romero, N.; East, J.; et al. Mitochondrial ATP fuels ABC transporter-mediated drug efflux in cancer chemoresistance. Nat. Commun. 2021, 12, 2804. [Google Scholar] [CrossRef]
- Fujiwara, M.; Tian, L.; Le, P.T.; DeMambro, V.E.; Becker, K.A.; Rosen, C.J.; Guntur, A.R. The mitophagy receptor Bcl-2-like protein 13 stimulates adipogenesis by regulating mitochondrial oxidative phosphorylation and apoptosis in mice. J. Biol. Chem. 2019, 294, 12683–12694. [Google Scholar] [CrossRef]
- Tsiriyotis, C.; Spandidos, D.A.; Sekeris, C.E. The mitochondrion as a primary site of action of glucocorticoids: Mitochondrial nucleotide sequences, showing similarity to hormone response elements, confer dexamethasone inducibility to chimaeric genes transfected in LATK- cells. Biochem. Biophys. Res. Commun. 1997, 235, 349–354. [Google Scholar] [CrossRef]
- Demonacos, C.V.; Karayanni, N.; Hatzoglou, E.; Tsiriyiotis, C.; Spandidos, D.A.; Sekeris, C.E. Mitochondrial genes as sites of primary action of steroid hormones. Steroids 1996, 61, 226–232. [Google Scholar] [CrossRef]
- Luan, G.; Li, G.; Ma, X.; Jin, Y.; Hu, N.; Li, J.; Wang, Z.; Wang, H. Dexamethasone-Induced Mitochondrial Dysfunction and Insulin Resistance-Study in 3T3-L1 Adipocytes and Mitochondria Isolated from Mouse Liver. Molecules 2019, 24, 1982. [Google Scholar] [CrossRef]
- Liu, J.; Peng, Y.; Wang, X.; Fan, Y.; Qin, C.; Shi, L.; Tang, Y.; Cao, K.; Li, H.; Long, J.; et al. Mitochondrial Dysfunction Launches Dexamethasone-Induced Skeletal Muscle Atrophy via AMPK/FOXO3 Signaling. Mol. Pharm. 2016, 13, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.D.; Agarwal, N.A.; Katyare, S.S. Effect of dexamethasone treatment on oxidative energy metabolism in rat liver mitochondria during postnatal developmental periods. Drug Chem. Toxicol. 2004, 27, 389–403. [Google Scholar] [CrossRef]
- Roussel, D.; Dumas, J.-F.; Augeraud, A.; Douay, O.; Foussard, F.; Malthiéry, Y.; Simard, G.; Ritz, P. Dexamethasone treatment specifically increases the basal proton conductance of rat liver mitochondria. FEBS Lett. 2003, 541, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Patil, A.J.; Mansoor, S.; Estrago-Franco, M.F.; Raymond, V.; Kenney, M.C.; Kuppermann, B.D. Effects of dexamethasone on human trabecular meshwork cells in vitro. Graefes Arch. Clin. Exp. Ophthalmol. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. 2013, 251, 1741–1746. [Google Scholar] [CrossRef] [PubMed]
- Filla, M.S.; Schwinn, M.K.; Nosie, A.K.; Clark, R.W.; Peters, D.M. Dexamethasone-associated cross-linked actin network formation in human trabecular meshwork cells involves β3 integrin signaling. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2952–2959. [Google Scholar] [CrossRef]
- Faralli, J.A.; Gagen, D.; Filla, M.S.; Crotti, T.N.; Peters, D.M. Dexamethasone increases αvβ3 integrin expression and affinity through a calcineurin/NFAT pathway. Biochim. Biophys. Acta 2013, 1833, 3306–3313. [Google Scholar] [CrossRef]
- Psarra, A.-M.G.; Sekeris, C.E. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: Role of the mitochondrial glucocorticoid receptor. Biochim. Biophys. Acta 2011, 1813, 1814–1821. [Google Scholar] [CrossRef]
- Hunter, R.G.; Seligsohn, M.; Rubin, T.G.; Griffiths, B.B.; Ozdemir, Y.; Pfaff, D.W.; Datson, N.A.; McEwen, B.S. Stress and corticosteroids regulate rat hippocampal mitochondrial DNA gene expression via the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2016, 113, 9099–9104. [Google Scholar] [CrossRef]
- Weber, K.; Brück, P.; Mikes, Z.; Küpper, J.-H.; Klingenspor, M.; Wiesner, R.J. Glucocorticoid hormone stimulates mitochondrial biogenesis specifically in skeletal muscle. Endocrinology 2002, 143, 177–184. [Google Scholar] [CrossRef]
- Sibayan, S.A.; Latina, M.A.; Sherwood, M.E.; Flotte, T.J.; White, K. Apoptosis and morphologic changes in drug-treated trabecular meshwork cells in vitro. Exp. Eye Res. 1998, 66, 521–529. [Google Scholar] [CrossRef]
- Watanabe, M.; Sato, T.; Tsugeno, Y.; Umetsu, A.; Suzuki, S.; Furuhashi, M.; Ida, Y.; Hikage, F.; Ohguro, H. Human Trabecular Meshwork (HTM) Cells Treated with TGF-β2 or Dexamethasone Respond to Compression Stress in Different Manners. Biomedicines 2022, 10, 1338. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Feng, H.; Li, S.; Meng, G.; Liu, S.; Tang, X.; Ma, Y.; Han, Y.; Xiao, Y.; Gu, Y.; et al. SIRT3 Mediates the Antioxidant Effect of Hydrogen Sulfide in Endothelial Cells. Antioxid. Redox Signal. 2016, 24, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, L.M.; Werthenbach, J.P.; Heintges-Kleinhofer, F.; Seeger, J.M.; Fritsch, M.; Günther, S.D.; Willenborg, S.; Brodesser, S.; Lucas, C.; Jüngst, C.; et al. Mitochondrial respiration controls neoangiogenesis during wound healing and tumour growth. Nat. Commun. 2020, 11, 3653. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 13106–13124. [Google Scholar] [CrossRef]
- Willems, P.H.G.M.; Rossignol, R.; Dieteren, C.E.J.; Murphy, M.P.; Koopman, W.J.H. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence | Reverse Sequence | Reference |
|---|---|---|---|
| GAPDH | ACAGTCAGCCGCATCTTCTT | GCAGGAGGCGTTGTCATT | [47] |
| MYOC | GGAAGAGAAGAAGCGACT | ATAAACTGGCTGATGAGGTC | [48] |
| PGC-1α | AAACAGCAGCAGAGACAAATGC | TTGGTTTGGCTTGTAAGTGTTGTG | [49] |
| TFAM | TGTTCACAATGGATAGGCAC | TCTGGGTTTTCCAAAGCAAG | [49] |
| SOD2 | CTGGACAAACCTCAGCCCT | CTGATTTGGACAAGCAGCAA | [49] |
| CATA | TGGAAAGAAGACTCCCATCG | CCAGAAGTCCCAGACCATGT | [49] |
| BGLOB | GGTGAGTCTATGGGACGCTT | GATCCTGAGACTTCCACACTGA | a |
| TMDQ | CCATCTTTGCAGGCACACTCATC | ATCCACCTCAACTGCCTGCTATG | [14] |
| Parameter Name | Control | 100 nM Dex | Units | p-Value |
|---|---|---|---|---|
| Mitochondrial O2 Basal Consumption | 21.9 ± 3.7 | 16.7 ± 4.1 | 0.0077 * | |
| Relative | 100 | 74.2 ± 10.6 | % | 0.052 |
| Mitochondrial O2 Maximal Consumption | 120 ± 26.8 | 74.9 ± 17.2 | 0.048 * | |
| Relative | 100 | 62.1 ± 5.6 | % | 0.007 * |
| O2 Consumption for ATP | 19.0 ± 3.8 | 12.8 ± 3.7 | 0.006 * | |
| Relative | 100 | 62 ± 4.8 | % | 0.032 * |
| Spare Respiratory Capacity | 98.39 ± 23.1 | 58.3 ± 13.2 | 0.065 | |
| Relative (to Basal) | 541.4 ± 30.0 | 459.8 ± 13.8 | % | 0.130 |
| Relative (to Control) | 100 | 85.4 ± 5 | % | 0.102 |
| Coupling Efficiency | 86.1 ± 3.0 | 75.7 ± 5.0 | % | 0.088 |
| Relative | 100 | 87.8 ± 7.1 | % | 0.097 |
| Proton Leak | 2.8 ± 0.2 | 3.8 ± 0.6 | 0.219 | |
| Relative | 100 | 138.0 ± 31 | % | 0.350 |
| Non-Mitochondrial O2 Consumption | 22.8 ± 4.0 | 17.0 ± 3.0 | 0.04 * | |
| Relative | 100 | 74.2 ± 5.0 | % | 0.012 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kennedy, S.; Williams, C.; Tsaturian, E.; Morgan, J.T. Dexamethasone Impairs ATP Production and Mitochondrial Performance in Human Trabecular Meshwork Cells. Curr. Issues Mol. Biol. 2024, 46, 9867-9880. https://doi.org/10.3390/cimb46090587
Kennedy S, Williams C, Tsaturian E, Morgan JT. Dexamethasone Impairs ATP Production and Mitochondrial Performance in Human Trabecular Meshwork Cells. Current Issues in Molecular Biology. 2024; 46(9):9867-9880. https://doi.org/10.3390/cimb46090587
Chicago/Turabian StyleKennedy, Shane, Clayton Williams, Emily Tsaturian, and Joshua T. Morgan. 2024. "Dexamethasone Impairs ATP Production and Mitochondrial Performance in Human Trabecular Meshwork Cells" Current Issues in Molecular Biology 46, no. 9: 9867-9880. https://doi.org/10.3390/cimb46090587