

Glioblastoma Tumor Microenvironment: An Important Modulator for Tumoral Progression and Therapy Resistance

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
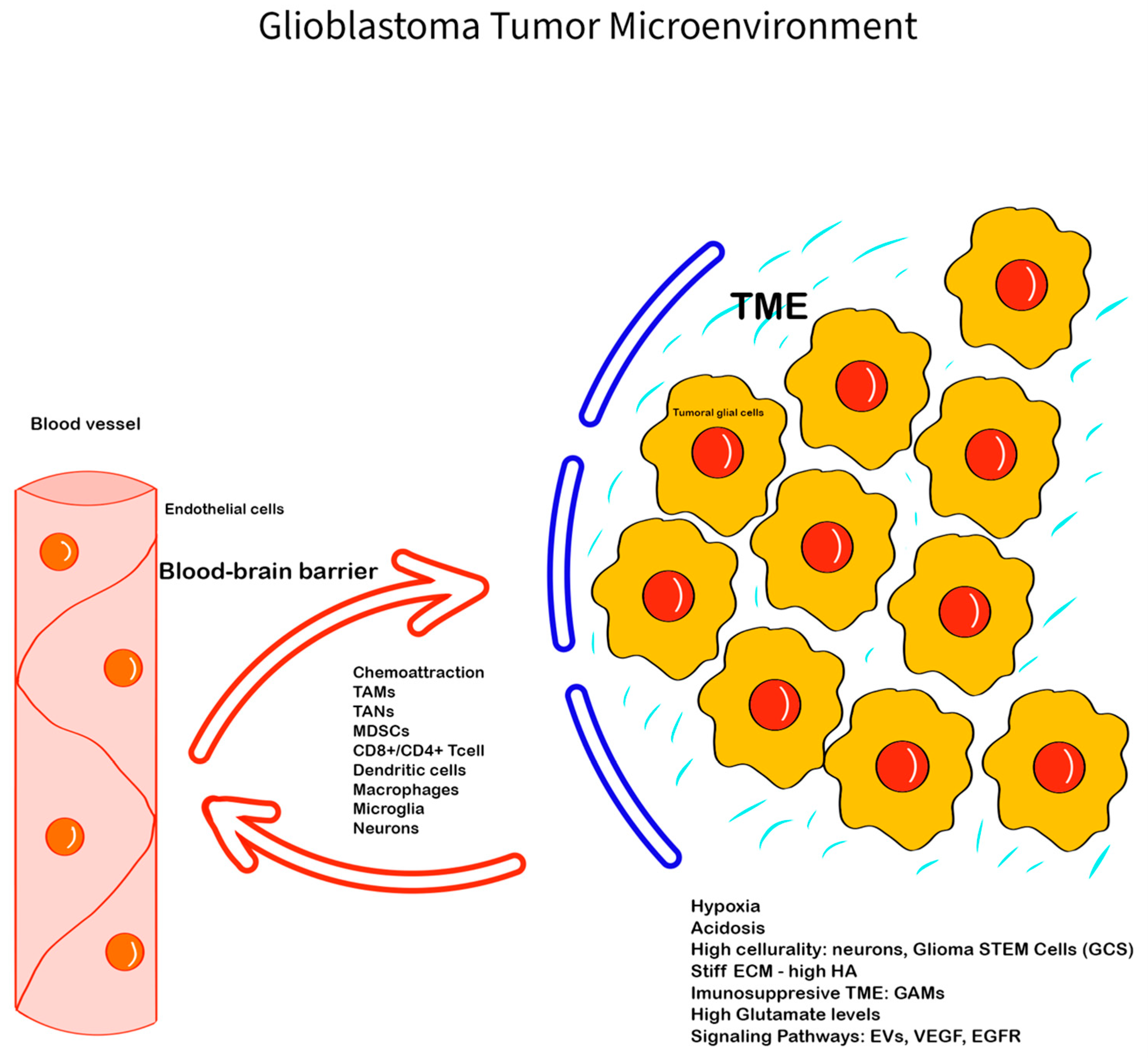
2. Glioblastoma Microenvironment—Main Features
2.1. The Extracellular Matrix of GBM TME
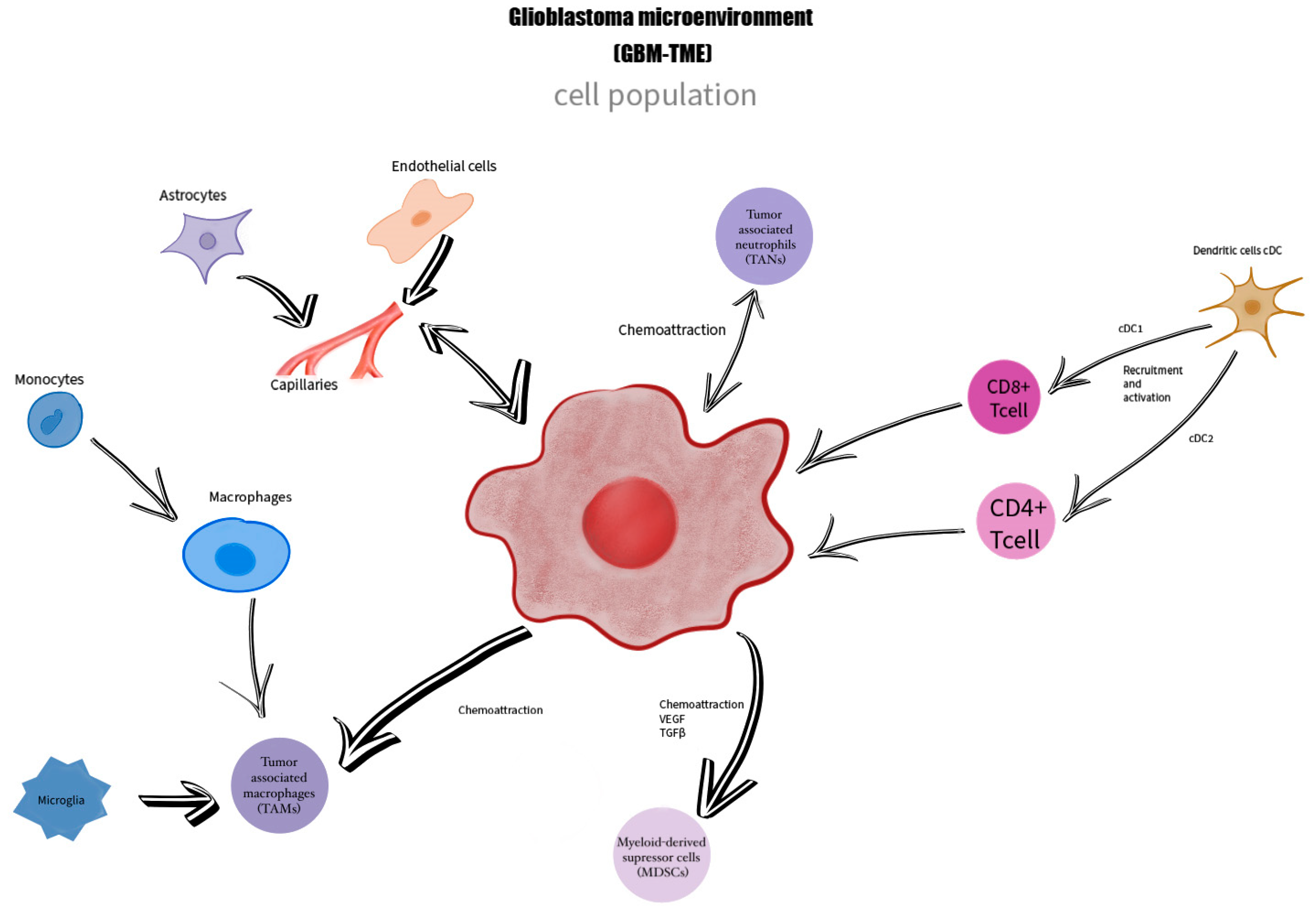
2.2. The Immune Component of GBM TME
2.3. Neuronal and Glial Component of GBM-TME
2.4. Communication Factors
2.5. The Perivascular Niche (PVN)
2.6. Hypoxia and Hypoxia-Inducible Factors (HIFs)
3. Targeted Therapies in GBM TME
Active Clinical Trials
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.W.; Vollmuth, P.; Foltyn-Dumitru, M.; Sahm, F.; Ahn, S.S.; Chang, J.H.; Kim, S.H. The 2021 WHO Classification for Gliomas and Implications on Imaging Diagnosis: Part 1-Key Points of the Fifth Edition and Summary of Imaging Findings on Adult-Type Diffuse Gliomas. J. Magn. Reson. Imaging 2023, 58, 677–689. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Barthel, L.; Hadamitzky, M.; Dammann, P.; Schedlowski, M.; Sure, U.; Thakur, B.K.; Hetze, S. Glioma: Molecular signature and crossroads with tumor microenvironment. Cancer Metastasis Rev. 2022, 41, 53–75. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Arismendi-Morillo, G.; Zuccoli, G.; Lee, D.C.; Duraj, T.; Elsakka, A.M.; Maroon, J.C.; Mukherjee, P.; Ta, L.; Shelton, L.; et al. Metabolic management of microenvironment acidity in glioblastoma. Front. Oncol. 2022, 12, 968351. [Google Scholar] [CrossRef]
- White, J.; White, M.P.J.; Wickremesekera, A.; Peng, L.; Gray, C. The tumour microenvironment, treatment resistance and recurrence in glioblastoma. J. Transl. Med. 2024, 22, 540. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neurooncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, A.; Aruta, G.; Rizzo, F.; Salvati, L.F.; Zeppa, P.; Garbossa, D.; Cofano, F. Systematic Review on Tumor Microenvironment in Glial Neoplasm: From Understanding Pathogenesis to Future Therapeutic Perspectives. Int. J. Mol. Sci. 2022, 23, 4166. [Google Scholar] [CrossRef]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019, 9, 1512. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Musatova, O.E.; Rubtsov, Y.P. Effects of glioblastoma-derived extracellular vesicles on the functions of immune cells. Front. Cell Dev. Biol. 2023, 11, 1060000. [Google Scholar] [CrossRef]
- Tiwari, A.; Trivedi, R.; Lin, S.Y. Tumor microenvironment: Barrier or opportunity towards effective cancer therapy. J. Biomed. Sci. 2022, 29, 83. [Google Scholar] [CrossRef]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, E.; Wakimoto, H. Extracellular matrix in glioblastoma: Opportunities for emerging therapeutic approaches. Am. J. Cancer Res. 2021, 11, 3742–3754. [Google Scholar]
- Marino, S.; Menna, G.; Di Bonaventura, R.; Lisi, L.; Mattogno, P.; Figà, F.; Bilgin, L.; D’Alessandris, Q.G.; Olivi, A.; Della Pepa, G.M. The Extracellular Matrix in Glioblastomas: A Glance at Its Structural Modifications in Shaping the Tumoral Microenvironment-A Systematic Review. Cancers 2023, 15, 1879. [Google Scholar] [CrossRef]
- Peng, D.H.; Rodriguez, B.L.; Diao, L.; Chen, L.; Wang, J.; Byers, L.A.; Wei, Y.; Chapman, H.A.; Yamauchi, M.; Behrens, C.; et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8+ T cell exhaustion. Nat. Commun. 2020, 11, 4520. [Google Scholar] [CrossRef]
- Yang, T.; Kong, Z.; Ma, W. PD-1/PD-L1 immune checkpoint inhibitors in glioblastoma: Clinical studies, challenges and potential. Hum. Vaccines Immunother. 2021, 17, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, C.; Anacker, J.; Ernestus, R.I.; Vince, G.H. A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J. Clin. Oncol. 2012, 3, 67–79. [Google Scholar] [CrossRef]
- Erices, J.I.; Bizama, C.; Niechi, I.; Uribe, D.; Rosales, A.; Fabres, K.; Navarro-Martínez, G.; Torres, Á.; San Martín, R.; Roa, J.C.; et al. Glioblastoma Microenvironment and Invasiveness: New Insights and Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 7047. [Google Scholar] [CrossRef] [PubMed]
- Groves, M.D.; Puduvalli, V.K.; Hess, K.R.; Jaeckle, K.A.; Peterson, P.; Yung, W.K.; Levin, V.A. Phase II trial of temozolomide plus the matrix metalloproteinase inhibitor, marimastat, in recurrent and progressive glioblastoma multiforme. J. Clin. Oncol. 2002, 20, 1383–1388. [Google Scholar] [CrossRef]
- Chitadze, G.; Lettau, M.; Luecke, S.; Wang, T.; Janssen, O.; Fürst, D.; Mytilineos, J.; Wesch, D.; Oberg, H.H.; Held-Feindt, J.; et al. NKG2D- and T-cell receptor-dependent lysis of malignant glioma cell lines by human γδ T cells: Modulation by temozolomide and A disintegrin and metalloproteases 10 and 17 inhibitors. Oncoimmunology 2016, 5, e1093276. [Google Scholar] [CrossRef]
- Han, J.; Jing, Y.; Han, F.; Sun, P. Comprehensive analysis of expression, prognosis and immune infiltration for TIMPs in glioblastoma. BMC Neurol. 2021, 21, 447. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.; Ghorpade, A. Tissue inhibitor of metalloproteinase (TIMP)-1: The TIMPed balance of matrix metalloproteinases in the central nervous system. J. Neurosci. Res. 2003, 74, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 1803, 55–71. [Google Scholar] [CrossRef]
- Liu, L.; Yang, S.; Lin, K.; Yu, X.; Meng, J.; Ma, C.; Wu, Z.; Hao, Y.; Chen, N.; Ge, Q.; et al. Sp1 induced gene TIMP1 is related to immune cell infiltration in glioblastoma. Sci. Rep. 2022, 12, 11181. [Google Scholar] [CrossRef]
- Logun, M.T.; Wynens, K.E.; Simchick, G.; Zhao, W.; Mao, L.; Zhao, Q.; Mukherjee, S.; Brat, D.J.; Karumbaiah, L. Surfen-mediated blockade of extratumoral chondroitin sulfate glycosaminoglycans inhibits glioblastoma invasion. Faseb J. 2019, 33, 11973–11992. [Google Scholar] [CrossRef]
- Kiyokawa, J.; Kawamura, Y.; Ghouse, S.M.; Acar, S.; Barçın, E.; Martínez-Quintanilla, J.; Martuza, R.L.; Alemany, R.; Rabkin, S.D.; Shah, K.; et al. Modification of Extracellular Matrix Enhances Oncolytic Adenovirus Immunotherapy in Glioblastoma. Clin. Cancer Res. 2021, 27, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Hosseinalizadeh, H.; Habibi Roudkenar, M.; Mohammadi Roushandeh, A.; Kuwahara, Y.; Tomita, K.; Sato, T. Natural killer cell immunotherapy in glioblastoma. Discov. Oncol. 2022, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Li, N.; Zhang, Z. Emerging therapies for glioblastoma: Current state and future directions. J. Exp. Clin. Cancer Res. 2022, 41, 142. [Google Scholar] [CrossRef]
- Sener, U.; Ruff, M.W.; Campian, J.L. Immunotherapy in Glioblastoma: Current Approaches and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 7046. [Google Scholar] [CrossRef]
- Jessen, N.A.; Munk, A.S.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef]
- Arrieta, V.A.; Dmello, C.; McGrail, D.J.; Brat, D.J.; Lee-Chang, C.; Heimberger, A.B.; Chand, D.; Stupp, R.; Sonabend, A.M. Immune checkpoint blockade in glioblastoma: From tumor heterogeneity to personalized treatment. J. Clin. Investig. 2023, 133, e163447. [Google Scholar] [CrossRef]
- Huang, J.; Liu, F.; Liu, Z.; Tang, H.; Wu, H.; Gong, Q.; Chen, J. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front. Pharmacol. 2017, 8, 242. [Google Scholar] [CrossRef]
- Driessens, G.; Kline, J.; Gajewski, T.F. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol. Rev. 2009, 229, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Wang, N.; Xu, C. Glioma-associated microglia/macrophages (GAMs) in glioblastoma: Immune function in the tumor microenvironment and implications for immunotherapy. Front. Immunol. 2023, 14, 1123853. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad—Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Persico, P.; Lorenzi, E.; Dipasquale, A.; Pessina, F.; Navarria, P.; Politi, L.S.; Santoro, A.; Simonelli, M. Checkpoint Inhibitors as High-Grade Gliomas Treatment: State of the Art and Future Perspectives. J. Clin. Med. 2021, 10, 1367. [Google Scholar] [CrossRef]
- Alban, T.J.; Bayik, D.; Otvos, B.; Rabljenovic, A.; Leng, L.; Jia-Shiun, L.; Roversi, G.; Lauko, A.; Momin, A.A.; Mohammadi, A.M.; et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front. Immunol. 2020, 11, 1191. [Google Scholar] [CrossRef]
- Dubinski, D.; Wölfer, J.; Hasselblatt, M.; Schneider-Hohendorf, T.; Bogdahn, U.; Stummer, W.; Wiendl, H.; Grauer, O.M. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro-Oncology 2016, 18, 807–818. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef]
- Locarno, C.V.; Simonelli, M.; Carenza, C.; Capucetti, A.; Stanzani, E.; Lorenzi, E.; Persico, P.; Della Bella, S.; Passoni, L.; Mavilio, D.; et al. Role of myeloid cells in the immunosuppressive microenvironment in gliomas. Immunobiology 2020, 225, 151853. [Google Scholar] [CrossRef]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Front. Immunol. 2019, 10, 2683. [Google Scholar] [CrossRef]
- Yan, D.; Li, W.; Liu, Q.; Yang, K. Advances in Immune Microenvironment and Immunotherapy of Isocitrate Dehydrogenase Mutated Glioma. Front. Immunol. 2022, 13, 914618. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Choudhury, A.; Keough, M.B.; Seo, K.; Ni, L.; Kakaizada, S.; Lee, A.; Aabedi, A.; Popova, G.; Lipkin, B.; et al. Glioblastoma remodelling of human neural circuits decreases survival. Nature 2023, 617, 599–607. [Google Scholar] [CrossRef]
- Xiong, J.; Zhou, L.; Yang, M.; Lim, Y.; Zhu, Y.H.; Fu, D.L.; Li, Z.W.; Zhong, J.H.; Xiao, Z.C.; Zhou, X.F. ProBDNF and its receptors are upregulated in glioma and inhibit the growth of glioma cells in vitro. Neuro-Oncology 2013, 15, 990–1007. [Google Scholar] [CrossRef] [PubMed]
- Assimakopoulou, M.; Kondyli, M.; Gatzounis, G.; Maraziotis, T.; Varakis, J. Neurotrophin receptors expression and JNK pathway activation in human astrocytomas. BMC Cancer 2007, 7, 202. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Rothstein, J.D.; Sontheimer, H. Compromised glutamate transport in human glioma cells: Reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J. Neurosci. 1999, 19, 10767–10777. [Google Scholar] [CrossRef] [PubMed]
- Marcus, H.J.; Carpenter, K.L.; Price, S.J.; Hutchinson, P.J. In vivo assessment of high-grade glioma biochemistry using microdialysis: A study of energy-related molecules, growth factors and cytokines. J. Neurooncol. 2010, 97, 11–23. [Google Scholar] [CrossRef]
- Ishiuchi, S.; Tsuzuki, K.; Yoshida, Y.; Yamada, N.; Hagimura, N.; Okado, H.; Miwa, A.; Kurihara, H.; Nakazato, Y.; Tamura, M.; et al. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002, 8, 971–978. [Google Scholar] [CrossRef]
- Rothstein, J.D. Paving new pathways. Nat. Med. 2002, 8, 938–940. [Google Scholar] [CrossRef]
- van Vuurden, D.G.; Yazdani, M.; Bosma, I.; Broekhuizen, A.J.; Postma, T.J.; Heimans, J.J.; van der Valk, P.; Aronica, E.; Tannous, B.A.; Würdinger, T.; et al. Attenuated AMPA receptor expression allows glioblastoma cell survival in glutamate-rich environment. PLoS ONE 2009, 4, e5953. [Google Scholar] [CrossRef]
- Sheta, M.; Taha, E.A.; Lu, Y.; Eguchi, T. Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology 2023, 12, 110. [Google Scholar] [CrossRef]
- Yekula, A.; Yekula, A.; Muralidharan, K.; Kang, K.; Carter, B.S.; Balaj, L. Extracellular Vesicles in Glioblastoma Tumor Microenvironment. Front. Immunol. 2019, 10, 3137. [Google Scholar] [CrossRef] [PubMed]
- Pavlyukov, M.S.; Yu, H.; Bastola, S.; Minata, M.; Shender, V.O.; Lee, Y.; Zhang, S.; Wang, J.; Komarova, S.; Wang, J.; et al. Apoptotic Cell-Derived Extracellular Vesicles Promote Malignancy of Glioblastoma Via Intercellular Transfer of Splicing Factors. Cancer Cell 2018, 34, 119–135.e110. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, A.; Wang, Y.; Nowicki, M.O.; Peruzzi, P.; Ansari, K.; Ogawa, D.; Balaj, L.; De Rienzo, G.; Mineo, M.; Nakano, I.; et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014, 74, 738–750. [Google Scholar] [CrossRef]
- Zhai, H.; Acharya, S.; Gravanis, I.; Mehmood, S.; Seidman, R.J.; Shroyer, K.R.; Hajjar, K.A.; Tsirka, S.E. Annexin A2 Promotes Glioma Cell Invasion and Tumor Progression. J. Neurosci. 2011, 31, 14346–14360. [Google Scholar] [CrossRef]
- Randles, A.; Wirsching, H.-G.; Dean, J.A.; Cheng, Y.-K.; Emerson, S.; Pattwell, S.S.; Holland, E.C.; Michor, F. Computational modelling of perivascular-niche dynamics for the optimization of treatment schedules for glioblastoma. Nat. Biomed. Eng. 2021, 5, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef]
- Chen, J.; Mao, S.; Li, H.; Zheng, M.; Yi, L.; Lin, J.M.; Lin, Z.X. The pathological structure of the perivascular niche in different microvascular patterns of glioblastoma. PLoS ONE 2017, 12, e0182183. [Google Scholar] [CrossRef]
- Popescu, A.M.; Alexandru, O.; Brindusa, C.; Purcaru, S.O.; Tache, D.E.; Tataranu, L.G.; Taisescu, C.; Dricu, A. Targeting the VEGF and PDGF signaling pathway in glioblastoma treatment. Int. J. Clin. Exp. Pathol. 2015, 8, 7825–7837. [Google Scholar]
- Bates, D.O. Vascular endothelial growth factors and vascular permeability. Cardiovasc. Res. 2010, 87, 262–271. [Google Scholar] [CrossRef]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef]
- Turkowski, K.; Brandenburg, S.; Mueller, A.; Kremenetskaia, I.; Bungert, A.D.; Blank, A.; Felsenstein, M.; Vajkoczy, P. VEGF as a modulator of the innate immune response in glioblastoma. Glia 2018, 66, 161–174. [Google Scholar] [CrossRef]
- Rodriguez, S.M.B.; Staicu, G.-A.; Sevastre, A.-S.; Baloi, C.; Ciubotaru, V.; Dricu, A.; Tataranu, L.G. Glioblastoma Stem Cells—Useful Tools in the Battle against Cancer. Int. J. Mol. Sci. 2022, 23, 4602. [Google Scholar] [CrossRef] [PubMed]
- Soubéran, A.; Brustlein, S.; Gouarné, C.; Chasson, L.; Tchoghandjian, A.; Malissen, M.; Rougon, G. Effects of VEGF blockade on the dynamics of the inflammatory landscape in glioblastoma-bearing mice. J. Neuroinflammation 2019, 16, 191. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Cardoso-Carneiro, D.; Valbom, I.; Paula Cury, F.; Aline Silva, V.; Granja, S.; Reis, R.M.; Baltazar, F.; Martinho, O. Metabolic alterations underlying Bevacizumab therapy in glioblastoma cells. Oncotarget 2017, 8, 103657–103670. [Google Scholar] [CrossRef]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef]
- Rodriguez, S.M.B.; Kamel, A.; Ciubotaru, G.V.; Onose, G.; Sevastre, A.-S.; Sfredel, V.; Danoiu, S.; Dricu, A.; Tataranu, L.G. An Overview of EGFR Mechanisms and Their Implications in Targeted Therapies for Glioblastoma. Int. J. Mol. Sci. 2023, 24, 11110. [Google Scholar] [CrossRef]
- Ezzati, S.; Salib, S.; Balasubramaniam, M.; Aboud, O. Epidermal Growth Factor Receptor Inhibitors in Glioblastoma: Current Status and Future Possibilities. Int. J. Mol. Sci. 2024, 25, 2316. [Google Scholar] [CrossRef]
- Mateos, M.E.; López-Laso, E.; Izquierdo, L.; Pérez-Navero, J.L.; García, S.; Garzás, C. Response to nimotuzumab in a child with a progressive diffuse intrinsic pontine glioma. Pediatr. Int. 2011, 53, 261–263. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, H.K. Current Understanding of Hypoxia in Glioblastoma Multiforme and Its Response to Immunotherapy. Cancers 2022, 14, 1176. [Google Scholar] [CrossRef]
- Muir, M.; Gopakumar, S.; Traylor, J.; Lee, S.; Rao, G. Glioblastoma multiforme: Novel therapeutic targets. Expert Opin. Ther. Targets 2020, 24, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Bianconi, A.; Palmieri, G.; Aruta, G.; Monticelli, M.; Zeppa, P.; Tartara, F.; Melcarne, A.; Garbossa, D.; Cofano, F. Updates in Glioblastoma Immunotherapy: An Overview of the Current Clinical and Translational Scenario. Biomedicines 2023, 11, 1520. [Google Scholar] [CrossRef] [PubMed]
- Swartz, A.M.; Li, Q.J.; Sampson, J.H. Rindopepimut: A promising immunotherapeutic for the treatment of glioblastoma multiforme. Immunotherapy 2014, 6, 679–690. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Malkki, H. Glioblastoma vaccine therapy disappointment in Phase III trial. Nat. Rev. Neurol. 2016, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, T.T.; Mulholland, P.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef]
- Wick, W.; Puduvalli, V.K.; Chamberlain, M.C.; van den Bent, M.J.; Carpentier, A.F.; Cher, L.M.; Mason, W.; Weller, M.; Hong, S.; Musib, L.; et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J. Clin. Oncol. 2010, 28, 1168–1174. [Google Scholar] [CrossRef]
- Galanis, E.; Buckner, J.C. Enzastaurin in the Treatment of Recurrent Glioblastoma: A Promise That Did Not Materialize. J. Clin. Oncol. 2010, 28, 1097–1098. [Google Scholar] [CrossRef]
- Dou, Z.; Lei, H.; Su, W.; Zhang, T.; Chen, X.; Yu, B.; Zhen, X.; Si, J.; Sun, C.; Zhang, H.; et al. Modification of BCLX pre-mRNA splicing has antitumor efficacy alone or in combination with radiotherapy in human glioblastoma cells. Cell Death Dis. 2024, 15, 160. [Google Scholar] [CrossRef]
- Wu, H.; Yang, L.; Liu, H.; Zhou, D.; Chen, D.; Zheng, X.; Yang, H.; Li, C.; Chang, J.; Wu, A.; et al. Exploring the efficacy of tumor electric field therapy against glioblastoma: An in vivo and in vitro study. CNS Neurosci. Ther. 2021, 27, 1587–1604. [Google Scholar] [CrossRef] [PubMed]
- Ghiaseddin, A.; Warren, S.; Allen, A.; Sampson, D.; Chen, D.; Sherman, A.; Greene, V.; Rahman, M.; Tran, D. CTIM-04. Updates for a Phase 2 Open-Labeled Study of PEMBROLIZUMAB Plus TTFIELDS Plus Maintenance Temozolomide in Patients with Newly Diagnosed Glioblastoma (2-the-Top). Neuro-Oncology 2020, 22, ii33. [Google Scholar] [CrossRef]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van den Bent, M.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro-Oncology 2023, 25, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Youssef, G.; Dietrich, J. Ipilimumab: An investigational immunotherapy for glioblastoma. Expert Opin. Investig. Drugs 2020, 29, 1187–1193. [Google Scholar] [CrossRef]
- Luksik, A.S.; Yazigi, E.; Shah, P.; Jackson, C.M. CAR T Cell Therapy in Glioblastoma: Overcoming Challenges Related to Antigen Expression. Cancers 2023, 15, 1414. [Google Scholar] [CrossRef]
- Jin, G.; Chang, Y.; Bao, X. Generation of chimeric antigen receptor macrophages from human pluripotent stem cells to target glioblastoma. Immunooncol. Technol. 2023, 20, 100409. [Google Scholar] [CrossRef]
- Chen, C.; Jing, W.; Chen, Y.; Wang, G.; Abdalla, M.; Gao, L.; Han, M.; Shi, C.; Li, A.; Sun, P.; et al. Intracavity generation of glioma stem cell-specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci. Transl. Med. 2022, 14, eabn1128. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Country | Title | Year |
|---|---|---|
| United States of America (North America) | A Phase 1b/2a Study of ACT001 and Anti-PD-1 in Patients with Surgically Accessible Recurrent Glioblastoma Multiforme | Started on 22 September 2021 |
| Germany (Europe) | Characterization of Metabolic Changes in the Glioma Tumor Tissue Induced by Transient Fasting (ERGO3) | Started on 19 August 2020 |
| China (Asia) | The Study of Microglia/Macrophages Involved Dynamic Evolution of Glioma Microenvironment and the Function and Visualization of Targeted Molecules of Glioma | Started on 9 November 2020 |
| United States of America (North America) | AB154 Combined With AB122 for Recurrent Glioblastoma | Started on 21 April 2021 |
| United States of America (North America) | Surgical Nivolumab And Ipilimumab For Recurrent GBM | Started on 1 February 2021 |
| China (Asia) | Testing the Addition of the Immune Therapy Drugs, Tocilizumab and Atezolizumab, to Radiation Therapy for Recurrent Glioblastoma | Started on 11 March 2022 |
| United Kingdom (Europe) | A Trial of Ipatasertib in Combination With Atezolizumab (IceCAP) | Started on 13 August 2018 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tataranu, L.G.; Turliuc, S.; Kamel, A.; Rizea, R.E.; Dricu, A.; Staicu, G.-A.; Baloi, S.C.; Rodriguez, S.M.B.; Manole, A.I.M. Glioblastoma Tumor Microenvironment: An Important Modulator for Tumoral Progression and Therapy Resistance. Curr. Issues Mol. Biol. 2024, 46, 9881-9894. https://doi.org/10.3390/cimb46090588
Tataranu LG, Turliuc S, Kamel A, Rizea RE, Dricu A, Staicu G-A, Baloi SC, Rodriguez SMB, Manole AIM. Glioblastoma Tumor Microenvironment: An Important Modulator for Tumoral Progression and Therapy Resistance. Current Issues in Molecular Biology. 2024; 46(9):9881-9894. https://doi.org/10.3390/cimb46090588
Chicago/Turabian StyleTataranu, Ligia Gabriela, Serban Turliuc, Amira Kamel, Radu Eugen Rizea, Anica Dricu, Georgiana-Adeline Staicu, Stefania Carina Baloi, Silvia Mara Baez Rodriguez, and Andrada Ioana Maria Manole. 2024. "Glioblastoma Tumor Microenvironment: An Important Modulator for Tumoral Progression and Therapy Resistance" Current Issues in Molecular Biology 46, no. 9: 9881-9894. https://doi.org/10.3390/cimb46090588
APA StyleTataranu, L. G., Turliuc, S., Kamel, A., Rizea, R. E., Dricu, A., Staicu, G.-A., Baloi, S. C., Rodriguez, S. M. B., & Manole, A. I. M. (2024). Glioblastoma Tumor Microenvironment: An Important Modulator for Tumoral Progression and Therapy Resistance. Current Issues in Molecular Biology, 46(9), 9881-9894. https://doi.org/10.3390/cimb46090588