
Mitochondrial Genome Characteristics and Comparative Genomic Analysis of Spartina alterniflora

Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Nucleotide Diversity Analysis
2.3. Intergenomic Sequence Transfers Analysis
2.4. Ka/Ks Analysis
2.5. Phylogenetic Tree Construction
2.6. Synteny Analysis
3. Results
3.1. Nucleotide Diversity of the S. alterniflora mt Genome
3.2. Intergenomic Sequence Transfers of S. alterniflora
3.3. Selective Pressure Analysis
3.4. Phylogenetic Inference
3.5. Sequence Collinearity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, D.; Hu, Y.; Liu, M.; Chang, Y.; Yan, X.; Bu, R.; Zhao, D.; Li, Z. Introduction and spread of an exotic plant, Spartina alterniflora, along coastal marshes of China. Wetlands 2017, 37, 1181–1193. [Google Scholar] [CrossRef]
- Cui, L.; Berger, U.; Cao, M.; Zhang, Y.; He, J.; Pan, L.; Jiang, J. Conservation and restoration of mangroves in response to invasion of Spartina alterniflora based on the MaxEnt model: A case study in China. Forests 2023, 14, 1220. [Google Scholar] [CrossRef]
- Zheng, J.; Wei, H.; Chen, R.; Liu, J.; Wang, L.; Gu, W. Invasive trends of Spartina Alterniflora in the southeastern Coast of China and potential distributional impacts on mangrove forests. Plants 2023, 12, 1923. [Google Scholar] [CrossRef] [PubMed]
- Hammani, K.; Giegé, P. RNA metabolism in plant mitochondria. Trends Plant Sci. 2014, 19, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Ramadani-Muja, J.; Ziomek, G.; Burgstaller, S.; Bischof, H.; Koshenov, Z.; Gottschalk, B.; Malli, R.; Graier, W.F. Tracking intra-and inter-organelle signaling of mitochondria. FEBS J. 2019, 286, 4378–4401. [Google Scholar] [CrossRef]
- Kong, J.; Wang, J.; Nie, L.; Tembrock, L.R.; Zou, C.; Kan, S.; Ma, X.; Wendel, J.F.; Wu, Z. Evolutionary dynamics of mitochondrial genomes and intracellular transfers among diploid and allopolyploid cotton species. BMC Biol. 2025, 23, 9. [Google Scholar] [CrossRef] [PubMed]
- Skippington, E.; Barkman, J.; Rice, D.W.; Palmer, J.D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. USA 2015, 112, E3515–E3524. [Google Scholar] [CrossRef] [PubMed]
- Putintseva, Y.A.; Bondar, E.I.; Simonov, E.P.; Sharov, V.V.; Oreshkova, N.V.; Kuzmin, D.A.; Konstantinov, Y.M.; Shmakov, V.N.; Belkov, V.I.; Sadovsky, M.G.; et al. Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genom. 2020, 21, 654. [Google Scholar] [CrossRef]
- Jackman, S.D.; Coombe, L.; Warren, R.L.; Kirk, H.; Trinh, E.; MacLeod, T.; Pleasance, S.; Pandoh, P.; Zhao, Y.; Coope, R.J.; et al. Complete mitochondrial genome of a gymnosperm, Sitka spruce (Picea sitchensis), indicates a complex physical structure. Genome Biol. Evol. 2020, 12, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, J.; He, W.; Kan, S.; Liao, X.; Jordan, D.R.; Mace, E.S.; Tao, Y.; Cruickshank, A.W.; Klein, R.; et al. Variation in mitogenome structural conformation in wild and cultivated lineages of sorghum corresponds with domestication history and plastome evolution. BMC Plant Biol. 2023, 23, 91. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kan, S.; Liao, X.; Zhou, J.; Tembrock, L.R.; Daniell, H.; Jin, S.; Wu, Z. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024, 29, 754–769. [Google Scholar] [CrossRef]
- Notsu, Y.; Masood, S.; Nishikawa, T.; Kubo, N.; Akiduki, G.; Nakazono, M.; Hirai, A.; Kadowaki, K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: Frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genom. 2002, 268, 434–445. [Google Scholar] [CrossRef]
- Cui, P.; Liu, H.; Lin, Q.; Ding, F.; Zhou, G.; Hu, S.; Liu, D.; Yang, D.; Zhan, K.; Zhang, A.; et al. A complete mitochondrial genome of wheat (Triticum aestivum cv. Chinese Yumai), and fast evolving mitochondrial genes in higher plants. J. Genet. 2009, 88, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Studer, B.; Byrne, S.L.; Farrell, J.D.; Panitz, F.; Bendixen, C.; Møller, I.M.; Asp, T. The genome and transcriptome of perennial ryegrass mitochondria. BMC Genom. 2013, 14, 202. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, K.; He, Y.; Wang, Y.; Qu, C.; Miao, J. The complete chloroplast genome of Spartina alterniflora. Mitochondrial DNA B 2020, 5, 2440–2441. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, W.; Cao, J.; He, Y.; Zhao, Y.; Qu, C.; Miao, J. The complete mitochondrial genome of Sporobolus alterniflorus (loisel.) PM Peterson & Saarela (Poaceae) and phylogenetic analysis. Mitochondrial DNA B 2021, 6, 1303–1305. [Google Scholar]
- Matheson, P.; McGaughran, A. Genomic data is missing for many highly invasive species, restricting our preparedness for escalating incursion rates. Sci. Rep. 2022, 12, 13987. [Google Scholar] [CrossRef]
- Li, C.; Wang, B.; Ji, Y.; Huang, L.; Wang, X.; Zhao, W.; Wang, H.; Yao, Y. Mitochondrial genome provides species-specific targets for the rapid detection of early invasive populations of Hylurgus ligniperda in China. BMC Genom. 2024, 25, 90. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinf. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doalla, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Chen, H.; Yang, D.; Liu, C. Diversity of mitochondrial plastid DNAs (MTPTs) in seed plants. Mitochondrial DNA A 2018, 29, 635–642. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Yang, J.; Park, S.; Gil, H.Y.; Pak, J.H.; Kim, S.C. Characterization and dynamics of intracellular gene transfer in plastid genomes of Viola (Violaceae) and order Malpighiales. Front. Plant Sci. 2021, 12, 678580. [Google Scholar] [CrossRef]
- Filip, E.; Skuza, L. Horizontal gene transfer involving chloroplasts. Int. J. Mol. Sci. 2021, 22, 4484. [Google Scholar] [CrossRef] [PubMed]
- Leister, D.; Kleine, T. Role of intercompartmental DNA transfer in producing genetic diversity. Int. Rev. Cell Mol. Biol. 2011, 291, 73–114. [Google Scholar]
- Martin, W. Gene transfer from organelles to the nucleus: Frequent and in big chunks. Proc. Natl. Acad. Sci. USA 2003, 100, 8612–8614. [Google Scholar] [CrossRef]
- Zhou, P.; Zhang, Q.; Li, F.; Huang, J.; Zhang, M. Assembly and comparative analysis of the complete mitochondrial genome of Ilex metabaptista (Aquifoliaceae), a Chinese endemic species with a narrow distribution. BMC Plant Biol. 2023, 23, 393. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Zhang, K.; Que, Y.; Li, Y. Assembly and analysis of the first complete mitochondrial genome of Punica granatum and the gene transfer from chloroplast genome. Front. Plant Sci. 2023, 14, 1132551. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, X.; Priyadarshani, S.V.G.N.; Wang, Y.; Ye, L.; Shi, C.; Ye, K.; Zhou, Q.; Luo, Z.; Deng, F.; et al. Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genom. 2021, 22, 167. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, X.; Li, Z.; Song, Y.; Sun, Z. Assembly and comparative analysis of the complete mitochondrial genome of Bupleurum chinense DC. BMC Genom. 2022, 23, 664. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Shen, A.; Liu, L.; Tan, Y.; Li, S.; Tan, Z. Assembly and comparative analysis of the complete multichromosomal mitochondrial genome of Cymbidium ensifolium, an orchid of high economic and ornamental value. BMC Plant Biol. 2024, 24, 255. [Google Scholar] [CrossRef]
- Jethva, J.; Lichtenauer, S.; Schmidt-Schippers, R.; Steffen-Heins, A.; Poschet, G.; Wirtz, M.; van Dongen, J.T.; Eirich, J.; Finkemeier, I.; Bilger, W.; et al. Mitochondrial alternative NADH dehydrogenases NDA1 and NDA2 promote survival of reoxygenation stress in Arabidopsis by safeguarding photosynthesis and limiting ROS generation. New Phytol. 2023, 238, 96–112. [Google Scholar] [CrossRef]
- Faivre-Nitschke, S.E.; Nazoa, P.; Gualberto, J.M.; Grienenberger, J.M.; Bonnard, G. Wheat mitochondria ccmB encodes the membrane domain of a putative ABC transporter involved in cytochrome c biogenesis. BBA-Gene Struct. Expr. 2001, 1519, 199–208. [Google Scholar] [CrossRef]

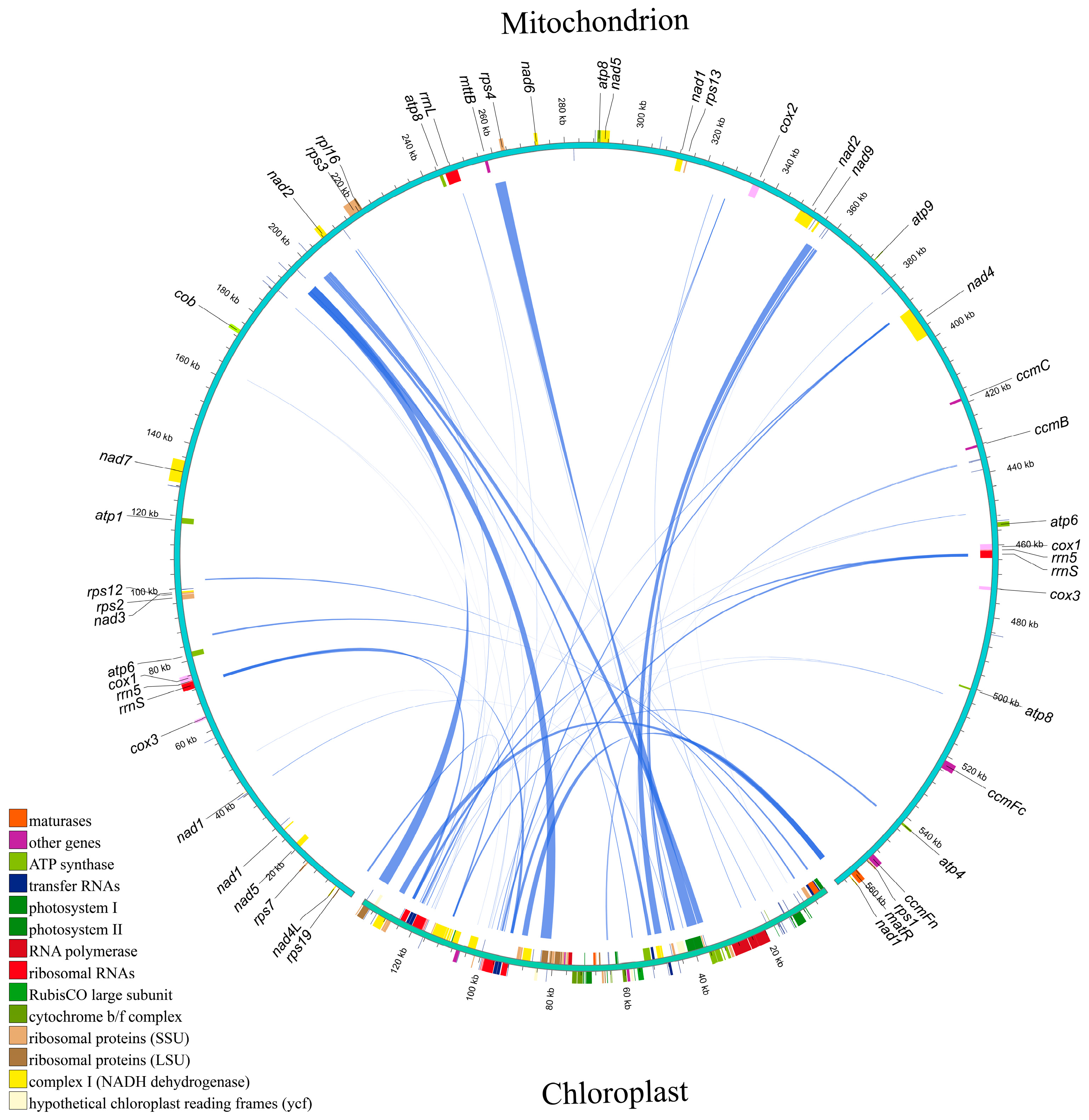
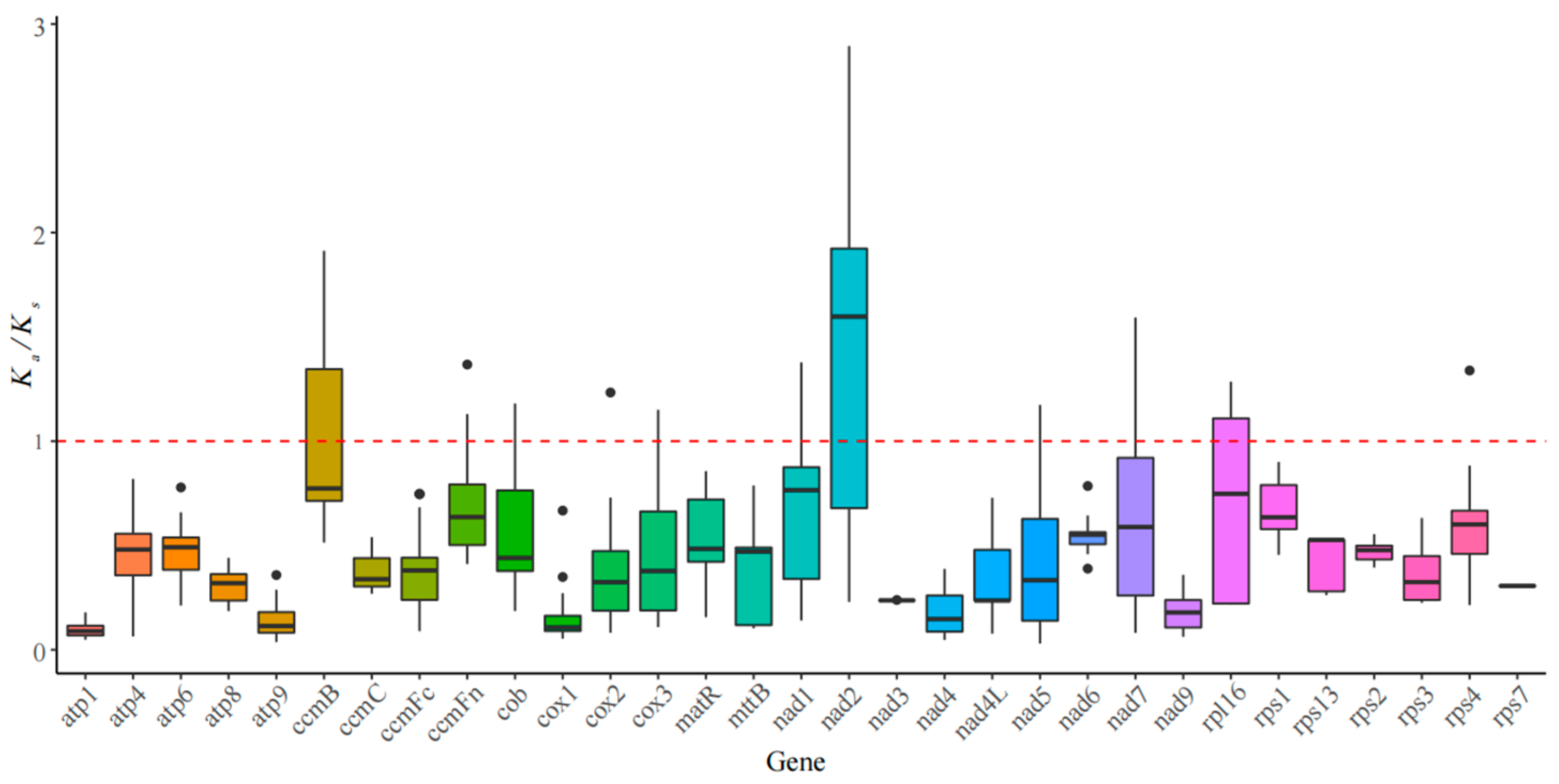
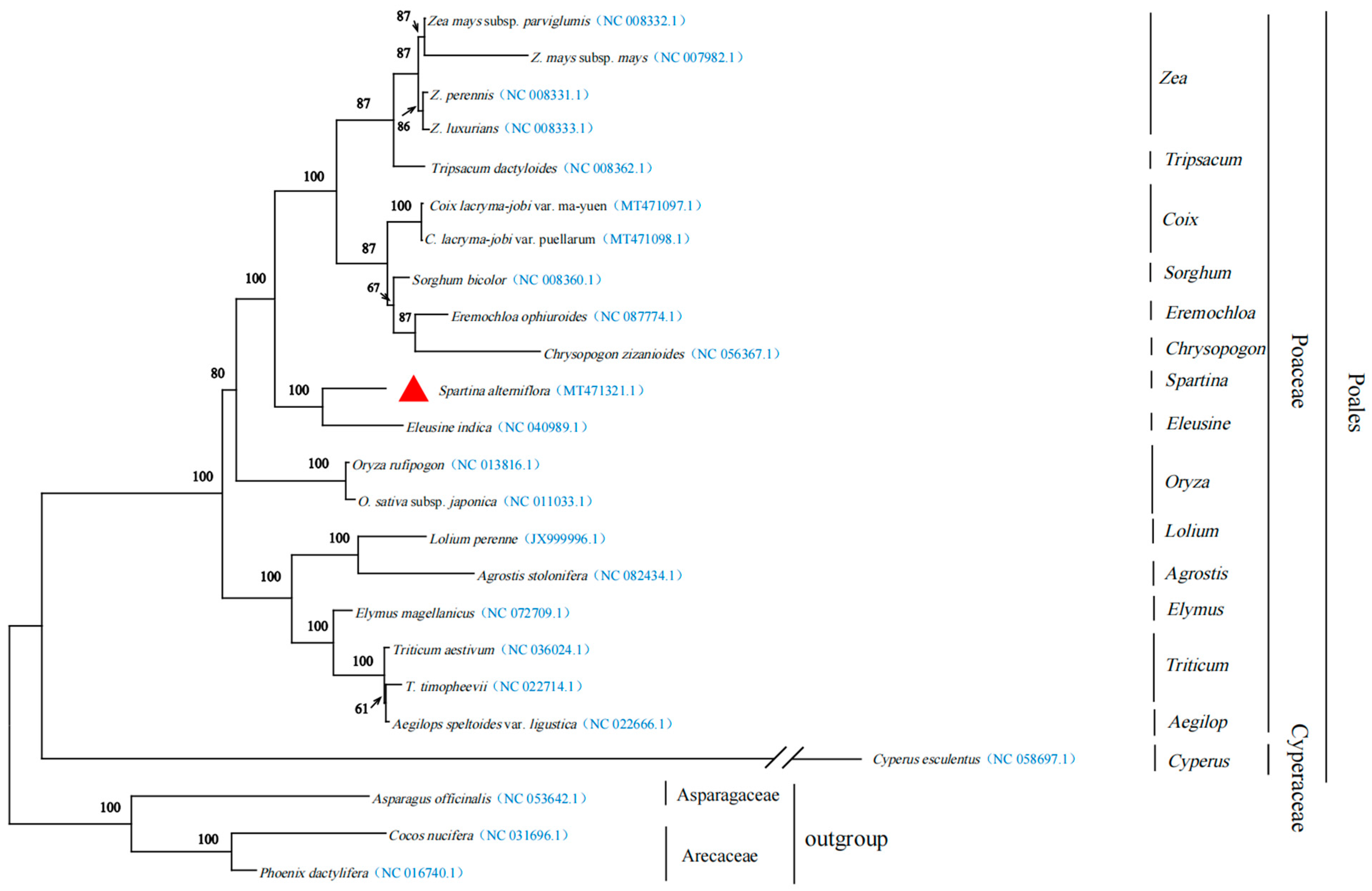
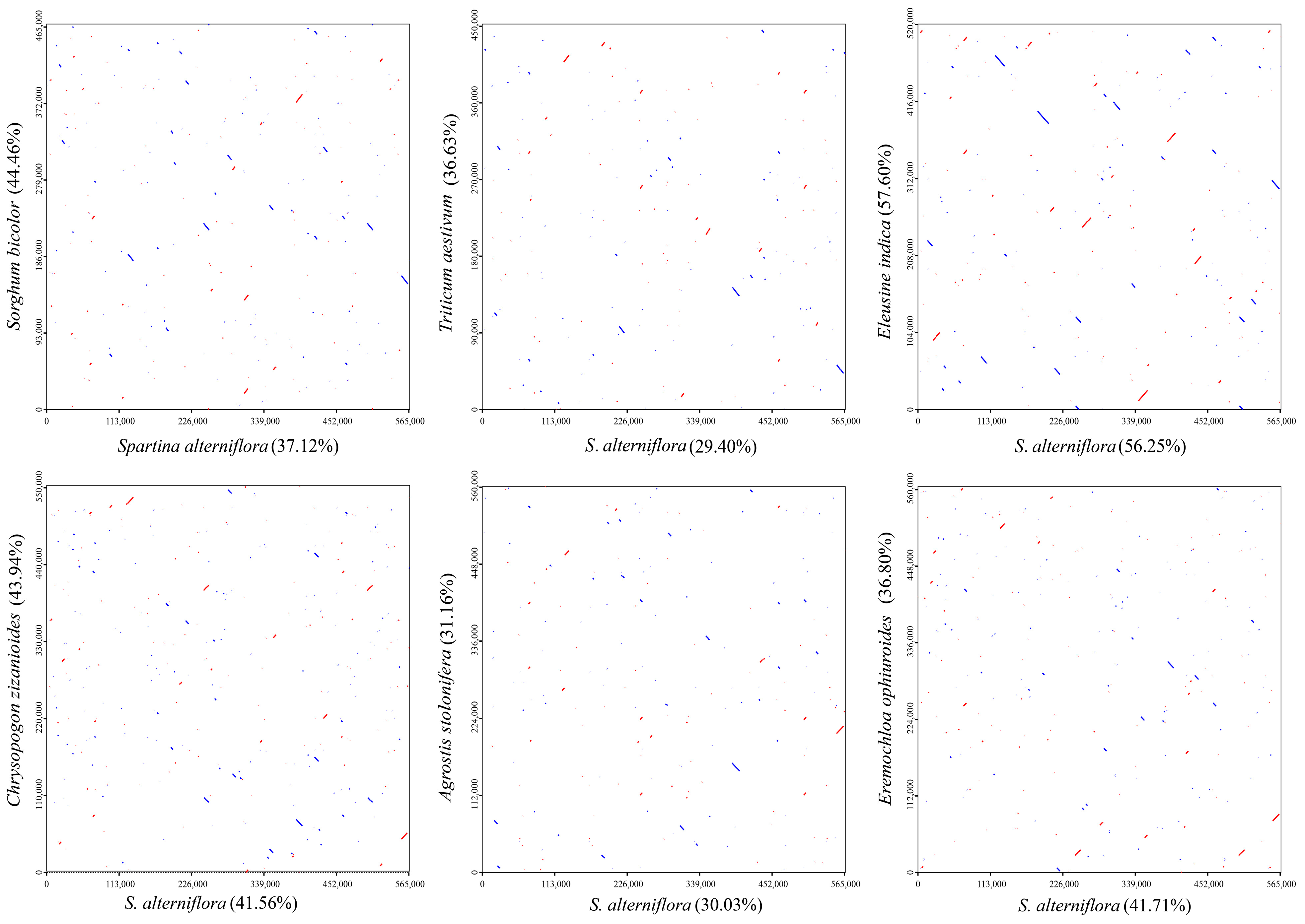

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Aligened Length (bp) | Sequence Identity (%) | Mismatches | Gap Opens | Chloroplast Genome (CP) | Mitochondrial Genome (MT) | Contained Genes | ||
|---|---|---|---|---|---|---|---|---|---|
| Start | End | Start | End | ||||||
| 1 | 3203 | 99.657 | 6 | 1 | 80,973 | 84,170 | 196,986 | 193,784 | trnM-CAT; trnH-GTG |
| 2 | 3203 | 99.657 | 6 | 1 | 132,219 | 135,416 | 193,784 | 196,986 | trnM-CAT; trnH-GTG |
| 3 | 3007 | 99.734 | 2 | 2 | 37,454 | 40,460 | 258,992 | 261,992 | |
| 4 | 1709 | 99.649 | 2 | 1 | 126,698 | 128,406 | 565,996 | 564,292 | |
| 5 | 1709 | 99.649 | 2 | 1 | 87,983 | 89,691 | 564,292 | 565,996 | |
| 6 | 2166 | 86.888 | 147 | 58 | 48,194 | 50,273 | 357,558 | 355,444 | trnF-GAA |
| 7 | 1297 | 98.227 | 20 | 2 | 35,467 | 36,761 | 203,241 | 201,946 | trnR-TCT |
| 8 | 1034 | 87.041 | 84 | 23 | 51,282 | 52,304 | 201,390 | 200,396 | |
| 9 | 585 | 99.658 | 1 | 1 | 95,399 | 95,982 | 389,831 | 389,247 | |
| 10 | 585 | 99.658 | 1 | 1 | 120,407 | 120,990 | 389,247 | 389,831 | |
| 11 | 825 | 84.848 | 70 | 23 | 44,149 | 44,951 | 359,302 | 358,511 | |
| 12 | 522 | 93.678 | 19 | 4 | 622 | 1137 | 85,462 | 85,975 | |
| 13 | 486 | 93.621 | 27 | 2 | 36,772 | 37,253 | 201,889 | 201,404 | |
| 14 | 532 | 87.030 | 51 | 7 | 110,463 | 110,982 | 592 | 67 | |
| 15 | 532 | 87.030 | 51 | 7 | 110,463 | 110,982 | 543,296 | 542,771 | |
| 16 | 453 | 87.417 | 41 | 10 | 5324 | 5768 | 101,801 | 102,245 | |
| 17 | 258 | 99.225 | 1 | 1 | 56,844 | 57,100 | 194,757 | 195,014 | |
| 18 | 410 | 81.463 | 36 | 17 | 45,103 | 45,487 | 358,312 | 357,918 | trnS-GGA |
| 19 | 238 | 89.916 | 11 | 8 | 64,331 | 64,567 | 436,134 | 436,359 | trnP-TGG |
| 20 | 889 | 73.566 | 180 | 41 | 123,201 | 124,064 | 73,559 | 72,701 | rrnS (partical:43.63%) |
| 21 | 889 | 73.566 | 180 | 41 | 92,325 | 93,188 | 72,701 | 73,559 | rrnS (partical:43.63%) |
| 22 | 889 | 73.566 | 180 | 41 | 92,325 | 93,188 | 463,240 | 462,382 | rrnS (partical:43.63%) |
| 23 | 889 | 73.566 | 180 | 41 | 123,201 | 124,064 | 462,382 | 463,240 | rrnS (partical:43.63%) |
| 24 | 155 | 97.419 | 3 | 1 | 41,618 | 41,772 | 258,998 | 258,845 | |
| 25 | 148 | 97.973 | 3 | 0 | 23,448 | 23,595 | 324,214 | 324,067 | |
| 26 | 226 | 86.726 | 29 | 1 | 97,053 | 97,277 | 327,985 | 327,760 | |
| 27 | 226 | 86.726 | 29 | 1 | 119,112 | 119,336 | 327,760 | 327,985 | |
| 28 | 145 | 93.793 | 8 | 1 | 45,141 | 45,284 | 40,212 | 40,356 | trnS-GGA (partical:86.52%) |
| 29 | 182 | 87.363 | 11 | 6 | 121,318 | 121,499 | 212,946 | 212,777 | |
| 30 | 182 | 87.363 | 11 | 6 | 94,890 | 95,071 | 212,777 | 212,946 | |
| 31 | 152 | 87.500 | 10 | 6 | 18,689 | 18,839 | 213,562 | 213,705 | trnC-GCA |
| 32 | 93 | 97.849 | 1 | 1 | 117,548 | 117,640 | 450,821 | 450,730 | |
| 33 | 93 | 97.849 | 1 | 1 | 98,749 | 98,841 | 450,730 | 450,821 | |
| 34 | 123 | 87.805 | 13 | 2 | 64,133 | 64,254 | 435,907 | 436,028 | trnW-CCA |
| 35 | 81 | 97.531 | 2 | 0 | 99,799 | 99,879 | 188,235 | 188,155 | trnN-GTT |
| 36 | 81 | 97.531 | 2 | 0 | 116,510 | 116,590 | 188,155 | 188,235 | trnN-GTT |
| 37 | 76 | 92.105 | 5 | 1 | 52,299 | 52,373 | 381,683 | 381,608 | trnM-CAT |
| 38 | 97 | 82.474 | 17 | 0 | 96,206 | 96,302 | 248,985 | 248,889 | rrnL (partical:2.73%) |
| 39 | 97 | 82.474 | 17 | 0 | 120,087 | 120,183 | 248,889 | 248,985 | rrnL (partical:2.73%) |
| 40 | 97 | 82.474 | 17 | 0 | 96,206 | 96,302 | 504,491 | 504,395 | |
| 41 | 97 | 82.474 | 17 | 0 | 120,087 | 120,183 | 504,395 | 504,491 | |
| 42 | 63 | 88.889 | 5 | 2 | 12,949 | 13,011 | 201,726 | 201,666 | |
| 43 | 32 | 100 | 0 | 0 | 99,400 | 99,431 | 162,713 | 162,682 | |
| 44 | 32 | 100 | 0 | 0 | 116,958 | 116,989 | 162,682 | 162,713 | |
| 45 | 35 | 97.143 | 0 | 1 | 103,130 | 103,163 | 48,940 | 48,974 | |
| 46 | 37 | 94.595 | 1 | 1 | 8068 | 8104 | 358,125 | 358,160 | trnS-GGA (partical:41.38%) |
| Total | 28,860 | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; Yue, C.; Li, H. Mitochondrial Genome Characteristics and Comparative Genomic Analysis of Spartina alterniflora. Curr. Issues Mol. Biol. 2025, 47, 107. https://doi.org/10.3390/cimb47020107
Zhu H, Yue C, Li H. Mitochondrial Genome Characteristics and Comparative Genomic Analysis of Spartina alterniflora. Current Issues in Molecular Biology. 2025; 47(2):107. https://doi.org/10.3390/cimb47020107
Chicago/Turabian StyleZhu, Hong, Chunlei Yue, and Hepeng Li. 2025. "Mitochondrial Genome Characteristics and Comparative Genomic Analysis of Spartina alterniflora" Current Issues in Molecular Biology 47, no. 2: 107. https://doi.org/10.3390/cimb47020107
APA StyleZhu, H., Yue, C., & Li, H. (2025). Mitochondrial Genome Characteristics and Comparative Genomic Analysis of Spartina alterniflora. Current Issues in Molecular Biology, 47(2), 107. https://doi.org/10.3390/cimb47020107