


In Silico Identification of Potential Antagonists Targeting the HPV16 E2-E1 Interaction: A Step Toward Novel Therapeutics for Cervical Cancer

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Binding Pocket
2.1.1. Previous Evidence on the Relevance of the TAD in the E2-E1 Interaction
2.1.2. Identification of Binding Site Using MOE
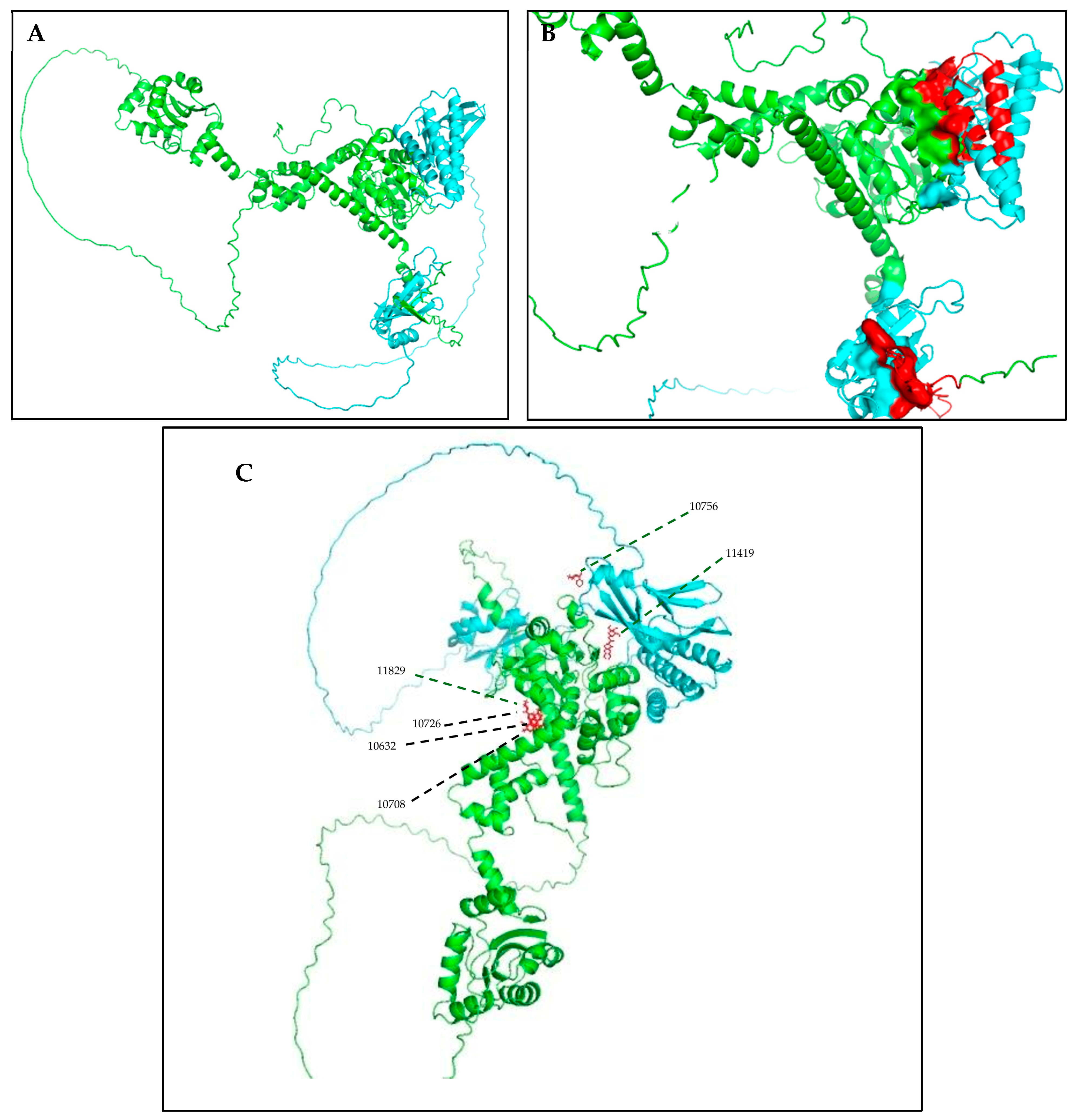
2.1.3. The Structural Conservation of the TAD Across Different HPV Types
2.2. Target Acquisition and Preparation
2.3. Ligand Selection and Preparation
2.4. Conformer Search
2.5. Molecular Recognition Simulations
- C: Average gain/loss in rotational and translational entropy.
- E(flex): Energy associated with loss of ligand flexibility.
- f(HB): Geometric imperfections in hydrogen bonds (value = 0.1).
- C(HB): Energy of ideal hydrogen bond.
- f(M): Geometric imperfections in metal–ligand interactions (value = 0.1).
- C(M): Energy of ideal metal–ligand bond.
- Di: Desolvation energy of atom i.
2.6. Docking Validation
2.7. Preprocessing and Ligand Optimization
- •
- Energy minimization with MMFF94 force field in Avogadro (version 1.2.0).
- •
- Protonation state correction at physiological pH using Epik server (Schrödinger).
2.8. Selection Criteria for Best Binding Poses
2.9. Search for TAD Sequences of HPV E2
2.10. Structural Modeling and Protein–Ligand Interaction Analysis
2.10.1. TAD Marking in E2
2.10.2. Ligand Selection, Evaluation, and Molecular Docking
3. Results
3.1. Target Preparation
3.2. Conformer Search
3.3. Molecular Recognition
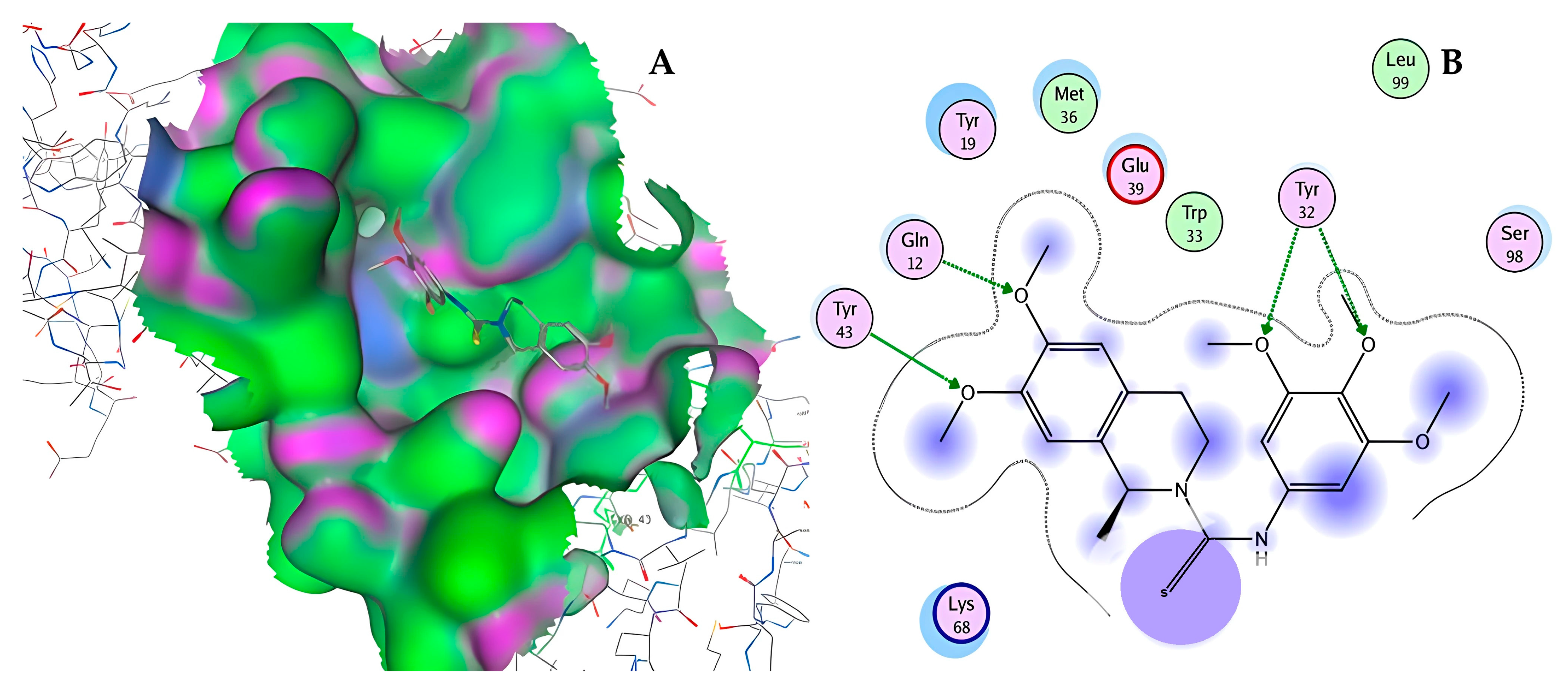
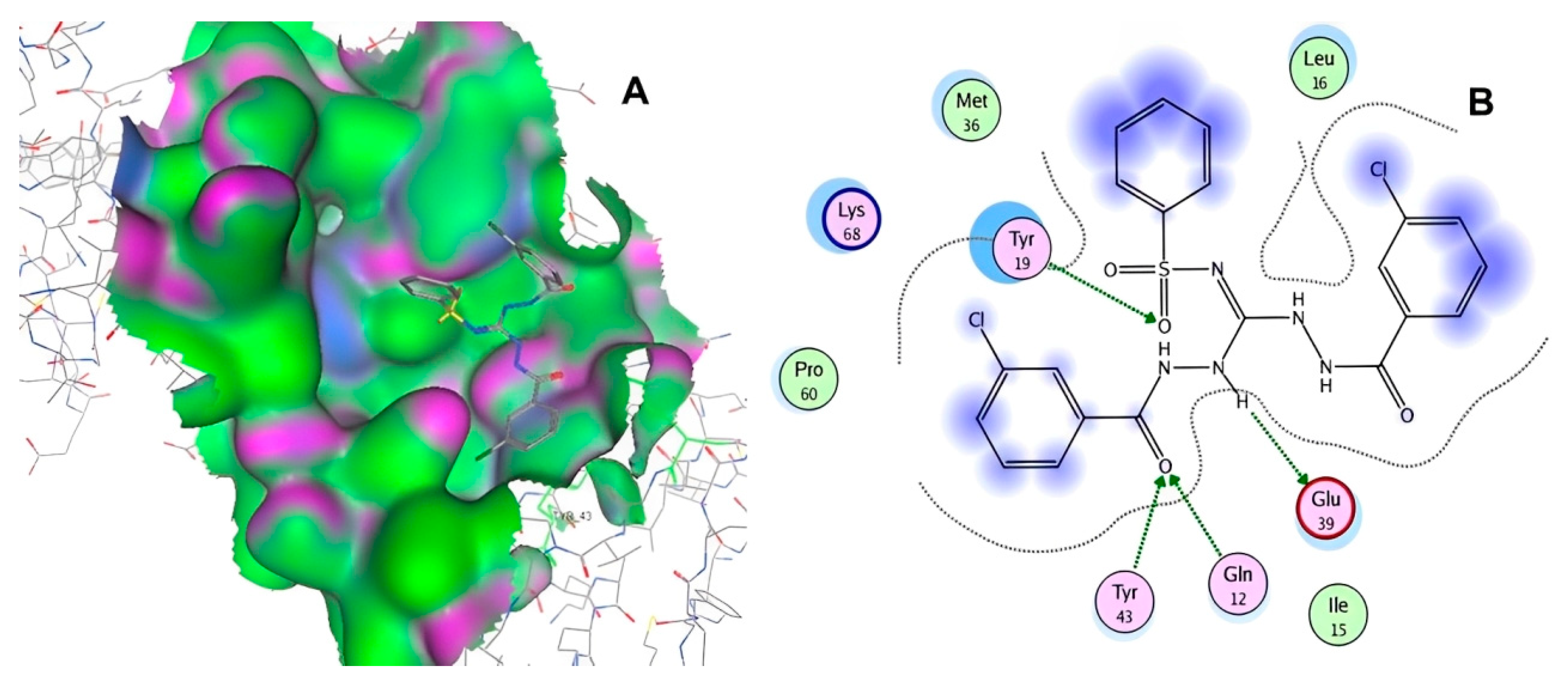
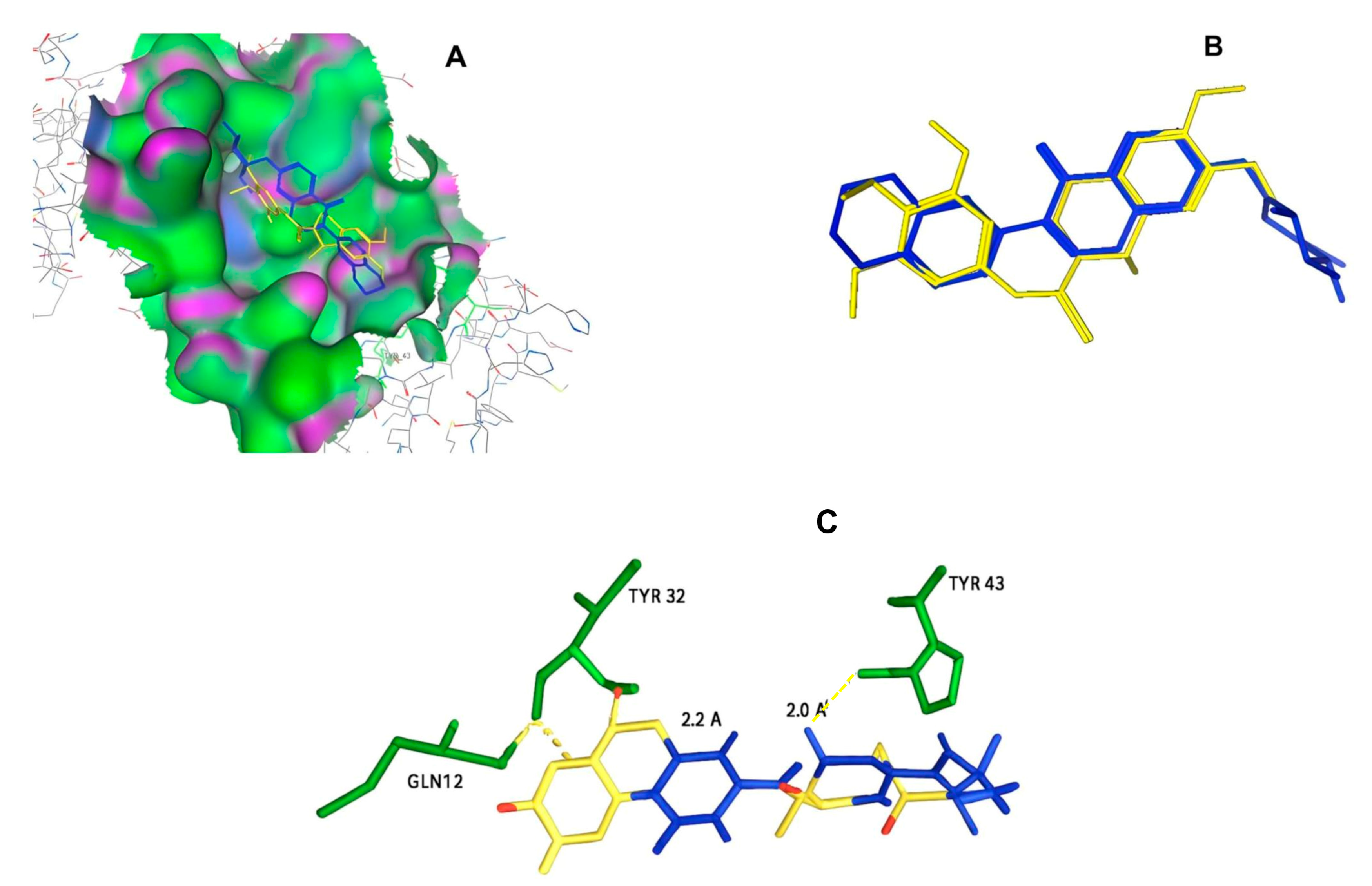
3.4. Structural Analysis
3.5. Sequence Alignment of HPV E2 TAD

3.6. Analysis of HPV16 E2 TAD and Ligand Interactions
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cherry, J.J.; Rietz, A.; Malinkevich, A.; Liu, Y.; Xie, M.; Bartolowits, M.; Davisson, V.J.; Baleja, J.D.; Androphy, E.J. Structure based identification and characterization of flavonoids that disrupt human papillomavirus-16 E6 function. PLoS ONE 2013, 8, e84506. [Google Scholar] [CrossRef] [PubMed]
- Plotzker, R.E.; Vaidya, A.; Pokharel, U.; Stier, E.A. Update in Epidemiology, Prevention, and Management. Infect. Dis. Clin. N. Am. 2023, 37, 289–310. [Google Scholar] [CrossRef]
- Cervical Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cervical-cancer (accessed on 24 November 2024).
- Human Papillomavirus and Related Diseases Report. Available online: https://hpvcentre.net/statistics/reports/MEX.pdf (accessed on 24 November 2024).
- Estadísticas a Propósito del día Mundial Contra el Cáncer (4 de Febrero). Available online: https://www.inegi.org.mx/contenidos/saladeprensa/aproposito/2015/cancer10.pdf (accessed on 24 November 2024).
- Mo, Y.; Ma, J.; Zhang, H.; Shen, J.; Chen, J.; Hong, J. Prophylactic and Therapeutic HPV Vaccines: Current Scenario and Perspectives. Front. Cell Infect. Microbiol. 2022, 12, 909223. [Google Scholar] [CrossRef] [PubMed]
- Hewavisenti, R.V.; Arena, J.; Ahlenstiel, C.L.; Sasson, S.C. Human papillomavirus in the setting of immunodeficiency: Pathogenesis and the emergence of next-generation therapies to reduce the high associated cancer risk. Front. Immunol. 2023, 14, 1112513. [Google Scholar] [CrossRef]
- Wang, R.; Huang, H.; Yu, C.; Li, X.; Wang, Y.; Xie, L. Current status and future directions for the development of human papillomavirus vaccines. Front. Immunol. 2024, 15, 1362770. [Google Scholar] [CrossRef] [PubMed]
- Fontan, C.T.; Das, D.; Bristol, M.L.; James, C.D.; Wang, X.; Lohner, H.; Atfi, A.; Morgan, I.M. Human Papillomavirus 16 (HPV16) E2 Repression of TWIST1 Transcription Is a Potential Mediator of HPV16 Cancer Outcomes. mSphere 2020, 5, e00981-20. [Google Scholar] [CrossRef]
- Antson, A.A.; Burns, J.E.; Moroz, O.V.; Scott, D.J.; Sanders, C.M.; Bronstein, I.B.; Dodson, G.G.; Wilson, K.S.; Maitland, N.J. Structure of the intact transactivation domain of the human papillomavirus E2 protein. Nature 2000, 403, 805–809. [Google Scholar] [CrossRef]
- Macalino, S.J.Y.; Basith, S.; Clavio, N.A.B.; Chang, H.; Kang, S.; Choi, S. Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery. Molecules 2018, 23, 1963. [Google Scholar] [CrossRef]
- Chang, Y.; Hawkins, B.A.; Du, J.J.; Groundwater, P.W.; Hibbs, D.E.; Lai, F. A Guide to In Silico Drug Design. Pharmaceutics 2022, 15, 49. [Google Scholar] [CrossRef]
- Gao, C.; Pan, M.M.; Lei, Y.J.; Tian, L.Q.; Jiang, H.Y.; Li, X.L.; Shi, Q.; Tian, C.; Yuan, Y.-K.; Fan, G.-X.; et al. A point mutation in the DNA-binding domain of HPV-2 E2 protein increases its DNA-binding capacity and reverses its transcriptional regulatory activity on the viral early promoter. BMC Mol. Biol. 2012, 13, 5. [Google Scholar] [CrossRef]
- DeSmet, M.; Kanginakudru, S.; Rietz, A.; Wu, W.H.; Roden, R.; Androphy, E.J. The Replicative Consequences of Papillomavirus E2 Protein Binding to the Origin Replication Factor ORC2. PLoS Pathog. 2016, 12, e1005934. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE). Available online: https://www.chemcomp.com/en/Products.htm (accessed on 24 November 2024).
- Maybridge HitFinder screening library. Available online: http://www.chem.maybridge.com/hitfinder/ (accessed on 24 November 2024).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.; DeLano, W. PyMOL 2020. Available online: http://www.pymol.org/pymol (accessed on 3 November 2024).
- RCSB Protein Data Bank.1DTO. Available online: https://www.rcsb.org/3d-view/1DTO/1 (accessed on 24 November 2024).
- Hegde, R.S.; Androphy, E.J. Crystal structure of the E2 DNA-binding domain from human papillomavirus type 16: Implications for its DNA binding-site selection mechanism. J. Mol. Biol. 1998, 284, 1479–1489. [Google Scholar] [CrossRef]
- Ferguson, M.K.; Botchan, M.R. Genetic analysis of the activation domain of bovine papillomavirus protein E2: Its role in transcription and replication. J. Virol. 1996, 70, 4193–4199. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Nishimura, A.; Tanaka, M.; Ueno, T.; Ishimoto, A.; Sakai, H. Modulation of the cell division cycle by human papillomavirus type 18 E4. J. Virol. 2002, 76, 10914–10920. [Google Scholar] [CrossRef]
- Cooper, C.S.; Upmeyer, S.N.; Winokur, P.L. Identification of single amino acids in the human papillomavirus 11 E2 protein critical for the transactivation or replication functions. Virology 1998, 241, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Kasukawa, H.; Howley, P.M.; Benson, J.D. A fifteen-amino-acid peptide inhibits human papillomavirus E1-E2 interaction and human papillomavirus DNA replication in vitro. J. Virol. 1998, 72, 8166–8173. [Google Scholar] [CrossRef]
- White, P.W.; Titolo, S.; Brault, K.; Thauvette, L.; Pelletier, A.; Welchner, E.; Bourgon, L.; Doyon, L.; Ogilvie, W.W.; Yoakim, C.; et al. Inhibition of human papillomavirus DNA replication by small molecule antagonists of the E1-E2 protein interaction. J. Biol. Chem. 2003, 278, 26765–26772. [Google Scholar] [CrossRef]
- Hernandez-Ramon, E.E.; Burns, J.E.; Zhang, W.; Walker, H.F.; Allen, S.; Antson, A.A.; Maitland, N.J. Dimerization of the human papillomavirus type 16 E2 N terminus results in DNA looping within the upstream regulatory region. J. Virol. 2008, 82, 4853–4861. [Google Scholar] [CrossRef]
- Abbate, E.A.; Berger, J.M.; Botchan, M.R. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes. Dev. 2004, 18, 1981–1996. [Google Scholar] [CrossRef]
- Davidson, W.; McGibbon, G.A.; White, P.W.; Yoakim, C.; Hopkins, J.L.; Guse, I.; Hambly, D.M.; Frego, L.; Ogilvie, W.W.; Lavallée, P.; et al. Characterization of the binding site for inhibitors of the HPV11 E1-E2 protein interaction on the E2 transactivation domain by photoaffinity labeling and mass spectrometry. Anal. Chem. 2004, 76, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- White, P.W.; Faucher, A.M.; Goudreau, N. Small molecule inhibitors of the human papillomavirus E1-E2 interaction. Curr. Top. Microbiol. Immunol. 2011, 348, 61–88. [Google Scholar] [CrossRef]
- Wang, Y.; Coulombe, R.; Cameron, D.R.; Thauvette, L.; Massariol, M.J.; Amon, L.M.; Fink, D.; Titolo, S.; Welchner, E.; Yoakim, C.; et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor. J. Biol. Chem. 2004, 279, 6976–6985. [Google Scholar] [CrossRef]
- Soumia, M.; Hajji, H.; El Mzibri, M.; Younes, F.Z.; Mohammed, B.; Mohamed, B.; Benaissa, M. In-Silico Molecular Modeling Studies to Identify Novel Potential Inhibitors of HPV E6 Protein. Vaccines 2022, 10, 1452. [Google Scholar] [CrossRef] [PubMed]
- Pappa, K.I.; Kontostathi, G.; Lygirou, V.; Zoidakis, J.; Anagnou, N.P. Novel structural approaches concerning HPV proteins: Insight into targeted therapies for cervical cancer (Review). Oncol. Rep. 2018, 39, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.; McIntosh, P.; Jackson, D.J.; Sorathia, R.; Miell, M.; Wang, Q.; Khan, J.; Soneji, Y.; Doorbar, J. A novel interaction between the human papillomavirus type 16 E2 and E1--E4 proteins leads to stabilization of E2. Virology 2009, 394, 266–275. [Google Scholar] [CrossRef]
- Breiner, B.; Preuss, L.; Roos, N.; Conrady, M.; Lilie, H.; Iftner, T.; Simon, C. Refolding and in vitro characterization of human papillomavirus 16 minor capsid protein L2. Biol. Chem. 2019, 400, 513–522. [Google Scholar] [CrossRef]
- Christensen, N.D.; Budgeon, L.R.; Cladel, N.M.; Hu, J. Recent advances in preclinical model systems for papillomaviruses. Virus Res. 2017, 231, 108–118. [Google Scholar] [CrossRef]
- Fradet-Turcotte, A.; Morin, G.; Lehoux, M.; Bullock, P.A.; Archambault, J. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology 2010, 399, 65–76. [Google Scholar] [CrossRef]
- Myers, J.E.; Guidry, J.T.; Scott, M.L.; Zwolinska, K.; Raikhy, G.; Prasai, K. Detecting episomal or integrated human papillomavirus 16 DNA using an exonuclease V-qPCR-based assay. Virology 2019, 537, 149–156. [Google Scholar] [CrossRef]
- Porter, S.S.; McBride, A.A. Human Papillomavirus Production and Infection of Primary Human Keratinocytes. Curr. Protoc. Microbiol. 2020, 57, e101. [Google Scholar] [CrossRef] [PubMed]
- Gissmann, L. HPV vaccines: Preclinical development. Arch. Med. Res. 2009, 40, 466–470. [Google Scholar] [CrossRef]
- Sable, R.; Jois, S. Surfing the Protein-Protein Interaction Surface Using Docking Methods: Application to the Design of PPI Inhibitors. Molecules 2015, 20, 11569–11603. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.H.; Christoffer, C.W.; Kihara, D. In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods 2017, 131, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Valkov, E.; Sharpe, T.; Marsh, M.; Greive, S.; Hyvönen, M. Targeting protein-protein interactions and fragment-based drug discovery. Top. Curr. Chem. 2012, 317, 145–179. [Google Scholar] [CrossRef]
- Rosell, M.; Fernández-Recio, J. Docking-based identification of small-molecule binding sites at protein-protein interfaces. Comput. Struct. Biotechnol. J. 2020, 18, 3750–3761. [Google Scholar] [CrossRef]
- Li, H.; Kasam, V.; Tautermann, C.S.; Seeliger, D.; Vaidehi, N. Computational method to identify druggable binding sites that target protein-protein interactions. J. Chem. Inf. Model. 2014, 54, 1391–1400. [Google Scholar] [CrossRef]
- Oliva, R.; Chermak, E.; Cavallo, L. Analysis and Ranking of Protein-Protein Docking Models Using Inter-Residue Contacts and Inter-Molecular Contact Maps. Molecules 2015, 20, 12045–12060. [Google Scholar] [CrossRef]
- Agu, P.C.; Afiukwa, C.A.; Orji, O.U.; Ezeh, E.M.; Ofoke, I.H.; Ogbu, C.O.; Ugwuja, E.I.; Aja, P.M. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep. 2023, 13, 13398. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Chemical Structure | Name | Binding Energy (Kcal/mol) |
|---|---|---|---|
| 11419 | 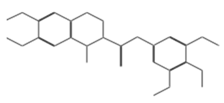 | N2(3,4,5-trimethoxyphenyl)-6,7-dimethoxy-1-methyl-1,2,3. | −14.3274 |
| 11829 |  | Beta-D-glucopyranose | −14.0887 |
| 10756 | 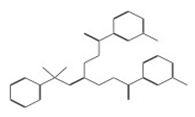 | N1-di[2-(3-chlorobenzoyl)hydrazino]methylene benzene-1-sulfonamide | −13.1464 |
| 11277 |  | N1-{4-methyl-2-[(3,4,5-trimethoxybenzylidene)amino]phenylacetamide | −13.0737 |
| 11667 | 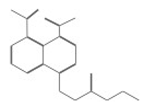 | 4-[(2-ethoxy-2-oxoethyl)thio]naphthalene-1,8-dicarboxylic acid | −12.9282 |
| 11048 |  | 1-(3,4-dimethoxybenzyl)-2-[(2-fluorophenyl)diazenyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline | −12.9047 |
| 11044 |  | N-(2-cyanoethyl)-3,4-dimethoxy-N-(tetrahydrofuran-2-ylmethyl)benzenesulfonamide | −12.8421 |
| 10635 | 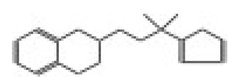 | N-(2,3-dihydro-1,4-benzodioxin-2-ylmethyl)-2-thiophenesulfonamide | −12.8407 |
| 11773 |  | 2-[2-(trifluoromethoxy)phenyl]-5-[3-(trifluoromethyl)phenyl]-1,3,4-oxadiazole | −12.8036 |
| 10708 |  | 2-({2-[(4-methoxy-1-methyl-1H-indazol-3-yl)amino]-2-oxoethyl}sulfinyl)acetic acid | −12.6922 |
| 10167 |  | N1-{2-[(2-cyanoethyl)thio]phenyl}-3,4,5-trimethoxybenzamide | −12.6907 |
| 11091 |  | 2-ethoxyethyl 2-{4-[(3,5-dichloro-2-pyridyl)oxy]phenoxy}propanoate | −12.6658 |
| 11744 | 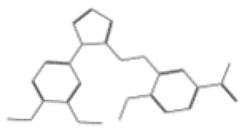 | 1-[3-({[1-(3,4-dimethoxyphenyl)-1H-1,2,3,4-tetrazol-5-yl]thio}methyl)-4-methoxyphenyl]ethanone | −12.6091 |
| 11080 | 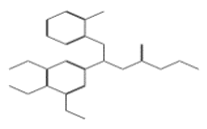 | Ethyl 3-[(2-aminophenyl)thio]-3-(3,4,5-trimethoxyphenyl)propanoate | −12.5635 |
| 10632 |  | N1-(3,4-dimethoxyphenyl)-2-cyano-3-(1H-pyrrol-2-yl)acrylamide | −12.5200 |
| 10006 | 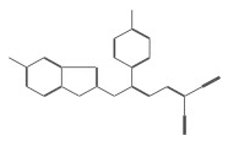 | 4-benzyl-2-({4-[5-(trifluoromethyl)-2-pyridyl]piperazin-1-yl}methyl)morpholine difumarate | −12.4922 |
| 11821 | 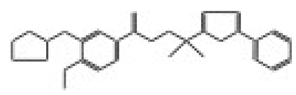 | N’-[3-(cyclopentyloxy)-4-methoxybenzoyl]-5-(2-pyridyl)-2-thiophenesulfonohydrazide | −12.4093 |
| 11987 |  | 4H-1,2,4-triazol-3-yl}sulfinyl}-1-(2-thienyl)-1-ethanone | −12.3364 |
| 10726 | 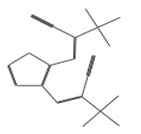 | 3-{2-[2-cyano-2-(methylsulfonyl)vinyl]-3-thienyl}-2-(methylsulfonyl)acrylonitrile | −12.3050 |
| 10007 | 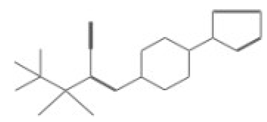 | 2-(tert-butylsulfonyl)-3-[4-(1H-pyrrol-1-yl)piperidino]acrylonitrile | −12.2512 |
| 11385 |  | 4-[3-(benzylamino)butyl]-2-methoxyphenol | −12.1643 |
| 10531 |  | Ethyl 2-methyl-5-(4-methylphenyl)-4-(morpholinosulfonyl)-3-furoate | −12.1102 |
| 11275 | 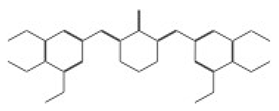 | 3,5-di(3,4,5-trimethoxybenzylidene)tetrahydro-2H-pyran-4-one | −12.0896 |
| 11821 | 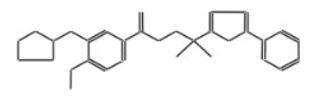 | 3-[2-(2-pyridyl)ethyl]-2-thioxoimidazolidin-4-one | −12.0887 |
| 10745 | 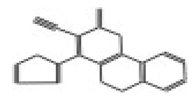 | 4-(2-furyl)-2-oxo-1,5-dihydro-2H-chromeno[4,3-b]pyridine-3-carbonitrile | −12.0696 |
| 11642 | 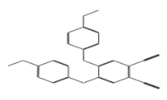 | 4,5-di(4-methoxyphenoxy)pentanedinitrile | −12.0528 |
| 11765 | 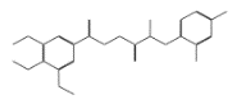 | N’1-[2-(2,4-dichlorophenoxy)propanol]-3,4,5-trimethoxybenzene-1-carbohydrazide | −12.0513 |
| 11688 |  | 4-(1,3-benzodioxol-5-ylmethyl)-N-[4-(trifluoromethoxy)phenyl]tetrahydro-1(2H)-pyrazinecarboxamide | −12.0467 |
| 11277 |  | N1-{4-methyl-2-[(3,4,5-trimethoxybenzylidene)amino]phenyl}acetamide | −12.0318 |
| 10711 |  | 2-[(carboxymethyl)(methyl)amino]-5-methoxybenzoic acid | −12.0258 |
| 10026 | 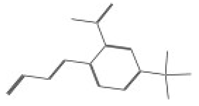 | 3-[2-nitro-4-(trifluoromethyl)phenyl]acrylonitrile | −12.0242 |
| No. | ID | Molecular Weight g/mol | Hydrogen Donors | Hydrogen Acceptors | log P |
|---|---|---|---|---|---|
| 1 | 11419 | 432.5380 | 1 | 7 | 2.8000 |
| 2 | 11829 | 458.4170 | 4 | 10 | 0.4600 |
| 3 | 10756 | 506.3680 | 4 | 9 | 3.7000 |
| 4 | 10708 | 309.3450 | 2 | 7 | 1.2700 |
| 5 | 10632 | 325.3660 | 2 | 6 | 1.0700 |
| 6 | 10726 | 342.4190 | 0 | 6 | 0.0000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandara-Mireles, J.A.; Loera Castañeda, V.; Grijalva Ávila, J.C.; Villanueva Fierro, I.; Muñoz, C.M.; Payan Gándara, H.; Loera Castañeda, G.A.; Patrón Romero, L.; Almanza Reyes, H. In Silico Identification of Potential Antagonists Targeting the HPV16 E2-E1 Interaction: A Step Toward Novel Therapeutics for Cervical Cancer. Curr. Issues Mol. Biol. 2025, 47, 288. https://doi.org/10.3390/cimb47040288
Gandara-Mireles JA, Loera Castañeda V, Grijalva Ávila JC, Villanueva Fierro I, Muñoz CM, Payan Gándara H, Loera Castañeda GA, Patrón Romero L, Almanza Reyes H. In Silico Identification of Potential Antagonists Targeting the HPV16 E2-E1 Interaction: A Step Toward Novel Therapeutics for Cervical Cancer. Current Issues in Molecular Biology. 2025; 47(4):288. https://doi.org/10.3390/cimb47040288
Chicago/Turabian StyleGandara-Mireles, Jesús Alonso, Verónica Loera Castañeda, Julio Cesar Grijalva Ávila, Ignacio Villanueva Fierro, Cynthia Mora Muñoz, Hugo Payan Gándara, Guadalupe Antonio Loera Castañeda, Leslie Patrón Romero, and Horacio Almanza Reyes. 2025. "In Silico Identification of Potential Antagonists Targeting the HPV16 E2-E1 Interaction: A Step Toward Novel Therapeutics for Cervical Cancer" Current Issues in Molecular Biology 47, no. 4: 288. https://doi.org/10.3390/cimb47040288
APA StyleGandara-Mireles, J. A., Loera Castañeda, V., Grijalva Ávila, J. C., Villanueva Fierro, I., Muñoz, C. M., Payan Gándara, H., Loera Castañeda, G. A., Patrón Romero, L., & Almanza Reyes, H. (2025). In Silico Identification of Potential Antagonists Targeting the HPV16 E2-E1 Interaction: A Step Toward Novel Therapeutics for Cervical Cancer. Current Issues in Molecular Biology, 47(4), 288. https://doi.org/10.3390/cimb47040288