
The Mechanical Role of YAP/TAZ in the Development of Diabetic Cardiomyopathy

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
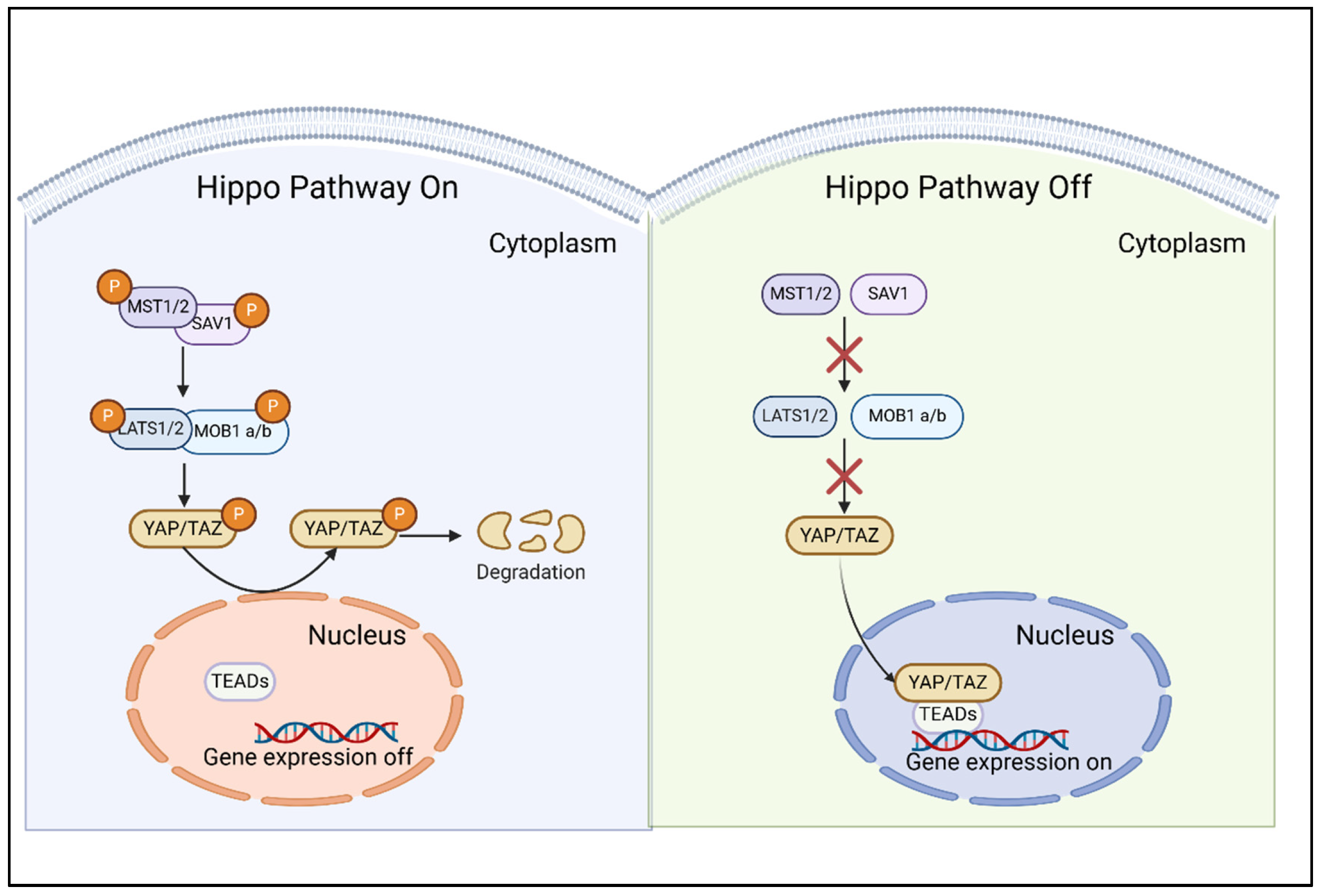
2. Characteristics of the Hippo/YAP Pathway
2.1. Composition and Intricate Regulation of the Hippo/YAP Pathway
2.2. YAP/TAZ: The Molecular Effectors and Mechanic-Transducers of the Hippo Pathway
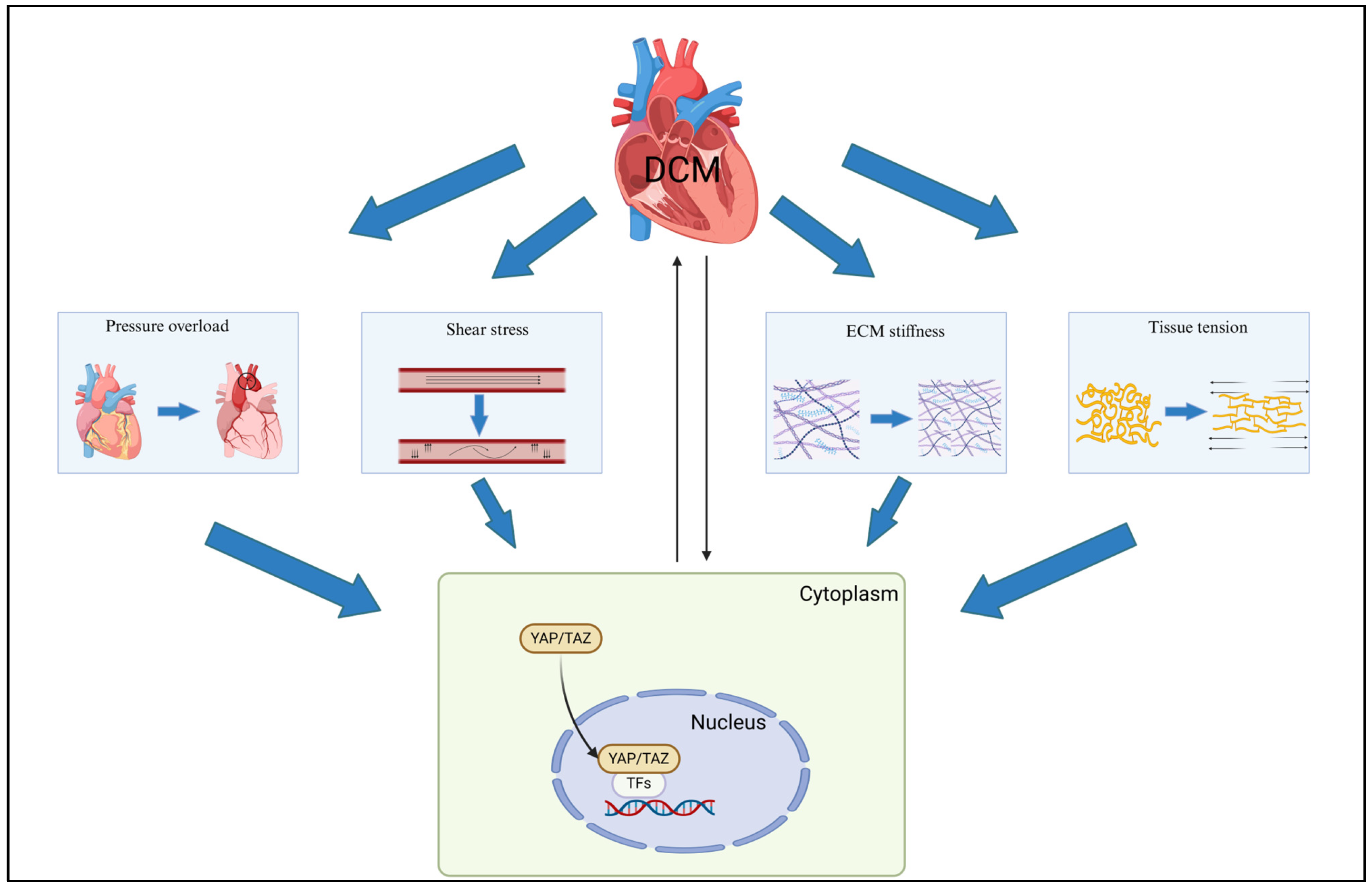
3. Mechanical Cues in Diabetic Cardiomyopathy
4. YAP/TAZ Mechanotransduction
4.1. Extracellular Matrix Properties
4.2. Pressure Overload and Hydrostatic Pressure
4.3. Shear Stress
4.4. Pathological Stretch and Tissue Tension
5. YAP/TAZ: Bridging the Interrelationship Between Diabetic Cardiomyopathy and Mechanotransduction
5.1. Cytoskeleton Protein
5.2. Extracellular Matrix Remodeling
6. YAP/TAZ in the Pathological Mechanisms of Diabetic Cardiomyopathy
6.1. Cardiac Fibrosis
6.2. Cardiac Hypertrophy
6.3. Myocardial Inflammation
6.4. Programmed Cell Death
6.5. Mitochondrial Dysfunction
7. YAP/TAZ: A Promising Therapeutic Target for Diabetic Cardiomyopathy
8. Conclusions and Prospect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DCM | Diabetic cardiomyopathy |
| HF | Heart failure |
| HFrEF | Heart Failure with Reduced Ejection Fraction |
| HFpEF | Heart Failure with Preserved Ejection Fraction |
| CKD | Chronic kidney disease |
| CAD | Coronary artery disease |
| ESC | European Society of Cardiology |
| LV | Left ventricular |
| ECM | Extracellular matrix |
| AGEs | Advanced glycation end products |
| YAP | Yes-associated protein |
| TAZ | Transcriptional co-activator with PDZ binding motif |
| TEADs | TEA domain transcription factor family |
| MST1/2 | Mammalian Ste20-like protein kinases 1/2 |
| SAV1 | Salvador family WW domain-containing protein 1 |
| LATS1/2 | Large tumor suppressor kinases 1/2 |
| MOB1a/b | MOB kinase activator 1A/B |
| CMR | Cardiac magnetic resonance |
| PO | Pressure overload |
| HFD | High-fat diet |
| TAC | Transverse aortic constriction |
| LVP | Left ventricular pressure |
| VSMCs | Vascular smooth muscle cells |
| MCs | Mesangial cells |
| ECs | Endothelial cells |
| CFs | Cardiac fibroblasts |
| F-actin | Filamentous actin |
| IFs | Intermediate filaments |
| STZ | Streptozotocin |
| α-SMA | α-smooth muscle actin |
| TRPV4 | Transient receptor potential vanilloid 4 |
| PCD | Programmed cell death |
| ECT | Engineered cardiac tissue |
| DRP1 | Dynamin-related protein 1 |
References
- Xu, Y.; Lu, J.; Li, M.; Wang, T.; Wang, K.; Cao, Q.; Ding, Y.; Xiang, Y.; Wang, S.; Yang, Q.; et al. Diabetes in China part 1: Epidemiology and risk factors. Lancet Public Health 2024, 9, e1089–e1097. [Google Scholar] [CrossRef]
- Pop-Busui, R.; Januzzi, J.L.; Bruemmer, D.; Butalia, S.; Green, J.B.; Horton, W.B.; Knight, C.; Levi, M.; Rasouli, N.; Richardson, C.R. Heart Failure: An Underappreciated Complication of Diabetes. A Consensus Report of the American Diabetes Association. Diabetes Care 2022, 45, 1670–1690. [Google Scholar] [CrossRef] [PubMed]
- Marwick, T.H.; Ritchie, R.; Shaw, J.E.; Kaye, D. Implications of Underlying Mechanisms for the Recognition and Management of Diabetic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 339–351. [Google Scholar] [CrossRef]
- Seferović, P.M.; Paulus, W.J.; Rosano, G.; Polovina, M.; Petrie, M.C.; Jhund, P.S.; Tschöpe, C.; Sattar, N.; Piepoli, M.; Papp, Z.; et al. Diabetic myocardial disorder. A clinical consensus statement of the Heart Failure Association of the ESC and the ESC Working Group on Myocardial & Pericardial Diseases. Eur. J. Heart Fail. 2024, 26, 1893–1903. [Google Scholar]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Januzzi, J.L.; Del Prato, S.; Rosenstock, J.; Butler, J.; Ezekowitz, J.; Ibrahim, N.E.; Lam, C.S.P.; Marwick, T.; Wilson Tang, W.H.; Liu, Y.; et al. Characterizing diabetic cardiomyopathy: Baseline results from the ARISE-HF trial. Cardiovasc. Diabetol. 2024, 23, 49. [Google Scholar] [CrossRef]
- Saucerman, J.J.; Tan, P.M.; Buchholz, K.S.; McCulloch, A.D.; Omens, J.H. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat. Rev. Cardiol. 2019, 16, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef]
- Wang, Y.; Chatterjee, E.; Li, G.; Xu, J.; Xiao, J. Force-sensing protein expression in response to cardiovascular mechanotransduction. EBioMedicine 2024, 110, 105412. [Google Scholar] [CrossRef]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Moya, I.M.; Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Driskill, J.H.; Pan, D. Control of stem cell renewal and fate by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 2023, 24, 895–911. [Google Scholar] [CrossRef]
- Maugeri-Sacca, M.; De Maria, R. The Hippo pathway in normal development and cancer. Pharmacol. Ther. 2018, 186, 60–72. [Google Scholar] [CrossRef]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef]
- Romanelli, G.; Villarreal, L.; Espasandin, C.; Benech, J.C. Diabetes induces modifications in costameric proteins and increases cardiomyocyte stiffness. Am. J. Physiol. Cell Physiol. 2024, 327, C1263–C1273. [Google Scholar] [CrossRef]
- Zhu, T.; Ye, Z.; Song, J.; Zhang, J.; Zhao, Y.; Xu, F.; Wang, J.; Huang, X.; Gao, B.; Li, F. Effect of extracellular matrix stiffness on efficacy of Dapagliflozin for diabetic cardiomyopathy. Cardiovasc. Diabetol. 2024, 23, 273. [Google Scholar] [CrossRef]
- Pofi, R.; Giannetta, E.; Galea, N.; Francone, M.; Campolo, F.; Barbagallo, F.; Gianfrilli, D.; Venneri, M.A.; Filardi, T.; Cristini, C.; et al. Diabetic Cardiomiopathy Progression is Triggered by miR122-5p and Involves Extracellular Matrix: A 5-Year Prospective Study. JACC Cardiovasc. Imaging 2021, 14, 1130–1142. [Google Scholar] [CrossRef]
- Shang, Y.; Zhang, X.; Leng, W.; Chen, L.; Lei, X.; Zhang, T.; Greiser, A.; Liang, Z.; Wang, J. Assessment of Diabetic Cardiomyopathy by Cardiovascular Magnetic Resonance T1 Mapping: Correlation with Left-Ventricular Diastolic Dysfunction and Diabetic Duration. J. Diabetes Res. 2017, 2017, 9584278. [Google Scholar] [CrossRef]
- Heegaard, B.; Deis, T.; Rossing, K.; Ersboll, M.; Kistorp, C.; Gustafsson, F. Diabetes mellitus and hemodynamics in advanced heart failure. Int. J. Cardiol. 2023, 379, 60–65. [Google Scholar] [CrossRef]
- Yeo, J.L.; Gulsin, G.S.; Brady, E.M.; Dattani, A.; Bilak, J.M.; Marsh, A.M.; Sian, M.; Athithan, L.; Parke, K.S.; Wormleighton, J.; et al. Association of ambulatory blood pressure with coronary microvascular and cardiac dysfunction in asymptomatic type 2 diabetes. Cardiovasc. Diabetol. 2022, 21, 85. [Google Scholar] [CrossRef]
- Ikeda, S.; Mukai, R.; Mizushima, W.; Zhai, P.; Oka, S.I.; Nakamura, M.; Del Re, D.P.; Sciarretta, S.; Hsu, C.P.; Shimokawa, H.; et al. Yes-Associated Protein (YAP) Facilitates Pressure Overload-Induced Dysfunction in the Diabetic Heart. JACC Basic. Transl. Sci. 2019, 4, 611–622. [Google Scholar] [CrossRef]
- Akhtar, M.S.; Pillai, K.K.; Hassan, M.Q.; Dhyani, N.; Ismail, M.V.; Najmi, A.K. Levosimendan reduces myocardial damage and improves cardiodynamics in streptozotocin induced diabetic cardiomyopathy via SERCA2a/NCX1 pathway. Life Sci. 2016, 153, 55–65. [Google Scholar] [CrossRef]
- Swiatlowska, P.; Tipping, W.; Marhuenda, E.; Severi, P.; Fomin, V.; Yang, Z.; Xiao, Q.; Graham, D.; Shanahan, C.; Iskratsch, T. Hypertensive Pressure Mechanosensing Alone Triggers Lipid Droplet Accumulation and Transdifferentiation of Vascular Smooth Muscle Cells to Foam Cells. Adv. Sci. (Weinh.) 2024, 11, e2308686. [Google Scholar] [CrossRef]
- Zheng, D.; Ma, J.; Yu, Y.; Li, M.; Ni, R.; Wang, G.; Chen, R.; Li, J.; Fan, G.C.; Lacefield, J.C.; et al. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia 2015, 58, 1949–1958. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Cooper-White, J.; Janmey, P.A.; Mooney, D.J.; Shenoy, V.B. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 2020, 584, 535–546. [Google Scholar] [CrossRef]
- Chen, G.; Deng, Y.; Xia, B.; Lv, Y. In Situ Regulation and Mechanisms of 3D Matrix Stiffness on the Activation and Reversion of Hepatic Stellate Cells. Adv. Healthc. Mater. 2023, 12, e2202560. [Google Scholar] [CrossRef]
- Cheng, B.; Li, M.; Wan, W.; Guo, H.; Genin, G.M.; Lin, M.; Xu, F. Predicting YAP/TAZ nuclear translocation in response to ECM mechanosensing. Biophys. J. 2023, 122, 43–53. [Google Scholar] [CrossRef]
- Fan, W.; Adebowale, K.; Vancza, L.; Li, Y.; Rabbi, M.F.; Kunimoto, K.; Chen, D.; Mozes, G.; Chiu, D.K.; Li, Y.; et al. Matrix viscoelasticity promotes liver cancer progression in the pre-cirrhotic liver. Nature 2024, 626, 635–642. [Google Scholar] [CrossRef]
- Ngai, D.; Mohabeer, A.L.; Mao, A.; Lino, M.; Bendeck, M.P. Stiffness-responsive feedback autoregulation of DDR1 expression is mediated by a DDR1-YAP/TAZ axis. Matrix Biol. 2022, 110, 129–140. [Google Scholar] [CrossRef]
- Park, M.; Jin, J.; An, D.Y.; Kim, D.H.; Lee, J.; Yun, J.W.; Hwang, I.; Park, J.S.; Kim, M.K.; Lee, Y.M.; et al. Targeting YAP Activity and Glutamine Metabolism Cooperatively Suppresses Tumor Progression by Preventing Extracellular Matrix Accumulation. Cancer Res. 2024, 84, 3388–3401. [Google Scholar] [CrossRef]
- Choi, S.; Hong, S.P.; Bae, J.H.; Suh, S.H.; Bae, H.; Kang, K.P.; Lee, H.J.; Koh, G.Y. Hyperactivation of YAP/TAZ Drives Alterations in Mesangial Cells through Stabilization of N-Myc in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2023, 34, 809–828. [Google Scholar] [CrossRef]
- Ikeda, S.; Mizushima, W.; Sciarretta, S.; Abdellatif, M.; Zhai, P.; Mukai, R.; Fefelova, N.; Oka, S.I.; Nakamura, M.; Del Re, D.P.; et al. Hippo Deficiency Leads to Cardiac Dysfunction Accompanied by Cardiomyocyte Dedifferentiation During Pressure Overload. Circ. Res. 2019, 124, 292–305. [Google Scholar] [CrossRef]
- Al-Nuaimi, D.A.; Rütsche, D.; Abukar, A.; Hiebert, P.; Zanetti, D.; Cesarovic, N.; Falk, V.; Werner, S.; Mazza, E.; Giampietro, C. Hydrostatic pressure drives sprouting angiogenesis via adherens junction remodelling and YAP signalling. Commun. Biol. 2024, 7, 940. [Google Scholar] [CrossRef]
- Park, J.; Jia, S.; Salter, D.; Bagnaninchi, P.; Hansen, C.G. The Hippo pathway drives the cellular response to hydrostatic pressure. EMBO J. 2022, 41, e108719. [Google Scholar] [CrossRef]
- Wang, Y.; Bai, B.; Hu, Y.; Wang, H.; Liu, N.; Li, Y.; Li, P.; Zhou, G.; Zhou, Q. Hydrostatic Pressure Modulates Intervertebral Disc Cell Survival and Extracellular Matrix Homeostasis via Regulating Hippo-YAP/TAZ Pathway. Stem Cells Int. 2021, 2021, 5626487. [Google Scholar] [CrossRef]
- Wang, K.C.; Yeh, Y.T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.L.; Li, Y.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef]
- Wang, L.; Luo, J.Y.; Li, B.; Tian, X.Y.; Chen, L.J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef]
- Kim, O.H.; Choi, Y.W.; Park, J.H.; Hong, S.A.; Hong, M.; Chang, I.H.; Lee, H.J. Fluid shear stress facilitates prostate cancer metastasis through Piezo1-Src-YAP axis. Life Sci. 2022, 308, 120936. [Google Scholar] [CrossRef]
- Solis, A.G.; Bielecki, P.; Steach, H.R.; Sharma, L.; Harman, C.C.D.; Yun, S.; de Zoete, M.R.; Warnock, J.N.; To, S.D.F.; York, A.G.; et al. Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity. Nature 2019, 573, 69–74. [Google Scholar] [CrossRef]
- Cai, X.; Wang, K.C.; Meng, Z. Mechanoregulation of YAP and TAZ in Cellular Homeostasis and Disease Progression. Front. Cell Dev. Biol. 2021, 9, 673599. [Google Scholar] [CrossRef]
- Byun, J.; Del Re, D.P.; Zhai, P.; Ikeda, S.; Shirakabe, A.; Mizushima, W.; Miyamoto, S.; Brown, J.H.; Sadoshima, J. Yes-associated protein (YAP) mediates adaptive cardiac hypertrophy in response to pressure overload. J. Biol. Chem. 2019, 294, 3603–3617. [Google Scholar] [CrossRef]
- Meng, X.; Zhu, Y.; Tan, H.; Daraqel, B.; Ming, Y.; Li, X.; Yang, G.; He, X.; Song, J.; Zheng, L. The cytoskeleton dynamics-dependent LINC complex in periodontal ligament stem cells transmits mechanical stress to the nuclear envelope and promotes YAP nuclear translocation. Stem Cell Res. Ther. 2024, 15, 284. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Lee, J.H.; Ahn, K.S.; Shim, H.W.; Yoon, J.Y.; Hyun, J.; Lee, J.H.; Jang, S.; Yoo, K.H.; Jang, Y.K.; et al. Cyclic Stretch Promotes Cellular Reprogramming Process through Cytoskeletal-Nuclear Mechano-Coupling and Epigenetic Modification. Adv. Sci. (Weinh.) 2023, 10, e2303395. [Google Scholar] [CrossRef]
- Wen, S.M.; Wen, W.C.; Chao, P.G. Zyxin and actin structure confer anisotropic YAP mechanotransduction. Acta Biomater. 2022, 152, 313–320. [Google Scholar] [CrossRef]
- Perestrelo, A.R.; Silva, A.C.; Oliver-De La Cruz, J.; Martino, F.; Horvath, V.; Caluori, G.; Polansky, O.; Vinarsky, V.; Azzato, G.; de Marco, G.; et al. Multiscale Analysis of Extracellular Matrix Remodeling in the Failing Heart. Circ. Res. 2021, 128, 24–38. [Google Scholar] [CrossRef]
- Niu, L.; Jia, Y.; Wu, M.; Liu, H.; Feng, Y.; Hu, Y.; Zhang, X.; Gao, D.; Xu, F.; Huang, G. Matrix stiffness controls cardiac fibroblast activation through regulating YAP via AT(1) R. J. Cell Physiol. 2020, 235, 8345–8357. [Google Scholar] [CrossRef] [PubMed]
- Moujaber, O.; Stochaj, U. The Cytoskeleton as Regulator of Cell Signaling Pathways. Trends Biochem. Sci. 2020, 45, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Seetharaman, S.; Etienne-Manneville, S. Cytoskeletal Crosstalk in Cell Migration. Trends Cell Biol. 2020, 30, 720–735. [Google Scholar] [CrossRef]
- Li, H.; Liu, Y.; Zhang, J.; Cai, M.; Cao, Z.; Gao, J.; Xu, H.; Shao, L.; Sun, J.; Shi, Y.; et al. Quantification of mechanical stimuli inducing nucleoplasmic translocation of YAP and its distribution mechanism using an AFM-dSTORM coupled technique. Nanoscale 2022, 14, 15516–15524. [Google Scholar] [CrossRef]
- Nikolajevic Starcevic, J.; Janic, M.; Sabovic, M. Molecular Mechanisms Responsible for Diastolic Dysfunction in Diabetes Mellitus Patients. Int. J. Mol. Sci. 2019, 20, 1197. [Google Scholar] [CrossRef]
- Benech, J.C.; Benech, N.; Zambrana, A.I.; Rauschert, I.; Bervejillo, V.; Oddone, N.; Damian, J.P. Diabetes increases stiffness of live cardiomyocytes measured by atomic force microscopy nanoindentation. Am. J. Physiol. Cell Physiol. 2014, 307, C910–C919. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, B.; Eggers, B.; Mattern, Y.; de Carvalho, T.S.; Marcus-Alic, K.; Tschoepe, D. Maladaptive response following glucose overload in GLUT4-overexpressing H9C2 cardiomyoblasts. Diabetes Obes. Metab. 2024, 26, 2379–2389. [Google Scholar] [CrossRef]
- Viola, H.; Johnstone, V.; Cserne Szappanos, H.; Richman, T.; Tsoutsman, T.; Filipovska, A.; Semsarian, C.; Hool, L. The L-type Ca(2+) channel facilitates abnormal metabolic activity in the cTnI-G203S mouse model of hypertrophic cardiomyopathy. J. Physiol. 2016, 594, 4051–4070. [Google Scholar] [CrossRef]
- Lee, J.Y.; Dominguez, A.A.; Nam, S.; Stowers, R.S.; Qi, L.S.; Chaudhuri, O. Identification of cell context-dependent YAP-associated proteins reveals beta(1) and beta(4) integrin mediate YAP translocation independently of cell spreading. Sci. Rep. 2019, 9, 17188. [Google Scholar]
- Wu, Y.; Li, L.; Li, W.; Li, N.; Zhang, X.; Zheng, L.; Zhong, S.; Lu, S.; Shu, X.; Zhou, J.; et al. Stretch-induced hepatic endothelial mechanocrine promotes hepatocyte proliferation. Hepatology 2024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, R.; Wang, Y.; Guo, S.; Wu, Z.; Liu, J.; Han, C.; Qiu, C.; Deng, G. Extracellular matrix stiffness mediates uterine repair via the Rap1a/ARHGAP35/RhoA/F-actin/YAP axis. Cell Commun. Signal. 2023, 21, 22. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhan, L.; Zhou, Q.Y.; Zhang, L.L.; Chen, X.M.; Hu, X.M.; Yuan, X.C. SIRT2 regulates microtubule stabilization in diabetic cardiomyopathy. Eur. J. Pharmacol. 2015, 764, 554–561. [Google Scholar] [CrossRef]
- Caporizzo, M.A.; Chen, C.Y.; Bedi, K.; Margulies, K.B.; Prosser, B.L. Microtubules Increase Diastolic Stiffness in Failing Human Cardiomyocytes and Myocardium. Circulation 2020, 141, 902–915. [Google Scholar] [CrossRef]
- Caporizzo, M.A.; Prosser, B.L. The microtubule cytoskeleton in cardiac mechanics and heart failure. Nat. Rev. Cardiol. 2022, 19, 364–378. [Google Scholar] [CrossRef]
- You, E.; Huh, Y.H.; Kwon, A.; Kim, S.H.; Chae, I.H.; Lee, O.J.; Ryu, J.H.; Park, M.H.; Kim, G.E.; Lee, J.S.; et al. SPIN90 Depletion and Microtubule Acetylation Mediate Stromal Fibroblast Activation in Breast Cancer Progression. Cancer Res. 2017, 77, 4710–4722. [Google Scholar] [CrossRef]
- Sousa, S.C.; Aroso, M.; Bessa, R.; Verissimo, E.; Ferreira da Silva, T.; Lopes, C.D.F.; Brites, P.; Vieira, J.; Vieira, C.P.; Aguiar, P.C.; et al. Stretch triggers microtubule stabilization and MARCKS-dependent membrane incorporation in the shaft of embryonic axons. Curr. Biol. 2024, 34, 4577–4588.e8. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Restle, D.; Janmey, P.A. Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys. J. 2014, 107, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Dawood, A.F.; Alzamil, N.M.; Hewett, P.W.; Momenah, M.A.; Dallak, M.; Kamar, S.S.; Abdel Kader, D.H.; Yassin, H.; Haidara, M.A.; Maarouf, A.; et al. Metformin Protects against Diabetic Cardiomyopathy: An Association between Desmin-Sarcomere Injury and the iNOS/mTOR/TIMP-1 Fibrosis Axis. Biomedicines 2022, 10, 984. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, B.; Engelbrecht, B.; Espelage, B.C.; Klusmeier, N.; Tiemann, J.; Gawlowski, T.; Mattern, Y.; Eisenacher, M.; Meyer, H.E.; Rabbani, N.; et al. Glyoxalase 1-knockdown in human aortic endothelial cells—Effect on the proteome and endothelial function estimates. Sci. Rep. 2016, 6, 37737. [Google Scholar] [CrossRef]
- Xie, J.; Hu, X.; Chen, L.; Piruska, A.; Zheng, Z.; Bao, M.; Huck, W.T.S. The Effect of Geometry and TGF-beta Signaling on Tumor Cell Migration from Free-Standing Microtissues. Adv. Healthc. Mater. 2022, 11, e2102696. [Google Scholar] [CrossRef]
- Atale, N.; Yadav, D.; Rani, V.; Jin, J.O. Pathophysiology, Clinical Characteristics of Diabetic Cardiomyopathy: Therapeutic Potential of Natural Polyphenols. Front. Nutr. 2020, 7, 564352. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, Y.; Zheng, R.; Xiong, Z.; Zhu, Z.; Gao, F.; Man, W.; Duan, Y.; Lin, J.; Zhang, X.; et al. Fibroblast-specific activation of Rnd3 protects against cardiac remodeling in diabetic cardiomyopathy via suppression of Notch and TGF-beta signaling. Theranostics 2022, 12, 7250–7266. [Google Scholar] [CrossRef]
- Widiapradja, A.; Kasparian, A.O.; McCaffrey, S.L.; Kolb, L.L.; Imig, J.D.; Lacey, J.L.; Melendez, G.C.; Levick, S.P. Replacement of Lost Substance P Reduces Fibrosis in the Diabetic Heart by Preventing Adverse Fibroblast and Macrophage Phenotype Changes. Cells 2021, 10, 2659. [Google Scholar] [CrossRef]
- Hutchinson, K.R.; Lord, C.K.; West, T.A.; Stewart, J.A., Jr. Cardiac fibroblast-dependent extracellular matrix accumulation is associated with diastolic stiffness in type 2 diabetes. PLoS ONE 2013, 8, e72080. [Google Scholar] [CrossRef]
- Akam-Baxter, E.A.; Bergemann, D.; Ridley, S.J.; To, S.; Andrea, B.; Moon, B.; Ma, H.; Zhou, Y.; Aguirre, A.; Caravan, P.; et al. Dynamics of collagen oxidation and cross linking in regenerating and irreversibly infarcted myocardium. Nat. Commun. 2024, 15, 4648. [Google Scholar] [CrossRef]
- Pan, K.-L.; Hsu, Y.-C.; Chang, S.-T.; Chung, C.-M.; Lin, C.-L. The Role of Cardiac Fibrosis in Diabetic Cardiomyopathy: From Pathophysiology to Clinical Diagnostic Tools. Int. J. Mol. Sci. 2023, 24, 8604. [Google Scholar] [CrossRef]
- Ebrahimighaei, R.; Sala-Newby, G.B.; Hudson, C.; Kimura, T.E.; Hathway, T.; Hawkins, J.; McNeill, M.C.; Richardson, R.; Newby, A.C.; Bond, M. Combined role for YAP-TEAD and YAP-RUNX2 signalling in substrate-stiffness regulation of cardiac fibroblast proliferation. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119329. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.C.; Abolghasemzade, S.; McKee, B.P.; Singh, I.; Pendyala, K.; Mohajeri, M.; Patel, H.; Shaji, A.; Kersey, A.L.; Harsh, K.; et al. Matrix stiffness drives drop like nuclear deformation and lamin A/C tension-dependent YAP nuclear localization. Nat. Commun. 2024, 15, 10151. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Xu, K.; He, Y.; Tao, B.; Yuan, Z.; Li, K.; Li, X.; Xia, Z.; Cai, K. A dynamic matrix potentiates mesenchymal stromal cell paracrine function via an effective mechanical dose. Biomater. Sci. 2020, 8, 4779–4791. [Google Scholar] [CrossRef]
- Li, X.; Liu, S.; Han, S.; Sun, Q.; Yang, J.; Zhang, Y.; Jiang, Y.; Wang, X.; Li, Q.; Wang, J. Dynamic Stiffening Hydrogel with Instructive Stiffening Timing Modulates Stem Cell Fate In Vitro and Enhances Bone Remodeling In Vivo. Adv. Healthc. Mater. 2023, 12, e2300326. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, X.; Tang, S.; Xue, L.; Wang, J.; Zhang, X. Extended preconditioning on soft matrices directs human mesenchymal stem cell fate via YAP transcriptional activity and chromatin organization. APL Bioeng. 2023, 7, 016110. [Google Scholar] [CrossRef]
- Huerta-Lopez, C.; Clemente-Manteca, A.; Velazquez-Carreras, D.; Espinosa, F.M.; Sanchez, J.G.; Martinez-Del-Pozo, A.; Garcia-Garcia, M.; Martin-Colomo, S.; Rodriguez-Blanco, A.; Esteban-Gonzalez, R.; et al. Cell response to extracellular matrix viscous energy dissipation outweighs high-rigidity sensing. Sci Adv. 2024, 10, eadf9758. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Gupta, A.; Najibi, A.J.; Seo, B.R.; Garry, R.; Tringides, C.M.; de Lazaro, I.; Darnell, M.; Gu, W.; Zhou, Q.; et al. Matrix viscoelasticity controls spatiotemporal tissue organization. Nat. Mater. 2023, 22, 117–127. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Yu, S.; Dong, X.; Yang, M.; Yu, Q.; Xiong, J.; Chen, J.; Dong, B.; Su, Q. (Pro)renin receptor involves in myocardial fibrosis and oxidative stress in diabetic cardiomyopathy via the PRR-YAP pathway. Sci. Rep. 2021, 11, 3259. [Google Scholar] [CrossRef]
- Hu, M.; Wang, H.; Li, S.; Yan, F.; Fu, C.; Li, L.; Yu, Y.; Xiong, J.; Dong, B. Yes-Associated Protein is Involved in Myocardial Fibrosis in Rats with Diabetic Cardiomyopathy. Diabetes Metab. Syndr. Obes. 2021, 14, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Ruan, S.; Li, J.; Lei, S.; Zhang, S.; Xu, D.; Zuo, A.; Li, L.; Guo, Y. Knockout of C1q/tumor necrosis factor-related protein-9 aggravates cardiac fibrosis in diabetic mice by regulating YAP-mediated autophagy. Front. Pharmacol. 2024, 15, 1407883. [Google Scholar] [CrossRef]
- Long, Y.; Niu, Y.; Liang, K.; Du, Y. Mechanical communication in fibrosis progression. Trends Cell Biol. 2022, 32, 70–90. [Google Scholar] [CrossRef]
- He, M.; Feng, L.; Chen, Y.; Gao, B.; Du, Y.; Zhou, L.; Li, F.; Liu, H. Polydatin attenuates tubulointerstitial fibrosis in diabetic kidney disease by inhibiting YAP expression and nuclear translocation. Front. Physiol. 2022, 13, 927794. [Google Scholar] [CrossRef]
- Lu, Q.B.; Ding, Y.; Liu, Y.; Wang, Z.C.; Wu, Y.J.; Niu, K.M.; Li, K.X.; Zhang, J.R.; Sun, H.J. Metrnl ameliorates diabetic cardiomyopathy via inactivation of cGAS/STING signaling dependent on LKB1/AMPK/ULK1-mediated autophagy. J. Adv. Res. 2023, 51, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.Y.; Liu, S.Q.; Zhang, T.; Shi, W.K.; Xing, Y.; Fang, W.X.; Zhang, M.; Chen, M.Y.; Xu, S.C.; Fan, M.Q.; et al. USP28 Serves as a Key Suppressor of Mitochondrial Morphofunctional Defects and Cardiac Dysfunction in the Diabetic Heart. Circulation 2024, 149, 684–706. [Google Scholar] [CrossRef]
- Yue, P.; Zhang, Y.; Liu, L.; Zhou, K.; Xia, S.; Peng, M.; Yan, H.; Tang, X.; Chen, Z.; Zhang, D.; et al. Yap1 modulates cardiomyocyte hypertrophy via impaired mitochondrial biogenesis in response to chronic mechanical stress overload. Theranostics 2022, 12, 7009–7031. [Google Scholar] [CrossRef] [PubMed]
- Kashihara, T.; Mukai, R.; Oka, S.I.; Zhai, P.; Nakada, Y.; Yang, Z.; Mizushima, W.; Nakahara, T.; Warren, J.S.; Abdellatif, M.; et al. YAP mediates compensatory cardiac hypertrophy through aerobic glycolysis in response to pressure overload. J. Clin. Investig. 2022, 132, e150595. [Google Scholar] [CrossRef]
- Adapala, R.K.; Katari, V.; Kanugula, A.K.; Ohanyan, V.; Paruchuri, S.; Thodeti, C.K. Deletion of Endothelial TRPV4 Protects Heart From Pressure Overload-Induced Hypertrophy. Hypertension 2023, 80, 2345–2356. [Google Scholar] [CrossRef]
- Sharma, S.; Goswami, R.; Zhang, D.X.; Rahaman, S.O. TRPV4 regulates matrix stiffness and TGFbeta1-induced epithelial-mesenchymal transition. J. Cell. Mol. Med. 2019, 23, 761–774. [Google Scholar] [CrossRef]
- Fang, T.; Wang, J.; Sun, S.; Deng, X.; Xue, M.; Han, F.; Sun, B.; Chen, L. JinLiDa granules alleviates cardiac hypertrophy and inflammation in diabetic cardiomyopathy by regulating TP53. Phytomedicine 2024, 130, 155659. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, S.; Corinaldesi, C.; Colletti, M.; Di Luigi, L.; Antinozzi, C.; Filardi, T.; Scolletta, S.; Basili, S.; Lenzi, A.; Morano, S.; et al. The phosphodiesterase 5 inhibitor sildenafil decreases the proinflammatory chemokine IL-8 in diabetic cardiomyopathy: In vivo and in vitro evidence. J. Endocrinol. Investig. 2019, 42, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Brady, E.M.; Gulsin, G.S.; Mirkes, E.M.; Parke, K.; Kanagala, P.; Ng, L.L.; Graham-Brown, M.P.M.; Athithan, L.; Henson, J.; Redman, E.; et al. Fibro-inflammatory recovery and type 2 diabetes remission following a low calorie diet but not exercise training: A secondary analysis of the DIASTOLIC randomised controlled trial. Diabet. Med. 2022, 39, e14884. [Google Scholar] [CrossRef]
- Che, H.; Wang, Y.; Li, Y.; Lv, J.; Li, H.; Liu, Y.; Dong, R.; Sun, Y.; Xu, X.; Zhao, J.; et al. Inhibition of microRNA-150-5p alleviates cardiac inflammation and fibrosis via targeting Smad7 in high glucose-treated cardiac fibroblasts. J. Cell. Physiol. 2020, 235, 7769–7779. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Saeed, A.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Guo, W.; Yang, C.; Zou, J.; Yu, T.; Li, M.; He, R.; Chen, K.; Hell, R.C.R.; Gross, E.R.; Zou, X.; et al. Interleukin-1beta polarization in M1 macrophage mediates myocardial fibrosis in diabetes. Int. Immunopharmacol. 2024, 131, 111858. [Google Scholar] [CrossRef]
- Mia, M.M.; Cibi, D.M.; Abdul Ghani, S.A.B.; Song, W.; Tee, N.; Ghosh, S.; Mao, J.; Olson, E.N.; Singh, M.K. YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction. PLoS Biol. 2020, 18, e3000941. [Google Scholar] [CrossRef]
- Meli, V.S.; Atcha, H.; Veerasubramanian, P.K.; Nagalla, R.R.; Luu, T.U.; Chen, E.Y.; Guerrero-Juarez, C.F.; Yamaga, K.; Pandori, W.; Hsieh, J.Y.; et al. YAP-mediated mechanotransduction tunes the macrophage inflammatory response. Sci Adv. 2020, 6, eabb8471. [Google Scholar] [CrossRef]
- Mei, F.; Guo, Y.; Wang, Y.; Zhou, Y.; Heng, B.C.; Xie, M.; Huang, X.; Zhang, S.; Ding, S.; Liu, F.; et al. Matrix stiffness regulates macrophage polarisation via the Piezo1-YAP signalling axis. Cell Prolif. 2024, 57, e13640. [Google Scholar] [CrossRef]
- Yuan, J.; Ofengeim, D. A guide to cell death pathways. Nat. Rev. Mol. Cell Biol. 2024, 25, 379–395. [Google Scholar] [CrossRef]
- Zhou, L.; Sun, J.; Gu, L.; Wang, S.; Yang, T.; Wei, T.; Shan, T.; Wang, H.; Wang, L. Programmed Cell Death: Complex Regulatory Networks in Cardiovascular Disease. Front. Cell Dev. Biol. 2021, 9, 794879. [Google Scholar] [CrossRef] [PubMed]
- Xuan, X.; Zhang, S. Targeting the programmed cell death (PCD) signaling mechanism with natural substances for the treatment of diabetic cardiomyopathy (DCM). Phytother. Res. 2023, 37, 5495–5508. [Google Scholar] [CrossRef]
- Gong, W.; Zhang, S.; Chen, Y.; Shen, J.; Zheng, Y.; Liu, X.; Zhu, M.; Meng, G. Protective role of hydrogen sulfide against diabetic cardiomyopathy via alleviating necroptosis. Free Radic. Biol. Med. 2022, 181, 29–42. [Google Scholar] [CrossRef]
- Liu, R.; Duan, T.; Yu, L.; Tang, Y.; Liu, S.; Wang, C.; Fang, W.J. Acid sphingomyelinase promotes diabetic cardiomyopathy via NADPH oxidase 4 mediated apoptosis. Cardiovasc. Diabetol. 2023, 22, 25. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Xiong, Y.; Xia, Z. Resveratrol ameliorates myocardial ischemia/reperfusion induced necroptosis through inhibition of the Hippo pathway. J. Bioenerg. Biomembr. 2023, 55, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, X.; Jing, H.; Ren, H.; Xu, S.; Guo, M. Trimethyltin induces apoptosis and necroptosis of mouse liver by oxidative stress through YAP phosphorylation. Ecotoxicol. Environ. Saf. 2022, 248, 114327. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e12. [Google Scholar] [CrossRef]
- Kawaue, T.; Yow, I.; Pan, Y.; Le, A.P.; Lou, Y.; Loberas, M.; Shagirov, M.; Teng, X.; Prost, J.; Hiraiwa, T.; et al. Inhomogeneous mechanotransduction defines the spatial pattern of apoptosis-induced compensatory proliferation. Dev. Cell 2023, 58, 267–277.e5. [Google Scholar] [CrossRef]
- Ke, W.; Liao, Z.; Liang, H.; Tong, B.; Song, Y.; Li, G.; Ma, L.; Wang, K.; Feng, X.; Li, S.; et al. Stiff Substrate Induces Nucleus Pulposus Cell Ferroptosis via YAP and N-Cadherin Mediated Mechanotransduction. Adv. Healthc. Mater. 2023, 12, e2300458. [Google Scholar] [CrossRef]
- Chen, Z.; Li, S.; Liu, M.; Yin, M.; Chen, J.; Li, Y.; Li, Q.; Zhou, Y.; Xia, Y.; Chen, A.; et al. Nicorandil alleviates cardiac microvascular ferroptosis in diabetic cardiomyopathy: Role of the mitochondria-localized AMPK-Parkin-ACSL4 signaling pathway. Pharmacol. Res. 2024, 200, 107057. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Zhou, W.; Men, H.; Bao, T.; Sun, Y.; Wang, Q.; Tan, Y.; Keller, B.B.; Tong, Q.; et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm. Sin. B 2022, 12, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, S.; Ouyang, X.; Wang, H.; Li, X.; Yao, Z.; Chen, S.; Fan, C. Glycolipotoxicity conferred tendinopathy through ferroptosis dictation of tendon-derived stem cells by YAP activation. IUBMB Life 2023, 75, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Cui, H.; Liu, Z.; Zhang, H.; Yang, Y.; Chen, X.; Zeng, Z.; Du, L.; Fu, M.; Yang, Z.; et al. Suppression of RCAN1 alleviated lipid accumulation and mitochondrial fission in diabetic cardiomyopathy. Metabolism 2024, 158, 155977. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Su, L.; Wu, K.; Mei, Y.; Liu, Z.; Chen, Y.; Zeng, W.; Xiao, Y.; Zhang, J.; Cai, G.; et al. USP7 promotes cardiometabolic disorders and mitochondrial homeostasis dysfunction in diabetic mice via stabilizing PGC1beta. Pharmacol. Res. 2024, 205, 107235. [Google Scholar] [CrossRef]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef]
- Wu, S.; Lu, Q.; Ding, Y.; Wu, Y.; Qiu, Y.; Wang, P.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Hyperglycemia-Driven Inhibition of AMP-Activated Protein Kinase α2 Induces Diabetic Cardiomyopathy by Promoting Mitochondria-Associated Endoplasmic Reticulum Membranes In Vivo. Circulation 2019, 139, 1913–1936. [Google Scholar] [CrossRef]
- Romani, P.; Benedetti, G.; Cusan, M.; Arboit, M.; Cirillo, C.; Wu, X.; Rouni, G.; Kostourou, V.; Aragona, M.; Giampietro, C.; et al. Mitochondrial mechanotransduction through MIEF1 coordinates the nuclear response to forces. Nat. Cell Biol. 2024, 26, 2046–2060. [Google Scholar] [CrossRef]
- Yu, Y.; Ma, M.; Li, C.; Dang, Q.; Lei, H.; Wang, G.; Su, J.; Li, Y. Irisin mitigates rheumatoid arthritis by suppressing mitochondrial fission via inhibiting YAP-Drp1 signaling pathway. Int. Immunopharmacol. 2024, 127, 111443. [Google Scholar] [CrossRef]
- Shi, C.; Cai, Y.; Li, Y.; Li, Y.; Hu, N.; Ma, S.; Hu, S.; Zhu, P.; Wang, W.; Zhou, H. Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 2018, 14, 59–71. [Google Scholar] [CrossRef]
- Garoffolo, G.; Casaburo, M.; Amadeo, F.; Salvi, M.; Bernava, G.; Piacentini, L.; Chimenti, I.; Zaccagnini, G.; Milcovich, G.; Zuccolo, E.; et al. Reduction of Cardiac Fibrosis by Interference With YAP-Dependent Transactivation. Circ. Res. 2022, 131, 239–257. [Google Scholar] [CrossRef]
- Song, S.; Li, Y.; Xu, Y.; Ma, L.; Pool Pizzi, M.; Jin, J.; Scott, A.W.; Huo, L.; Wang, Y.; Lee, J.H.; et al. Targeting Hippo coactivator YAP1 through BET bromodomain inhibition in esophageal adenocarcinoma. Mol. Oncol. 2020, 14, 1410–1426. [Google Scholar] [CrossRef]
- Mu, J.; Zhang, D.; Tian, Y.; Xie, Z.; Zou, M.H. BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J. Mol. Cell. Cardiol. 2020, 149, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Kim, Y.J.; Choi, S.H.; Lee, C.S.; Yoon, J.S. Yes-Associated Protein Mediates the Transition from Inflammation to Fibrosis in Graves’ Orbitopathy. Thyroid 2023, 33, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Sabe, S.A.; Harris, D.D.; Broadwin, M.; Xu, C.M.; Sabra, M.; Banerjee, D.; Abid, M.R.; Sellke, F.W. Comparative effects of canagliflozin and sitagliptin in chronically ischemic myocardium. Vessel Plus 2024, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Chen, Y.; Li, N.; Yang, X.; Zhou, L.; Li, H.; Wang, T.; Xie, M.; Liu, H. Dapagliflozin delays renal fibrosis in diabetic kidney disease by inhibiting YAP/TAZ activation. Life Sci. 2023, 322, 121671. [Google Scholar] [CrossRef]
- Kang, L.; Yi, J.; Lau, C.W.; He, L.; Chen, Q.; Xu, S.; Li, J.; Xia, Y.; Zhang, Y.; Huang, Y.; et al. AMPK-Dependent YAP Inhibition Mediates the Protective Effect of Metformin against Obesity-Associated Endothelial Dysfunction and Inflammation. Antioxidants 2023, 12, 1681. [Google Scholar] [CrossRef]
- Santos, D.M.; Pantano, L.; Pronzati, G.; Grasberger, P.; Probst, C.K.; Black, K.E.; Spinney, J.J.; Hariri, L.P.; Nichols, R.; Lin, Y.; et al. Screening for YAP Inhibitors Identifies Statins as Modulators of Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 479–492. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef]
- Wang, S.; Tian, C.; Gao, Z.; Zhang, B.; Zhao, L. Research status and trends of the diabetic cardiomyopathy in the past 10 years (2012-2021): A bibliometric analysis. Front. Cardiovasc. Med. 2022, 9, 1018841. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, J.-X.; Zhang, L.; Liu, H.-H.; Zhang, Z.-Y.; Zhao, N.; Zhou, J.-B.; Qian, L.-L.; Wang, R.-X. The Mechanical Role of YAP/TAZ in the Development of Diabetic Cardiomyopathy. Curr. Issues Mol. Biol. 2025, 47, 297. https://doi.org/10.3390/cimb47050297
Shen J-X, Zhang L, Liu H-H, Zhang Z-Y, Zhao N, Zhou J-B, Qian L-L, Wang R-X. The Mechanical Role of YAP/TAZ in the Development of Diabetic Cardiomyopathy. Current Issues in Molecular Biology. 2025; 47(5):297. https://doi.org/10.3390/cimb47050297
Chicago/Turabian StyleShen, Jun-Xian, Ling Zhang, Huan-Huan Liu, Zhen-Ye Zhang, Ning Zhao, Jia-Bin Zhou, Ling-Ling Qian, and Ru-Xing Wang. 2025. "The Mechanical Role of YAP/TAZ in the Development of Diabetic Cardiomyopathy" Current Issues in Molecular Biology 47, no. 5: 297. https://doi.org/10.3390/cimb47050297
APA StyleShen, J.-X., Zhang, L., Liu, H.-H., Zhang, Z.-Y., Zhao, N., Zhou, J.-B., Qian, L.-L., & Wang, R.-X. (2025). The Mechanical Role of YAP/TAZ in the Development of Diabetic Cardiomyopathy. Current Issues in Molecular Biology, 47(5), 297. https://doi.org/10.3390/cimb47050297