
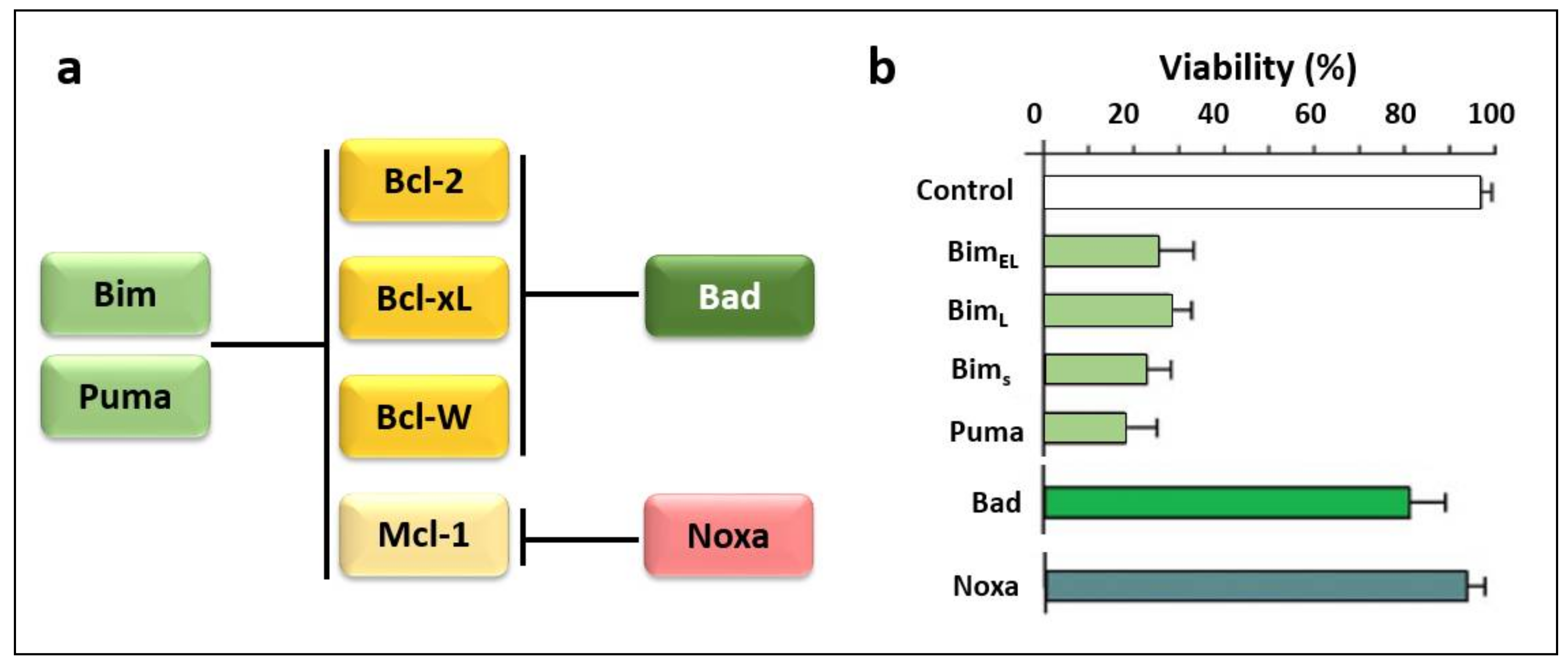
BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
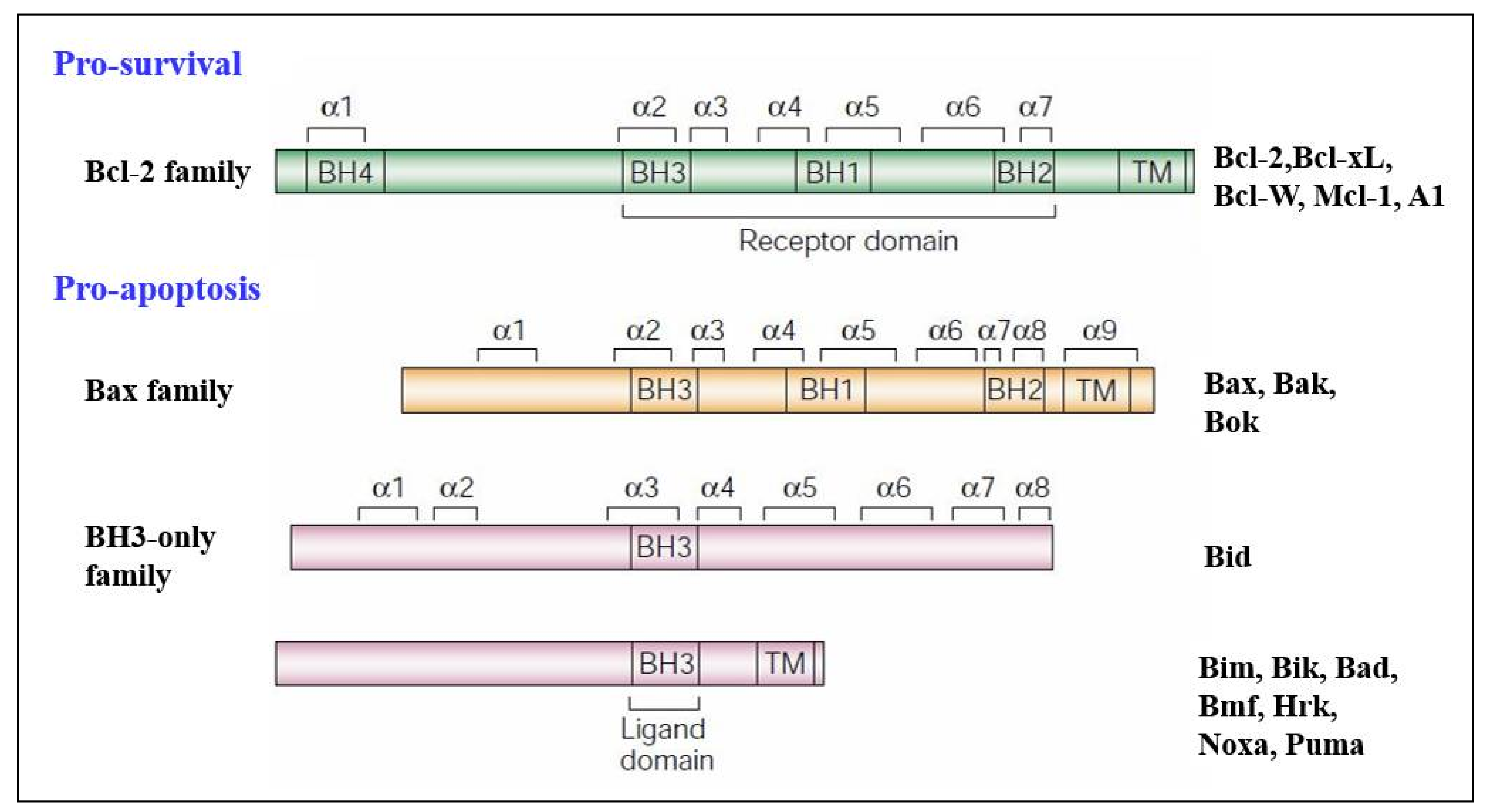
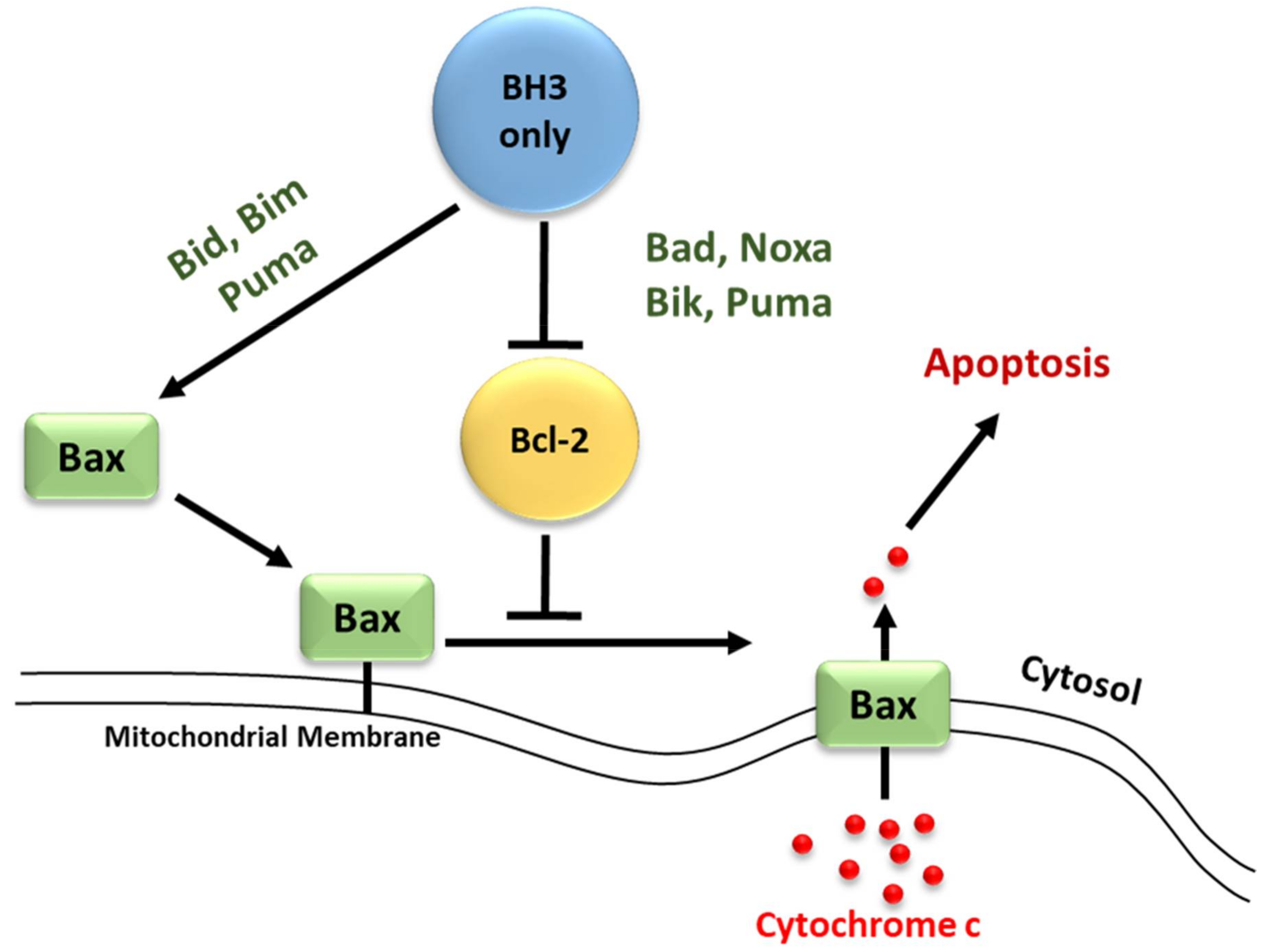
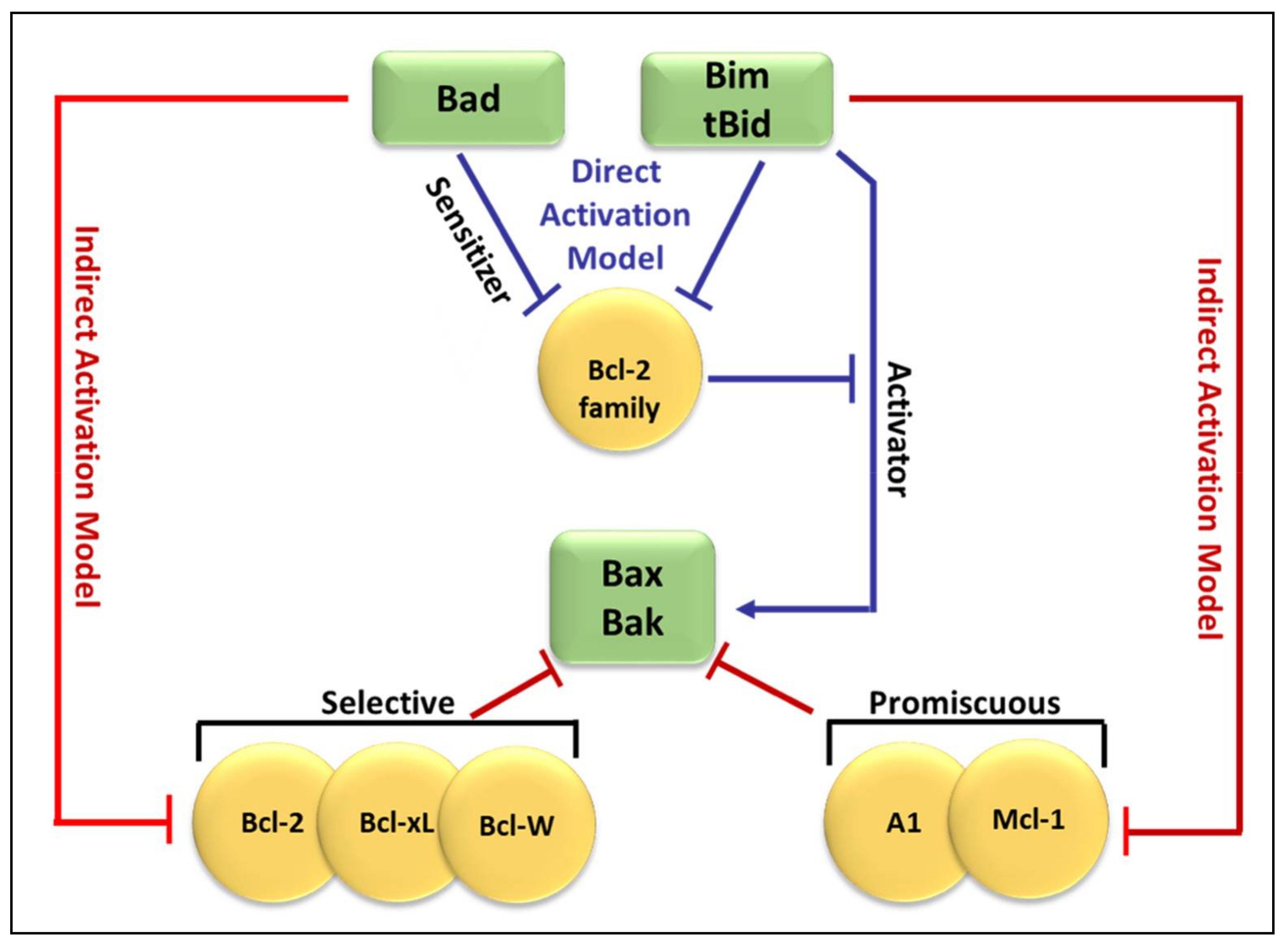
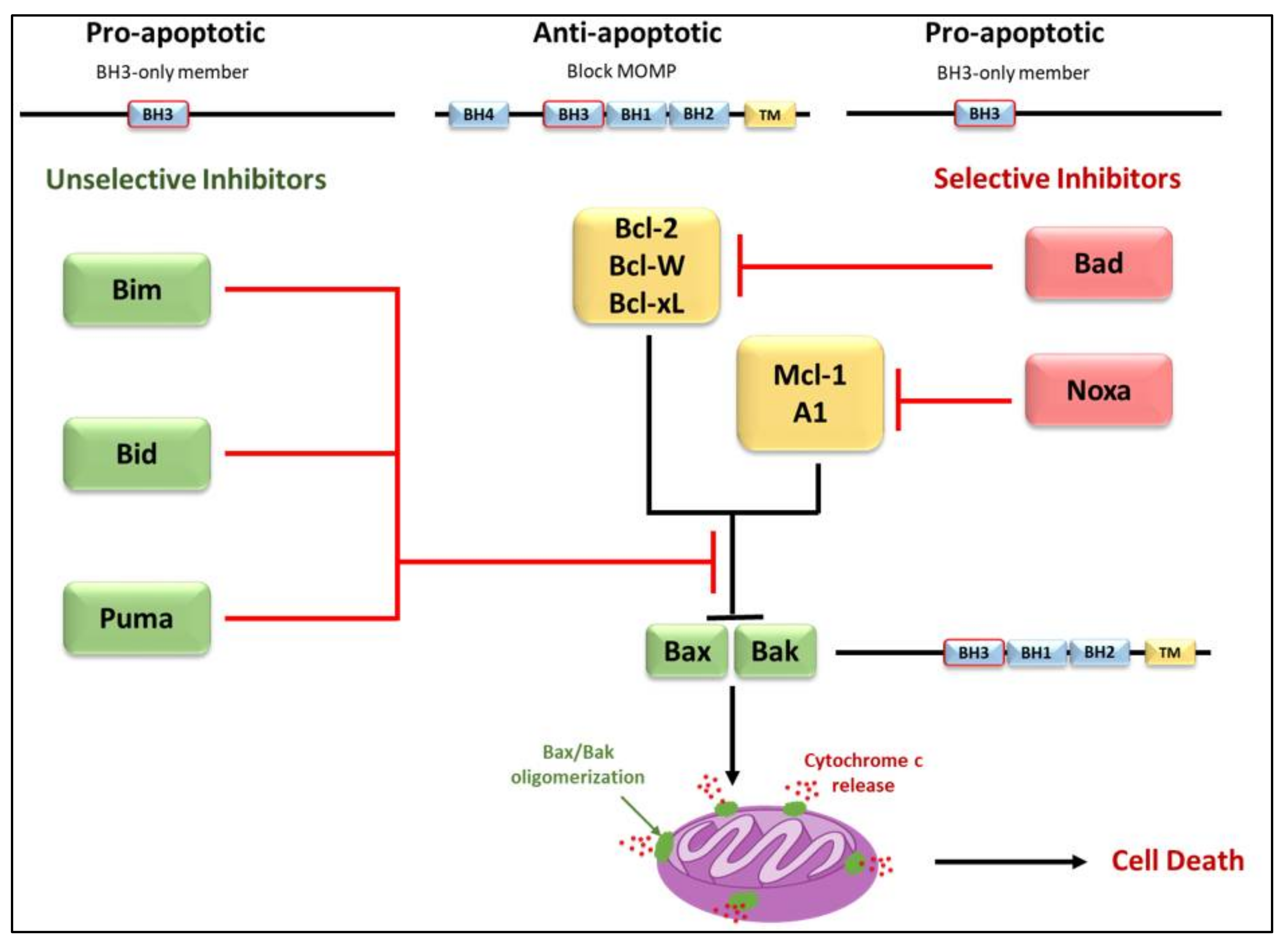
2. Bcl-2 Proteins and the Regulation of Mitochondrial Outer Membrane Permeability
3. Regulation of Apoptosis
3.1. Regulators of Cell Death: Caspases
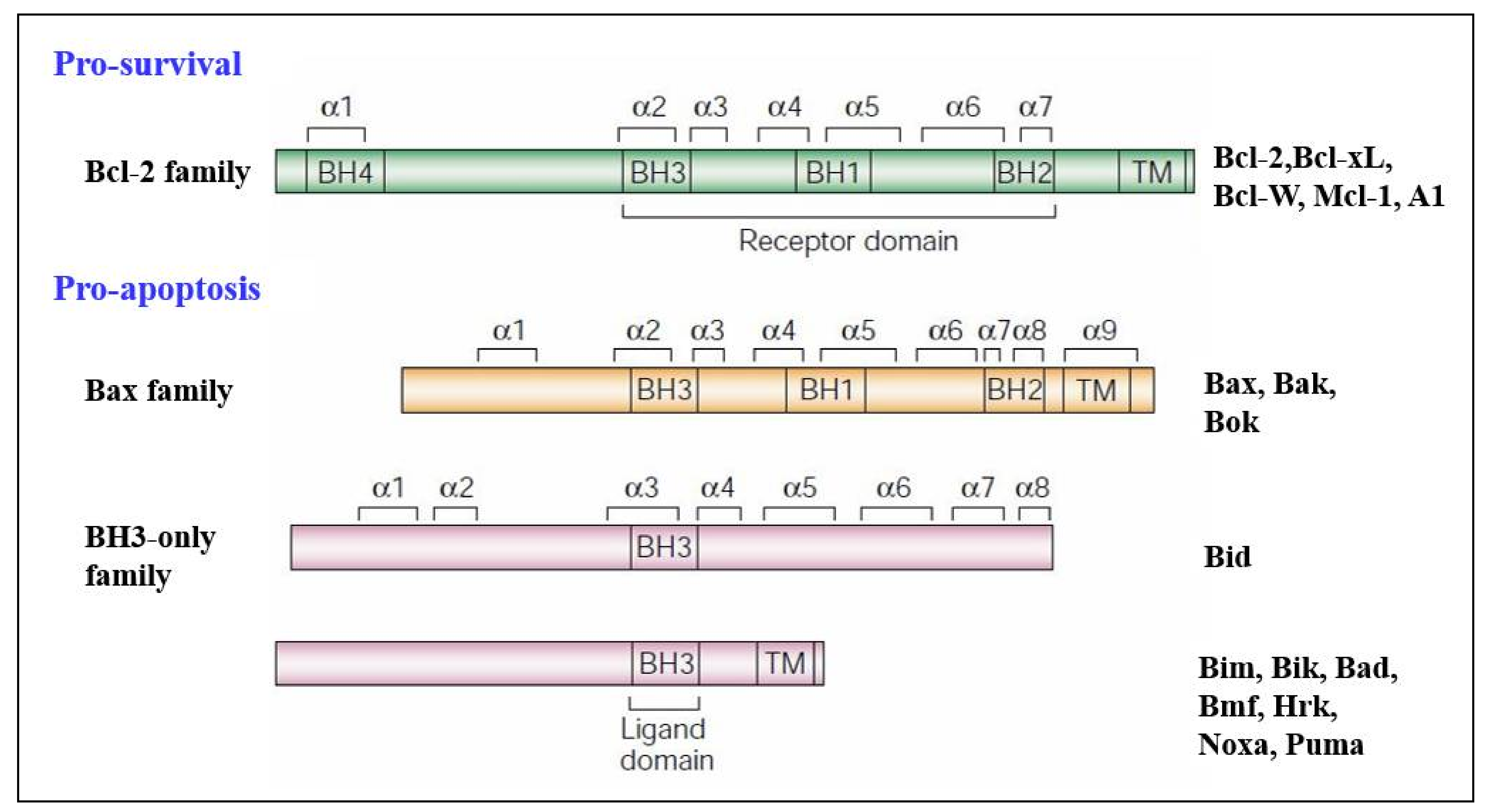
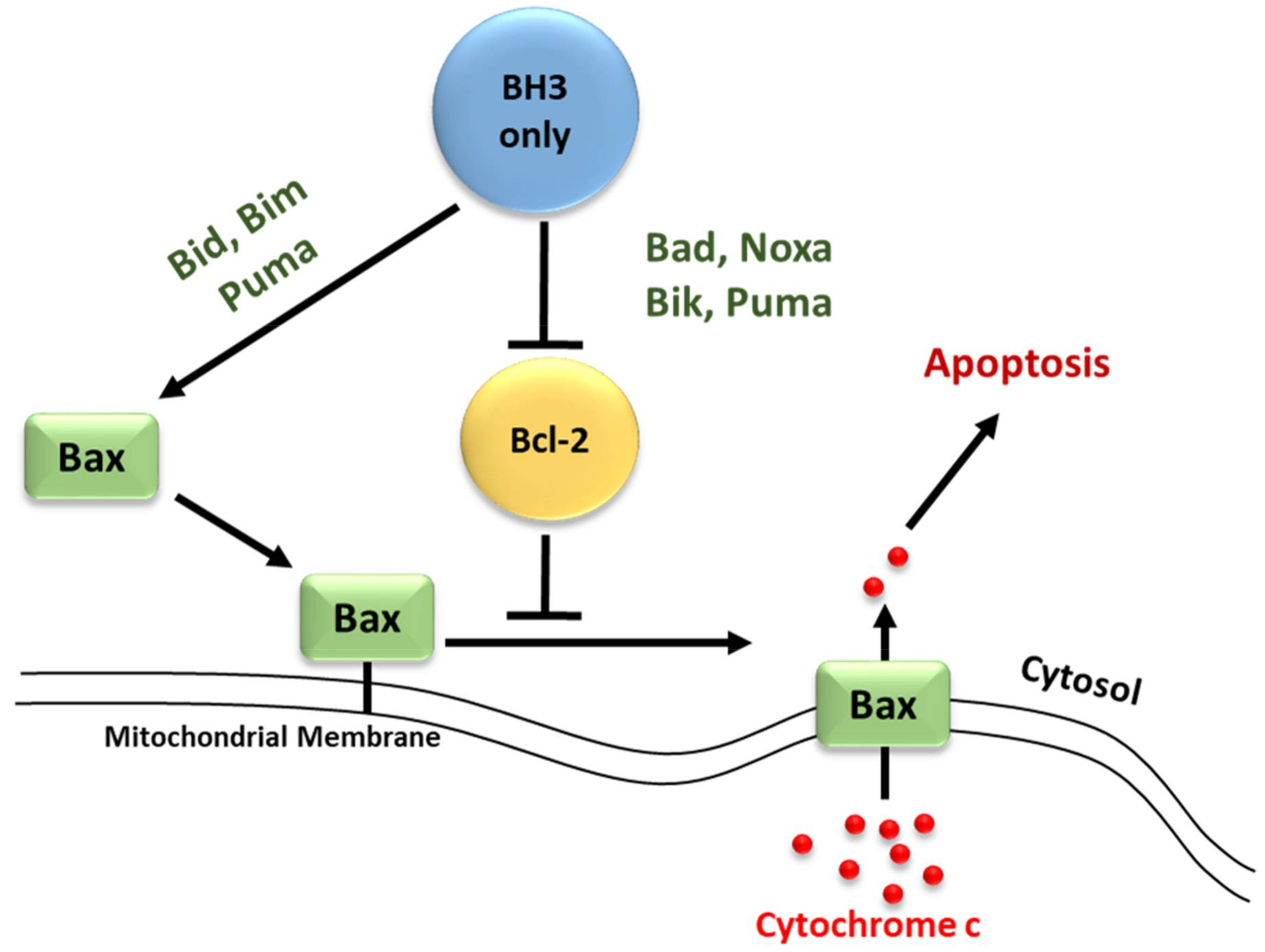
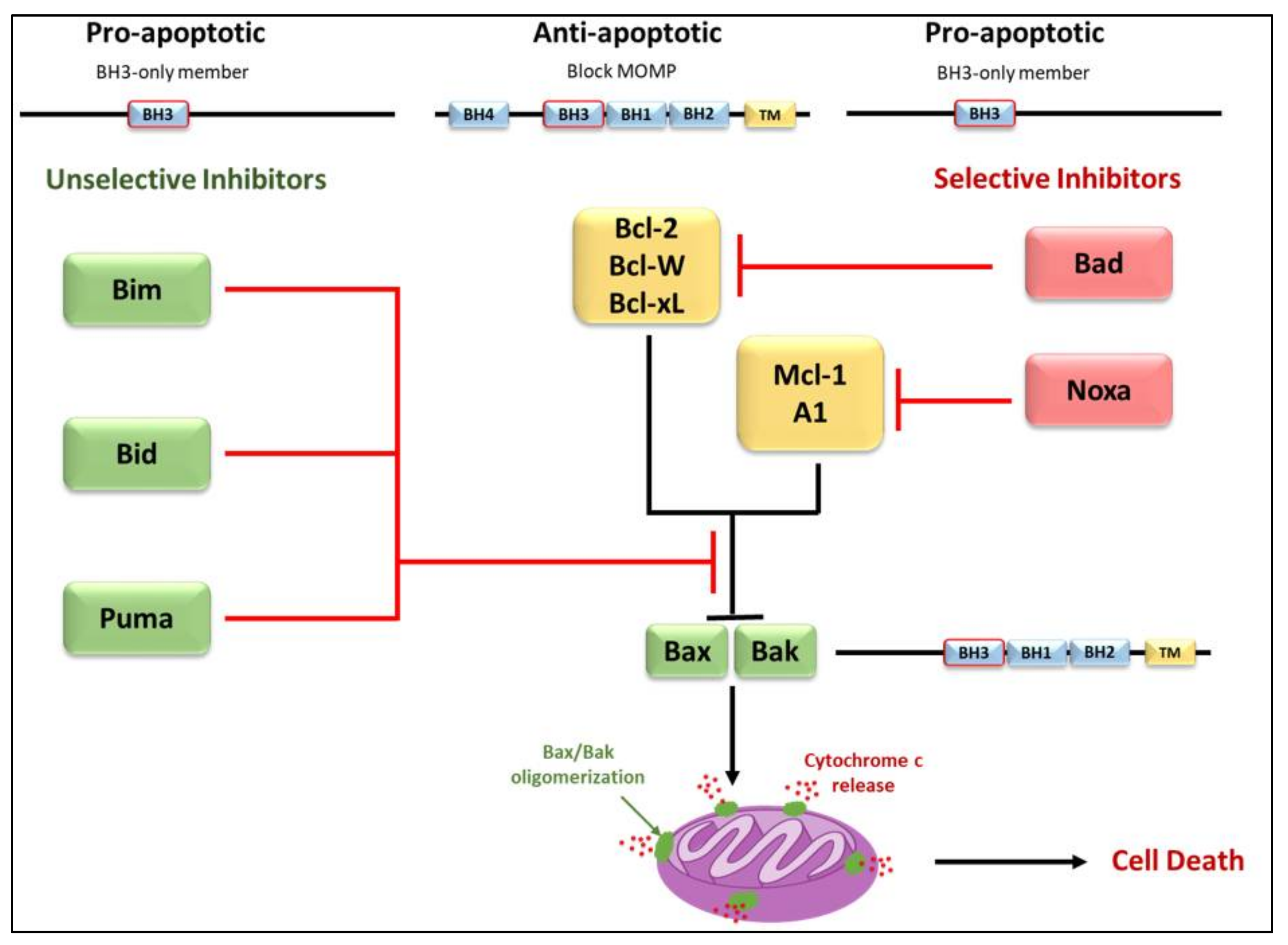
3.2. Regulators of Cell Death: Bcl-2 Family Proteins
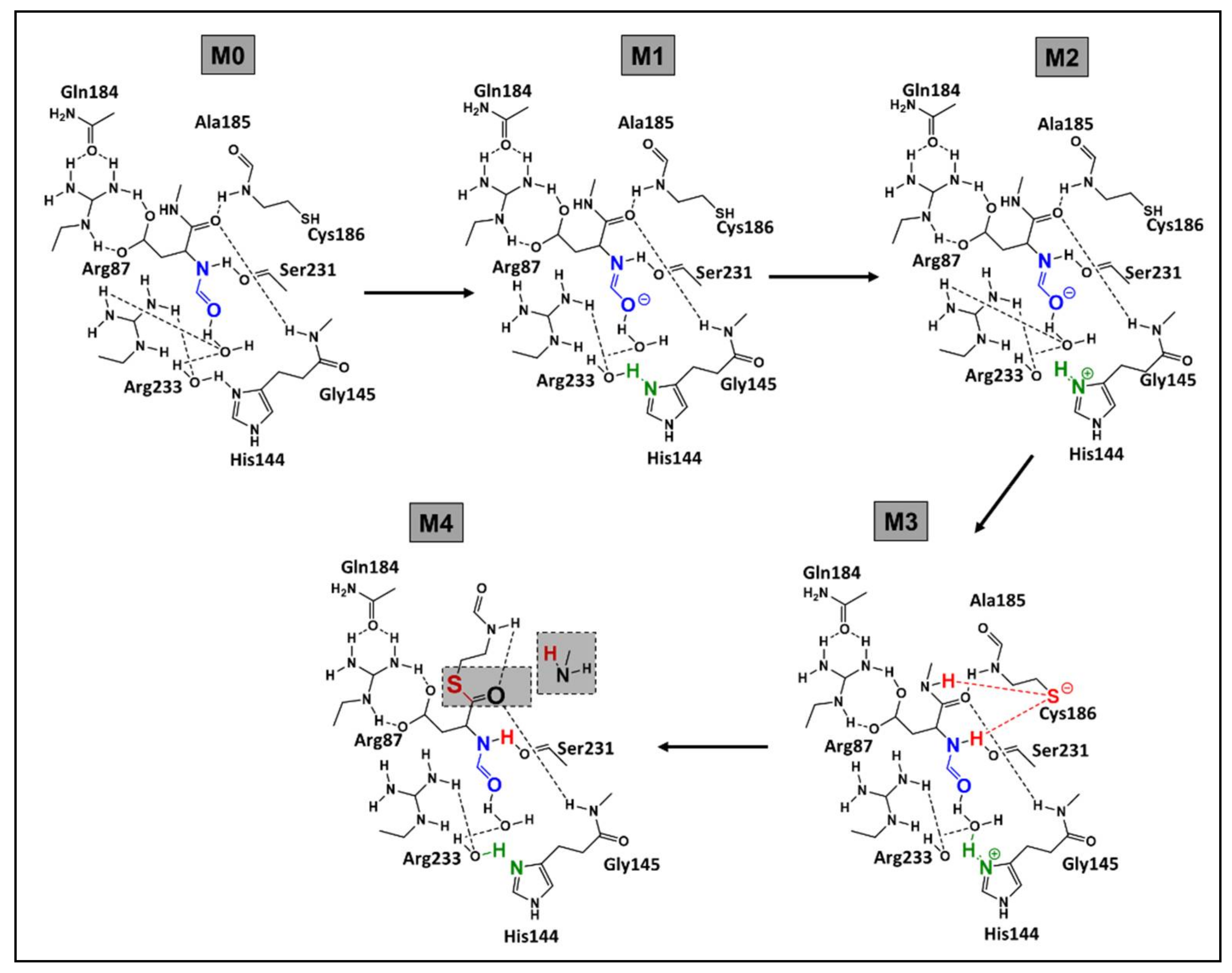
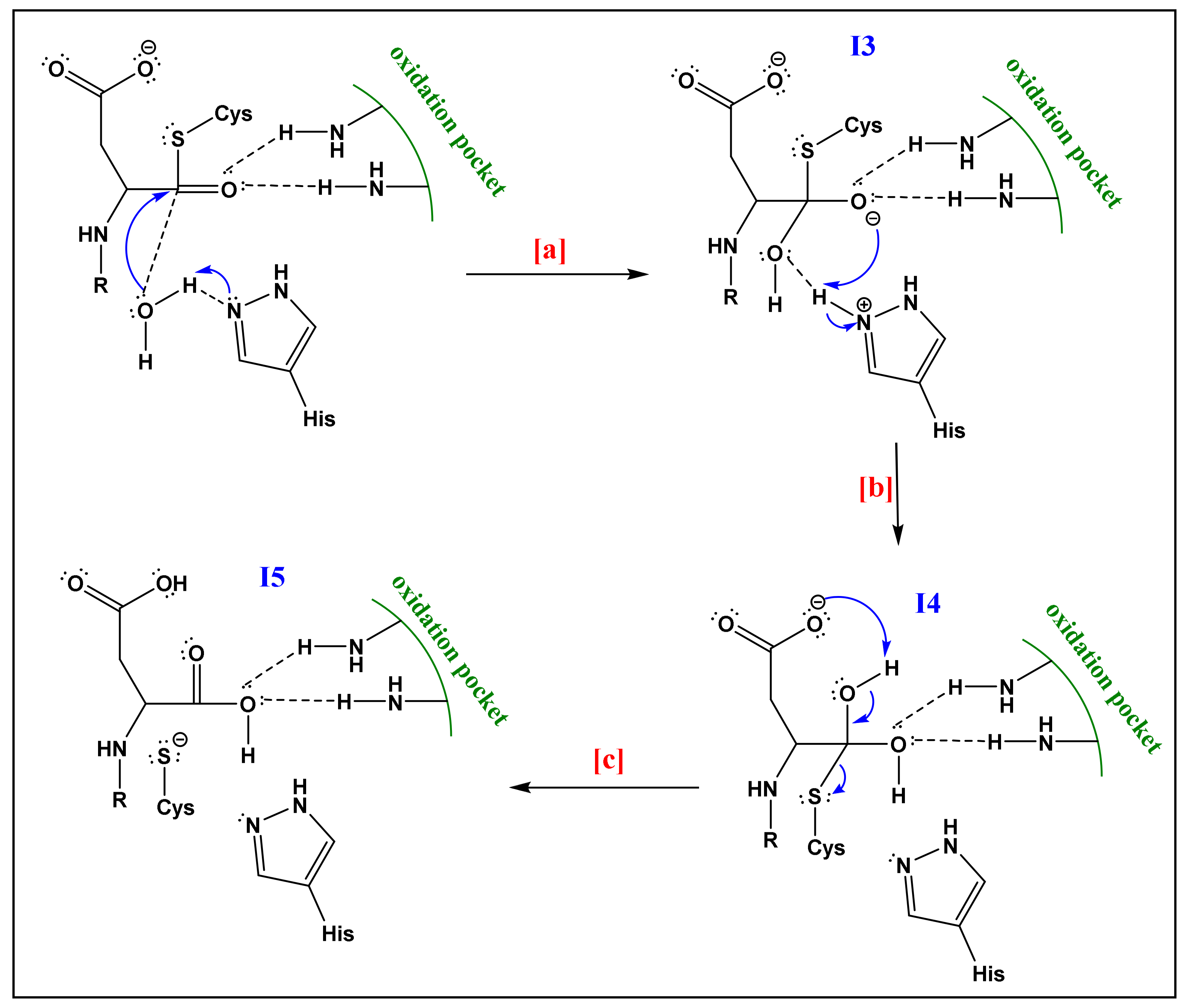
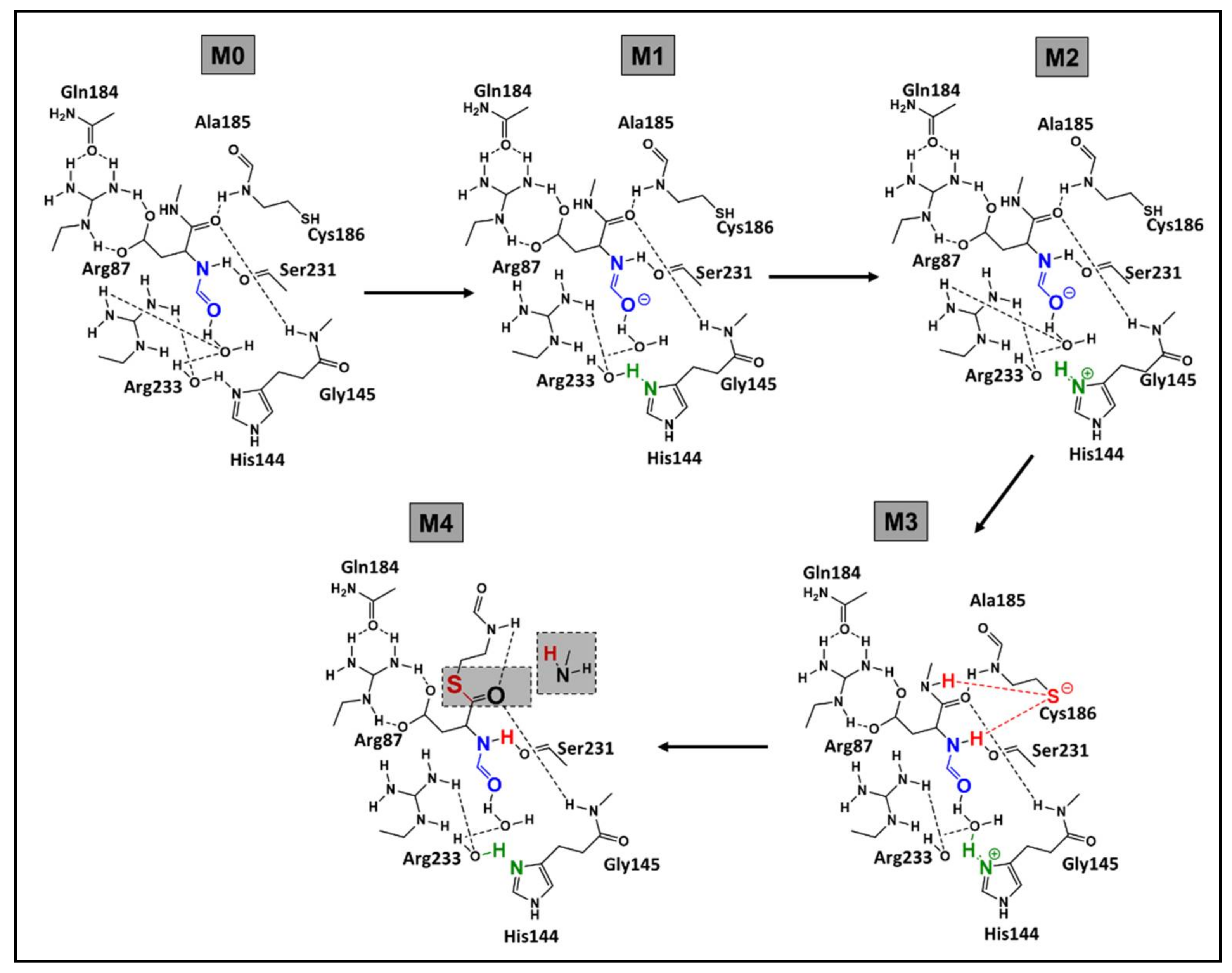
3.3. Elucidation of the Caspases Reaction Mechanism
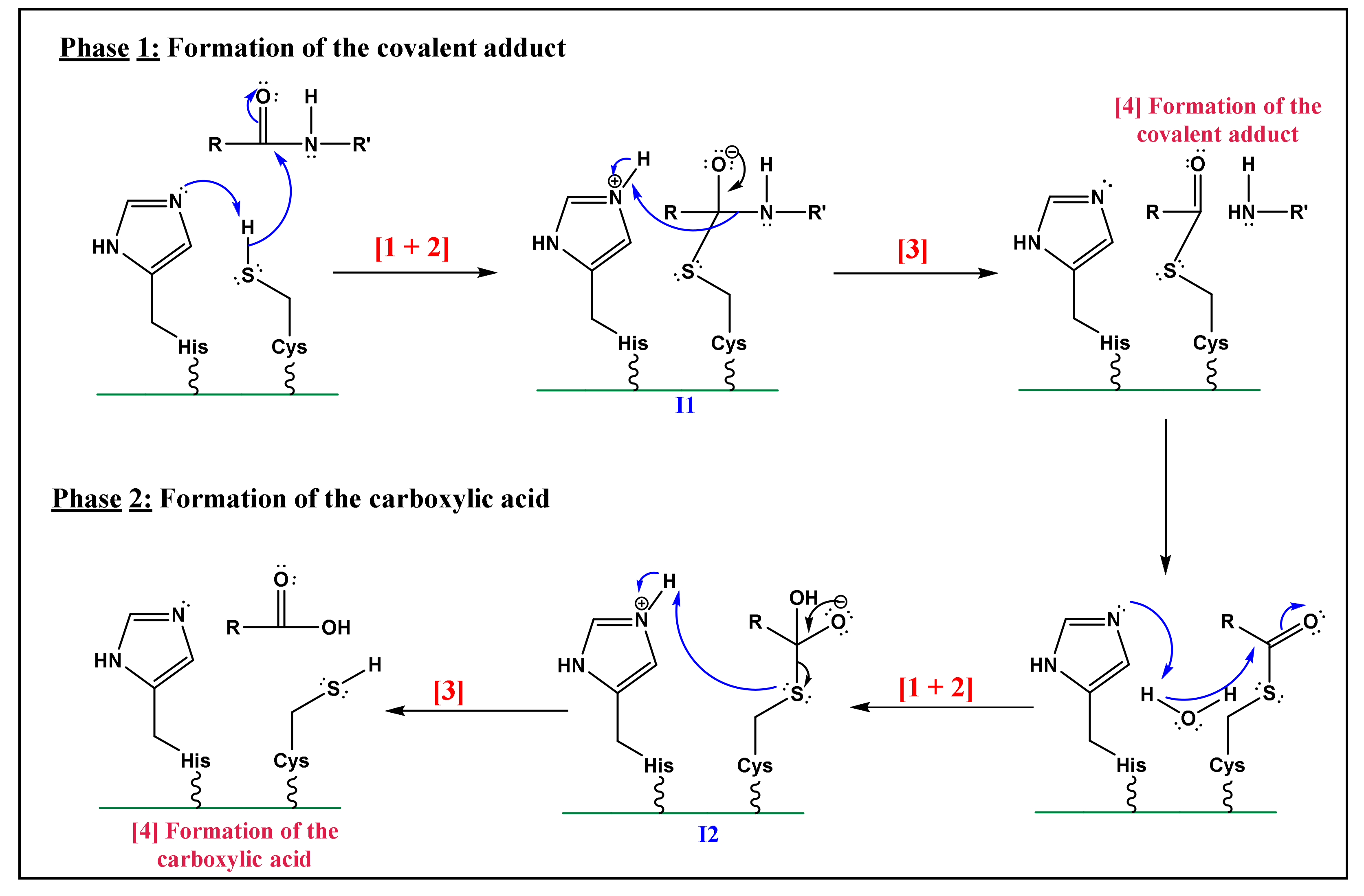
- Nucleophilic activation: the alkaline property of one of the nitrogen atoms, in the imidazolic part of Histidine (His), deprotonates the hydrogen in the thiol (-SH) group of the Cys residue, yielding a thiolate.
- Thiolate nucleophilic attack on carbonyl: the carbonyl group of the aspartic peptide bond undergoes a nucleophilic attack by the yielded thiolate in the latter step. This contributes to the formation of a first tetrahedral intermediate (I1; Figure 5).
- α-amino protonation: the amine group of I1 constitutes a good leaving group. This will enhance the possibility of the protonation of the α-amino moiety by the previous protonated nitrogen of the His residue.
- Formation of the covalent adduct: the acyl-enzyme complex and the cleavage of the peptide bond.
- Once, again the alkaline property of one of the nitrogen atoms in the imidazolic part of His deprotonates a water molecule.
- This deprotonation contributes to the formation of a hydroxide. The strong alkaline and nucleophilic property of the hydroxide contributes to the attack of the electrophilic site of the carbonyl function. This yields a second tetrahedral intermediate (I2; Figure 5).
- α-thio protonation: Similarly, to the third step of phase 1, the sulfur in I2 constitutes a good leaving group. This enhances the possibility of protonation of the α-thio moiety by the previously protonated nitrogen of the His residue.
- Formation of the carboxylic acid by regeneration of His and Cys counterparts.
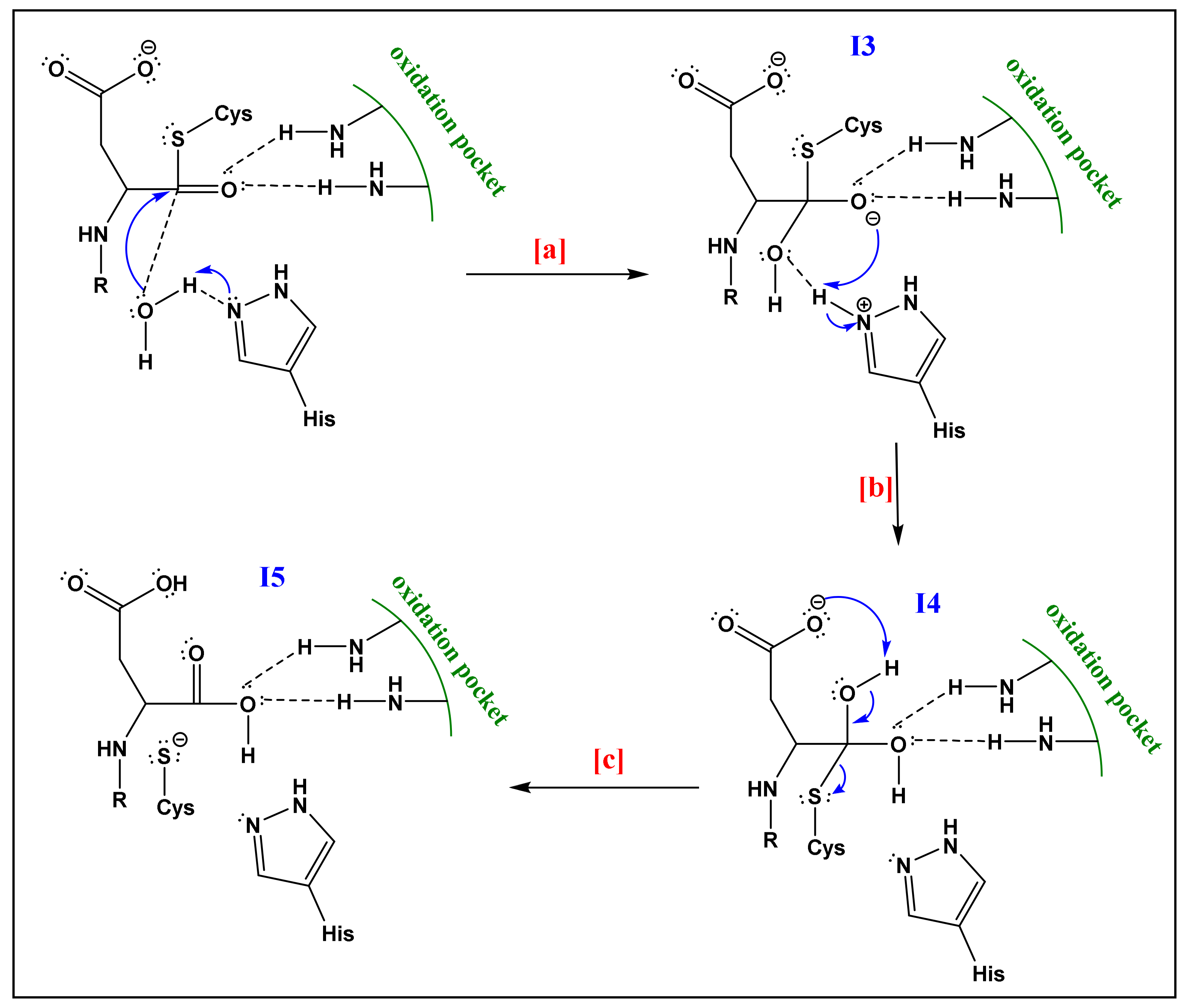
- The already formed hydrogen bond between His and the first water molecule will favor the deprotonation of the latter. The yielded hydroxide attacks the acyl-enzyme complex on the carbonyl moiety.
- The yielded alkoxide in I3 will attack the proton already captured by the His part in (a). This will form a germinal diol (I4; Figure 6).
- The carboxylate function of the side-chain aspartate will attack one of the hydrogens of the diol. The thiol, acting as a good leaving group, will enhance the possibility of the formation of a carbonyl bond; thereby a carboxylic acid and a thiolate (I5; Figure 6). The computational investigation shows that for the attack of the water molecule, a free energy barrier of about 19 ± 4 kcal/mol must be overcome, these trends are following the experimental results of Sulpizi et al. [88].
4. BH3-Only Protein Noxa
4.1. Discovery
4.2. General Features and Transcript Variants
4.3. Regulation of Noxa Expression and Post-Translational Modification
4.4. Subcellular Localization and Association with Bcl-2-like Proteins
5. BH3-Only Protein Puma
5.1. Regulation of the BH3-Only Protein Puma
5.2. p53-Dependent Apoptosis
5.3. p53-Independent Apoptosis
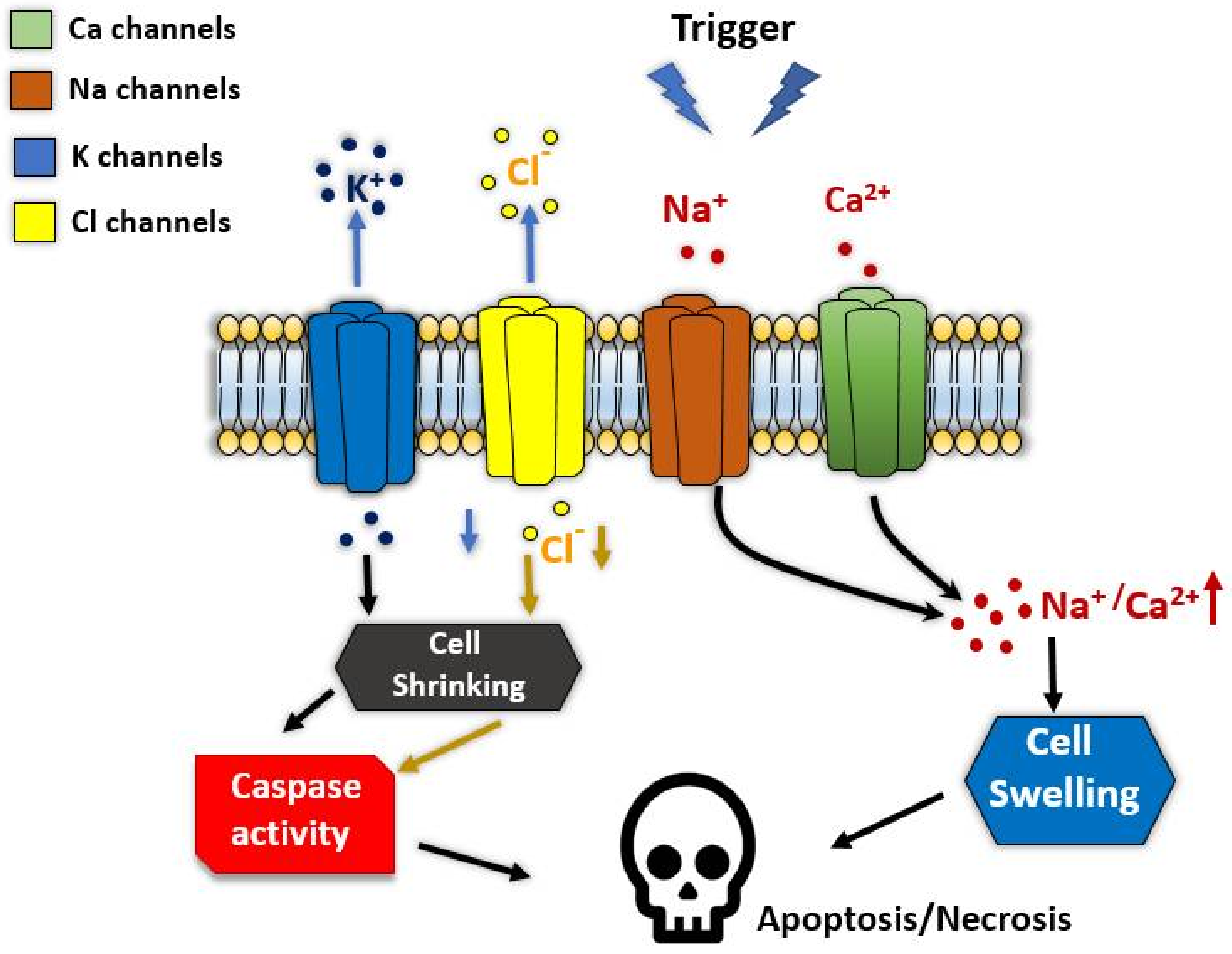
6. Ion Channels in Regulated Cell Death
7. Heat Shock Response to Puma and Noxa Proteins Expression In Vitro
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cui, Z.-G.; Piao, J.-L.; Kondo, T.; Ogawa, R.; Tsuneyama, K.; Zhao, Q.-L.; Feril, L.B., Jr.; Inadera, H. Molecular Mechanisms of Hyperthermia-Induced Apoptosis Enhanced by Docosahexaenoic Acid: Implication for Cancer Therapy. Chem. Biol. Interact. 2014, 215, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F. A Histochemical Study of Hypertrophy and Ischaemic Injury of Rat Liver with Special Reference to Changes in Lysosomes. J. Pathol. Bacteriol. 1965, 90, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled Demolition at the Cellular Level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Kihlmark, M.; Imreh, G.; Hallberg, E. Sequential Degradation of Proteins from the Nuclear Envelope during Apoptosis. J. Cell Sci. 2001, 114, 3643–3653. [Google Scholar] [CrossRef]
- Yoo, H.; Cha, H.J.; Lee, J.; Yu, E.-O.; Bae, S.; Jung, J.H.; Sohn, I.; Lee, S.-J.; Yang, K.-H.; Woo, S.-H. Specific Proteolysis of the A-Kinase-Anchoring Protein 149 at the Asp582 Residue by Caspases during Apoptosis. Oncol. Rep. 2008, 19, 1577–1582. [Google Scholar] [PubMed]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 Gene Promotes Haemopoietic Cell Survival and Cooperates with c-Myc to Immortalize Pre-B Cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Liadis, N.; Murakami, K.; Eweida, M.; Elford, A.R.; Sheu, L.; Gaisano, H.Y.; Hakem, R.; Ohashi, P.S.; Woo, M. Caspase-3-Dependent β-Cell Apoptosis in the Initiation of Autoimmune Diabetes Mellitus. Mol. Cell. Biol. 2005, 25, 3620–3629. [Google Scholar] [CrossRef] [Green Version]
- Rohn, T.T.; Head, E. Caspases as Therapeutic Targets in Alzheimer’s Disease: Is It Time to “Cut” to the Chase? Int. J. Clin. Exp. Pathol. 2009, 2, 108. [Google Scholar] [PubMed]
- Soto-Mercado, V.; Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. (−)-Epigallocatechin-3-Gallate Diminishes Intra-and Extracellular Amyloid-Induced Cytotoxic Effects on Cholinergic-like Neurons from Familial Alzheimer’s Disease PSEN1 E280A. Biomolecules 2021, 11, 1845. [Google Scholar] [CrossRef] [PubMed]
- Townsend, P.A.; Kozhevnikova, M.V.; Cexus, O.N.; Zamyatnin, A.A.; Soond, S.M. BH3-Mimetics: Recent Developments in Cancer Therapy. J. Exp. Clin. Cancer Res. 2021, 40, 355. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, M.; Ohsumi, Y. Isolation and Characterization of Autophagy-Defective Mutants of Saccharomyces Cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Klionsky, D.J. Development by Self-Digestion: Molecular Mechanisms and Biological Functions of Autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Sinha, S.; Levine, B. The Autophagy Effector Beclin 1: A Novel BH3-Only Protein. Oncogene 2008, 27, S137–S148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, S.W.; Lin, A.W. Apoptosis in Cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.Y.; Ryan, K.M. Autophagy and Cancer–Issues We Need to Digest. J. Cell Sci. 2012, 125, 2349–2358. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Debatin, K.-M. Extrinsic versus Intrinsic Apoptosis Pathways in Anticancer Chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, Y.; Torres, A.; Milián, M. Apoptosis’ Activation Associated to BH3 Only Domain and BCL-2 Homology Domain Proteins: New Way to Design Anti-Cancer Drugs. J. Cancer Prev. Curr. Res. 2019, 10, 54–59. [Google Scholar] [CrossRef]
- D’Orsi, B.; Mateyka, J.; Prehn, J.H. Control of Mitochondrial Physiology and Cell Death by the Bcl-2 Family Proteins Bax and Bok. Neurochem. Int. 2017, 109, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of Apoptosis in Health and Disease: The Balancing Act of BCL-2 Family Proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Hatok, J.; Racay, P. Bcl-2 Family Proteins: Master Regulators of Cell Survival. Biomol. Concepts 2016, 7, 259–270. [Google Scholar] [CrossRef]
- Opferman, J.T.; Kothari, A. Anti-Apoptotic BCL-2 Family Members in Development. Cell Death Differ. 2018, 25, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Greaves, G.; Milani, M.; Butterworth, M.; Carter, R.J.; Byrne, D.P.; Eyers, P.A.; Luo, X.; Cohen, G.M.; Varadarajan, S. BH3-Only Proteins Are Dispensable for Apoptosis Induced by Pharmacological Inhibition of Both MCL-1 and BCL-X L. Cell Death Differ. 2019, 26, 1037–1047. [Google Scholar] [CrossRef]
- Huang, K.; O’Neill, K.L.; Li, J.; Zhou, W.; Han, N.; Pang, X.; Wu, W.; Struble, L.; Borgstahl, G.; Liu, Z. BH3-Only Proteins Target BCL-XL/MCL-1, Not BAX/BAK, to Initiate Apoptosis. Cell Res. 2019, 29, 942–952. [Google Scholar] [CrossRef]
- Glab, J.A.; Mbogo, G.W.; Puthalakath, H. BH3-Only Proteins in Health and Disease. In International Review of Cell and Molecular Biology; Elsevier: Oxford, UK, 2017; Volume 328, pp. 163–196. [Google Scholar]
- Happo, L.; Strasser, A.; Cory, S. BH3-Only Proteins in Apoptosis at a Glance. J. Cell Sci. 2012, 125, 1081–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, H.; Ding, H.; Peterson, K.L.; Meng, X.W.; Schneider, P.A.; Knorr, K.L.; Kaufmann, S.H. Measurement of BH3-Only Protein Tolerance. Cell Death Differ. 2018, 25, 282–293. [Google Scholar] [CrossRef]
- Ludwig, L.M.; Roach, L.E.; Fisher, J.K.; Walensky, L.D.; LaBelle, J.L. Regulation of Immune Homeostasis by Direct Activator BH3-Only Proteins; AACR: New Orleans, LA, USA, 2016. [Google Scholar]
- Chen, A.; Madu, C.O.; Lu, Y. The Functional Role of Bcl-2 Family of Proteins in the Immune System and Cancer. Oncomedicine 2019, 4, 17–26. [Google Scholar] [CrossRef]
- Guikema, J.E.; Amiot, M.; Eldering, E. Exploiting the Pro-Apoptotic Function of NOXA as a Therapeutic Modality in Cancer. Expert Opin. Ther. Targets 2017, 21, 767–779. [Google Scholar] [CrossRef]
- Shukla, S.; Saxena, S.; Singh, B.K.; Kakkar, P. BH3-Only Protein BIM: An Emerging Target in Chemotherapy. Eur. J. Cell Biol. 2017, 96, 728–738. [Google Scholar] [CrossRef]
- Warren, C.F.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 Family Isoforms in Apoptosis and Cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.-W.; Ho, C.-J.; Chen, H.-W.; Pao, Y.-H.; Chen, L.-E.; Yang, M.-C.; Huang, S.-B.; Wang, S.; Chen, C.-H.; Wang, C. P53 Enhances Apoptosis Induced by Doxorubicin Only under Conditions of Severe DNA Damage. Cell Cycle 2018, 17, 2175–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial Dysfunction in Neurodegenerative Diseases and Drug Targets via Apoptotic Signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Cowan, K.; Anichtchik, O.; Luo, S. Mitochondrial Integrity in Neurodegeneration. CNS Neurosci. Ther. 2019, 25, 825–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Thomas, H.R.; Li, Z.; Yeo, N.C.F.; Scott, H.E.; Dang, N.; Hossain, M.I.; Andrabi, S.A.; Parant, J.M. Puma, Noxa, P53, and P63 Differentially Mediate Stress Pathway Induced Apoptosis. Cell Death Dis. 2021, 12, 659. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of Apoptosis by the BCL-2 Protein Family: Implications for Physiology and Therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Cossman, J.; Jaffe, E.; Croce, C.M. Involvement of the Bcl-2 Gene in Human Follicular Lymphoma. Science 1985, 228, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; O’Connor, L.; Dixit, V.M. Apoptosis Signaling. Annu. Rev. Biochem. 2000, 69, 217–245. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. Life-or-Death Decisions by the Bcl-2 Protein Family. Trends Biochem. Sci. 2001, 26, 61–66. [Google Scholar] [CrossRef]
- Roufayel, R. Regulation of Stressed-Induced Cell Death by the Bcl-2 Family of Apoptotic Proteins. Mol. Membr. Biol. 2016, 33, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Borner, C. The Bcl-2 Protein Family: Sensors and Checkpoints for Life-or-Death Decisions. Mol. Immunol. 2003, 39, 615–647. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 Family Reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Er, E.; Oliver, L.; Cartron, P.-F.; Juin, P.; Manon, S.; Vallette, F.M. Mitochondria as the Target of the Pro-Apoptotic Protein Bax. Biochim. Biophys. Acta (BBA)-Bioenerg. 2006, 1757, 1301–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Strasser, A. The BCL-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 Apoptotic Switch in Cancer Development and Therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.C.; Strasser, A. BH3-Only Proteins—Essential Initiators of Apoptotic Cell Death. Cell 2000, 103, 839–842. [Google Scholar] [CrossRef] [Green Version]
- Antonsson, B. Bax and Other Pro-Apoptotic Bcl-2 Family “Killer-Proteins” and Their Victim the Mitochondrion. Cell Tissue Res. 2001, 306, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Giam, M.; Huang, D.C.S.; Bouillet, P. BH3-Only Proteins and Their Roles in Programmed Cell Death. Oncogene 2008, 27, S128–S136. [Google Scholar] [CrossRef] [PubMed]
- Muchmore, S.W.; Sattler, M.; Liang, H.; Meadows, R.P.; Harlan, J.E.; Yoon, H.S.; Nettesheim, D.; Chang, B.S.; Thompson, C.B.; Wong, S.-L. X-Ray and NMR Structure of Human Bcl-x L, an Inhibitor of Programmed Cell Death. Nature 1996, 381, 335–341. [Google Scholar] [CrossRef]
- Hikisz, P.; Kiliańska, Z. PUMA, a Critical Mediator of Cell Death—One Decade on from Its Discovery. Cell. Mol. Biol. Lett. 2012, 17, 646–669. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Yang, H.; Fairlie, W.D.; Czabotar, P.E.; Fischer, S.F.; Perugini, M.A.; Huang, D.C.S.; Colman, P.M. Vaccinia Virus Anti-Apoptotic F1L Is a Novel Bcl-2-like Domain-Swapped Dimer That Binds a Highly Selective Subset of BH3-Containing Death Ligands. Cell Death Differ. 2008, 15, 1564–1571. [Google Scholar] [CrossRef] [Green Version]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the Rules of Programmed Cell Death to Improve Therapy of Cancer and Other Diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar] [CrossRef] [PubMed]
- Antignani, A.; Youle, R.J. How Do Bax and Bak Lead to Permeabilization of the Outer Mitochondrial Membrane? Curr. Opin. Cell Biol. 2006, 18, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Lomonosova, E.; Chinnadurai, G. BH3-Only Proteins in Apoptosis and beyond: An Overview. Oncogene 2008, 27, S2–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
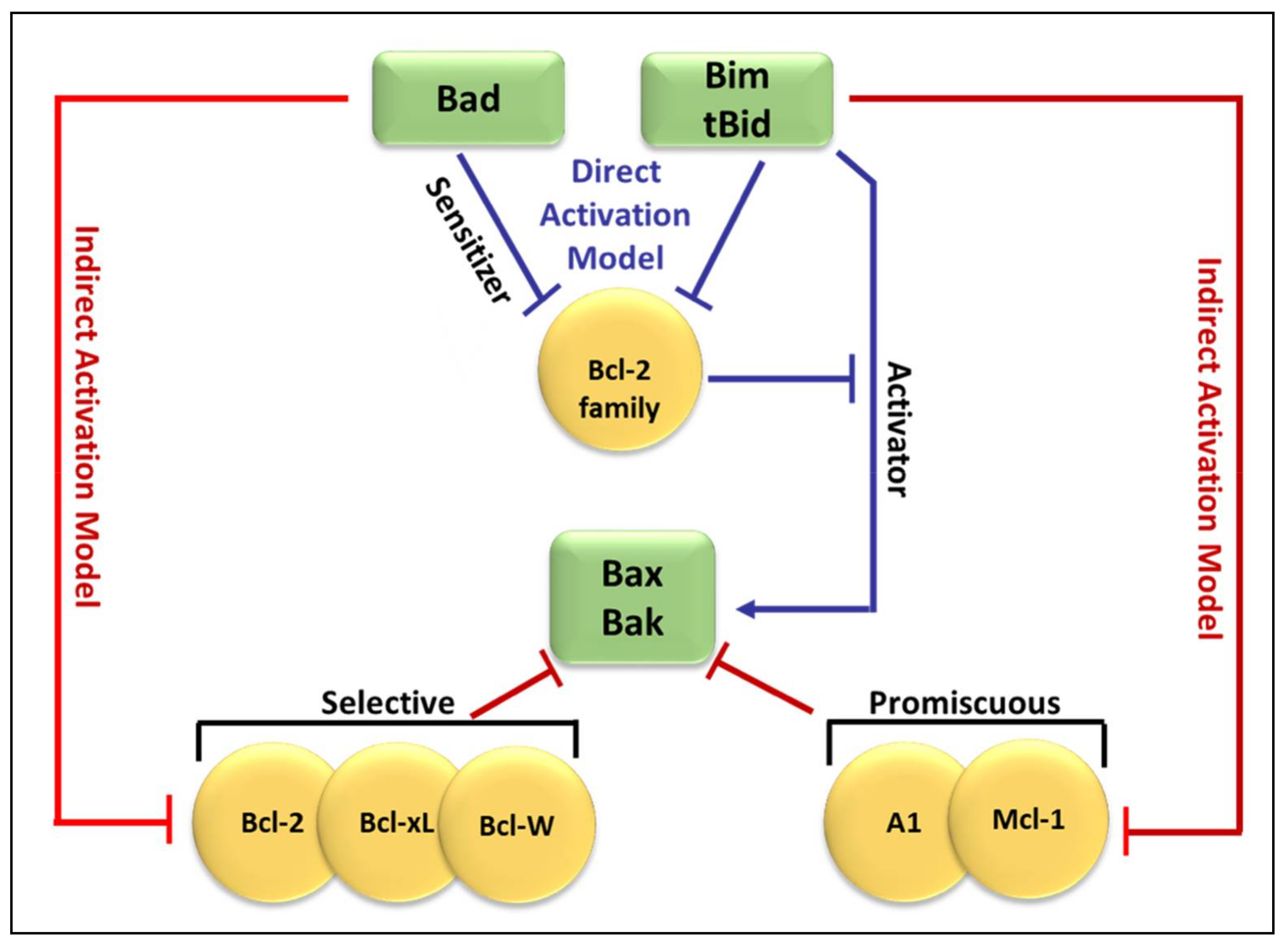
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P. Apoptosis Initiated When BH3 Ligands Engage Multiple Bcl-2 Homologs, Not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J. Structure of Bcl-XL-Bak Peptide Complex: Recognition between Regulators of Apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 Domains of BH3-Only Proteins Differentially Regulate Bax-Mediated Mitochondrial Membrane Permeabilization Both Directly and Indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef]
- Kim, H.; Rafiuddin-Shah, M.; Tu, H.-C.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. Hierarchical Regulation of Mitochondrion-Dependent Apoptosis by BCL-2 Subfamilies. Nat. Cell Biol. 2006, 8, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. How Do BCL-2 Proteins Induce Mitochondrial Outer Membrane Permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Tu, H.-C.; Ren, D.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. Stepwise Activation of BAX and BAK by TBID, BIM, and PUMA Initiates Mitochondrial Apoptosis. Mol. Cell 2009, 36, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soond, S.M.; Kozhevnikova, M.V.; Savvateeva, L.V.; Townsend, P.A.; Zamyatnin, A.A. Intrinsically Connected: Therapeutically Targeting the Cathepsin Proteases and the Bcl-2 Family of Protein Substrates as Co-Regulators of Apoptosis. Int. J. Mol. Sci. 2021, 22, 4669. [Google Scholar] [CrossRef] [PubMed]
- Gavathiotis, E.; Suzuki, M.; Davis, M.L.; Pitter, K.; Bird, G.H.; Katz, S.G.; Tu, H.-C.; Kim, H.; Cheng, E.H.-Y.; Tjandra, N. BAX Activation Is Initiated at a Novel Interaction Site. Nature 2008, 455, 1076–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golla, C.; Bilal, M.; Dwucet, A.; Bader, N.; Anthonymuthu, J.; Heiland, T.; Pruss, M.; Westhoff, M.-A.; Siegelin, M.D.; Capanni, F. Photodynamic Therapy Combined with Bcl-2/Bcl-XL Inhibition Increases the Noxa/Mcl-1 Ratio Independent of Usp9X and Synergistically Enhances Apoptosis in Glioblastoma. Cancers 2021, 13, 4123. [Google Scholar] [CrossRef]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak Is Sequestered by Mcl-1 and Bcl-XL, but Not Bcl-2, until Displaced by BH3-Only Proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Crawford, E.D.; Wells, J.A. Caspase Substrates and Cellular Remodeling. Annu. Rev. Biochem. 2011, 80, 1055–1087. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.D.; Weil, M.; Raff, M.C. Programmed Cell Death in Animal Development. Cell 1997, 88, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Vaux, D.; Strasser, A. The Molecular Biology of Apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 2239–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mintoo, M.; Khan, S.; Wani, A.; Malik, S.; Bhurta, D.; Bharate, S.; Malik, F.; Mondhe, D. A Rohitukine Derivative IIIM-290 Induces P53 Dependent Mitochondrial Apoptosis in Acute Lymphoblastic Leukemia Cells. Mol. Carcinog. 2021, 60, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Renault, T.T.; Chipuk, J.E. Death upon a Kiss: Mitochondrial Outer Membrane Composition and Organelle Communication Govern Sensitivity to BAK/BAX-Dependent Apoptosis. Chem. Biol. 2014, 21, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Busche, S.; John, K.; Wandrer, F.; Vondran, F.W.; Lehmann, U.; Wedemeyer, H.; Essmann, F.; Schulze-Osthoff, K.; Bantel, H. BH3-Only Protein Expression Determines Hepatocellular Carcinoma Response to Sorafenib-Based Treatment. Cell Death Dis. 2021, 12, 736. [Google Scholar] [CrossRef]
- Shiozaki, E.N.; Shi, Y. Caspases, IAPs and Smac/DIABLO: Mechanisms from Structural Biology. Trends Biochem. Sci. 2004, 29, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Roufayel, R.; Kadry, S. Expression of MiR-23a by Apoptotic Regulators in Human Cancer: A Review. Cancer Biol. Ther. 2017, 18, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouquette-Jazdanian, A.K.; Kortum, R.L.; Li, W.; Merrill, R.K.; Nguyen, P.H.; Samelson, L.E.; Sommers, C.L. MiR-155 Controls Lymphoproliferation in LAT Mutant Mice by Restraining T-Cell Apoptosis via SHIP-1/MTOR and PAK1/FOXO3/BIM Pathways. PLoS ONE 2015, 10, e0131823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.; Lazebnik, Y. Caspase-9 and APAF-1 Form an Active Holoenzyme. Genes Dev. 1999, 13, 3179–3184. [Google Scholar] [CrossRef] [Green Version]
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-Dimensional Structure of the Apoptosome: Implications for Assembly, Procaspase-9 Binding, and Activation. Mol. Cell 2002, 9, 423–432. [Google Scholar] [CrossRef]
- Green, D.R.; Evan, G.I. A Matter of Life and Death. Cancer Cell 2002, 1, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The Multiple Functions of Cytochrome c and Their Regulation in Life and Death Decisions of the Mammalian Cell: From Respiration to Apoptosis. Mitochondrion 2011, 11, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Z.-G.; Piao, J.-L.; Rehman, M.U.; Ogawa, R.; Li, P.; Zhao, Q.-L.; Kondo, T.; Inadera, H. Molecular Mechanisms of Hyperthermia-Induced Apoptosis Enhanced by Withaferin A. Eur. J. Pharmacol. 2014, 723, 99–107. [Google Scholar] [CrossRef]
- Stankiewicz, A.R.; Livingstone, A.M.; Mohseni, N.; Mosser, D.D. Regulation of Heat-Induced Apoptosis by Mcl-1 Degradation and Its Inhibition by Hsp70. Cell Death Differ. 2009, 16, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miscione, G.P.; Calvaresi, M.; Bottoni, A. Computational Evidence for the Catalytic Mechanism of Caspase-7. A DFT Investigation. J. Phys. Chem. B 2010, 114, 4637–4645. [Google Scholar] [CrossRef] [PubMed]
- Brady, K.D.; Giegel, D.A.; Grinnell, C.; Lunney, E.; Talanian, R.V.; Wong, W.; Walker, N. A Catalytic Mechanism for Caspase-1 and for Bimodal Inhibition of Caspase-1 by Activated Aspartic Ketones. Bioorg. Med. Chem. 1999, 7, 621–631. [Google Scholar] [CrossRef]
- Stennicke, H.R.; Salvesen, G.S. Catalytic Properties of the Caspases. Cell Death Differ. 1999, 6, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Chéreau, D.; Kodandapani, L.; Tomaselli, K.J.; Spada, A.P.; Wu, J.C. Structural and Functional Analysis of Caspase Active Sites. Biochemistry 2003, 42, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- Sulpizi, M.; Rothlisberger, U.; Carloni, P. Molecular Dynamics Studies of Caspase-3. Biophys. J. 2003, 84, 2207–2215. [Google Scholar] [CrossRef] [Green Version]
- Sulpizi, M.; Laio, A.; VandeVondele, J.; Cattaneo, A.; Rothlisberger, U.; Carloni, P. Reaction Mechanism of Caspases: Insights from QM/MM Car–Parrinello Simulations. Proteins Struct. Funct. Bioinform. 2003, 52, 212–224. [Google Scholar] [CrossRef]
- Fuentes-Prior, P.; Salvesen, G.S. The Protein Structures That Shape Caspase Activity, Specificity, Activation and Inhibition. Biochem. J. 2004, 384, 201–232. [Google Scholar] [CrossRef] [Green Version]
- Mikolajczyk, J.; Scott, F.L.; Krajewski, S.; Sutherlin, D.P.; Salvesen, G.S. Activation and Substrate Specificity of Caspase-14. Biochemistry 2004, 43, 10560–10569. [Google Scholar] [CrossRef]
- Assadi, M.H.N.; Hanaor, D.A. Theoretical Study on Copper’s Energetics and Magnetism in TiO2 Polymorphs. J. Appl. Phys. 2013, 113, 233913. [Google Scholar] [CrossRef] [Green Version]
- Kassis, S.; Grondin, M.; Averill-Bates, D.A. Heat Shock Increases Levels of Reactive Oxygen Species, Autophagy and Apoptosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2021, 1868, 118924. [Google Scholar] [CrossRef]
- Hijikata, M.; Kato, N.; Sato, T.; Kagami, Y.; Shimotohno, K. Molecular Cloning and Characterization of a CDNA for a Novel Phorbol-12-Myristate-13-Acetate-Responsive Gene That Is Highly Expressed in an Adult T-Cell Leukemia Cell Line. J. Virol. 1990, 64, 4632–4639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploner, C.; Kofler, R.; Villunger, A. Noxa: At the Tip of the Balance between Life and Death. Oncogene 2009, 27, S84. [Google Scholar] [CrossRef] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-Only Member of the Bcl-2 Family and Candidate Mediator of P53-Induced Apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef]
- Seo, Y.-W.; Shin, J.N.; Ko, K.H.; Cha, J.H.; Park, J.Y.; Lee, B.R.; Yun, C.-W.; Kim, Y.M.; Seol, D.; Kim, D. The Molecular Mechanism of Noxa-Induced Mitochondrial Dysfunction in P53-Mediated Cell Death. J. Biol. Chem. 2003, 278, 48292–48299. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, Y. Identification and Characterization of Two Splicing Variants of Human Noxa. Anticancer Res. 2008, 28, 1667–1674. [Google Scholar]
- Fei, P.; Bernhard, E.J.; El-Deiry, W.S. Tissue-Specific Induction of P53 Targets in Vivo. Cancer Res. 2002, 62, 7316–7327. [Google Scholar]
- Saha, M.N.; Jiang, H.; Mukai, A.; Chang, H. RITA Inhibits Multiple Myeloma Cell Growth through Induction of P53-Mediated Caspase-Dependent Apoptosis and Synergistically Enhances Nutlin-Induced Cytotoxic Responses. Mol. Cancer Ther. 2010, 9, 3041–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghavami, S.; Mutawe, M.M.; Sharma, P.; Yeganeh, B.; McNeill, K.D.; Klonisch, T.; Unruh, H.; Kashani, H.H.; Schaafsma, D.; Los, M.; et al. Mevalonate cascade regulation of airway mesenchymal cell autophagy and apoptosis: A dual role for p53. PLoS ONE 2011, 6, e16523. [Google Scholar]
- Raats, D.A.; de Bruijn, M.T.; Steller, E.J.; Emmink, B.L.; Borel-Rinkes, I.H.; Kranenburg, O. Synergistic Killing of Colorectal Cancer Cells by Oxaliplatin and ABT-737. Cell. Oncol. 2011, 34, 307–313. [Google Scholar] [CrossRef]
- Qin, J.-Z.; Stennett, L.; Bacon, P.; Bodner, B.; Hendrix, M.J.; Seftor, R.E.; Seftor, E.A.; Margaryan, N.V.; Pollock, P.M.; Curtis, A. P53-Independent NOXA Induction Overcomes Apoptotic Resistance of Malignant Melanomas. Mol. Cancer Ther. 2004, 3, 895–902. [Google Scholar]
- Flinterman, M.; Guelen, L.; Ezzati-Nik, S.; Killick, R.; Melino, G.; Tominaga, K.; Mymryk, J.S.; Gäken, J.; Tavassoli, M. E1A Activates Transcription of P73 and Noxa to Induce Apoptosis. J. Biol. Chem. 2005, 280, 5945–5959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aikawa, T.; Shinzawa, K.; Tanaka, N.; Tsujimoto, Y. Noxa Is Necessary for Hydrogen Peroxide-Induced Caspase-Dependent Cell Death. FEBS Lett. 2010, 584, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Valis, K.; Prochazka, L.; Boura, E.; Chladova, J.; Obsil, T.; Rohlena, J.; Truksa, J.; Dong, L.-F.; Ralph, S.J.; Neuzil, J. Hippo/Mst1 Stimulates Transcription of the Proapoptotic Mediator NOXA in a FoxO1-Dependent Manner. Cancer Res. 2011, 71, 946–954. [Google Scholar] [CrossRef] [Green Version]
- Alves, N.L.; Derks, I.A.; Berk, E.; Spijker, R.; van Lier, R.A.; Eldering, E. The Noxa/Mcl-1 Axis Regulates Susceptibility to Apoptosis under Glucose Limitation in Dividing T Cells. Immunity 2006, 24, 703–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, L.M.S.; Nugent, J.K.; Zhao, X.; Irwin, M.S. HDM2 Antagonist Nutlin-3 Disrupts P73-HDM2 Binding and Enhances P73 Function. Oncogene 2008, 27, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, A.G.; Trama, J.; Crighton, D.; Ryan, K.M.; Fearnhead, H.O. Activation of P73 and Induction of Noxa by DNA Damage Requires NF-Kappa B. Aging 2009, 1, 335. [Google Scholar] [CrossRef]
- Santidrián, A.F.; González-Gironès, D.M.; Iglesias-Serret, D.; Coll-Mulet, L.; Cosialls, A.M.; de Frias, M.; Campàs, C.; González-Barca, E.; Alonso, E.; Labi, V. AICAR Induces Apoptosis Independently of AMPK and P53 through Up-Regulation of the BH3-Only Proteins BIM and NOXA in Chronic Lymphocytic Leukemia Cells. Blood 2010, 116, 3023–3032. [Google Scholar] [CrossRef] [Green Version]
- Iglesias-Serret, D.; Piqué, M.; Barragán, M.; Cosialls, A.M.; Santidrián, A.F.; González-Gironès, D.M.; Coll-Mulet, L.; de Frias, M.; Pons, G.; Gil, J. Aspirin Induces Apoptosis in Human Leukemia Cells Independently of NF-ΚB and MAPKs through Alteration of the Mcl-1/Noxa Balance. Apoptosis 2010, 15, 219–229. [Google Scholar] [CrossRef]
- Sheridan, C.; Brumatti, G.; Elgendy, M.; Brunet, M.; Martin, S.J. An ERK-Dependent Pathway to Noxa Expression Regulates Apoptosis by Platinum-Based Chemotherapeutic Drugs. Oncogene 2010, 29, 6428–6441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Tabe, Y.; Kojima, K.; Zhou, Y.; Pittaluga, S.; Konopleva, M.; Miida, T.; Raffeld, M. MDM2 Antagonist Nutlin-3 Enhances Bortezomib-Mediated Mitochondrial Apoptosis in TP53-Mutated Mantle Cell Lymphoma. Cancer Lett. 2010, 299, 161–170. [Google Scholar] [CrossRef]
- DiFeo, A.; Huang, F.; Sangodkar, J.; Terzo, E.A.; Leake, D.; Narla, G.; Martignetti, J.A. KLF6-SV1 Is a Novel Antiapoptotic Protein That Targets the BH3-Only Protein NOXA for Degradation and Whose Inhibition Extends Survival in an Ovarian Cancer Model. Cancer Res. 2009, 69, 4733–4741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In Vitro and in Vivo Selective Antitumor Activity of a Novel Orally Bioavailable Proteasome Inhibitor MLN9708 against Multiple Myeloma Cells. Clin. Cancer Res. 2011, 17, 5311–5321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, Y.; Verhaegen, M.; Miller, T.P.; Rush, J.L.; Steiner, P.; Opipari, A.W.; Lowe, S.W.; Soengas, M.S. Differential Regulation of Noxa in Normal Melanocytes and Melanoma Cells by Proteasome Inhibition: Therapeutic Implications. Cancer Res. 2005, 65, 6294–6304. [Google Scholar] [CrossRef] [Green Version]
- Fribley, A.M.; Evenchik, B.; Zeng, Q.; Park, B.K.; Guan, J.Y.; Zhang, H.; Hale, T.J.; Soengas, M.S.; Kaufman, R.J.; Wang, C.-Y. Proteasome Inhibitor PS-341 Induces Apoptosis in Cisplatin-Resistant Squamous Cell Carcinoma Cells by Induction of Noxa. J. Biol. Chem. 2006, 281, 31440–31447. [Google Scholar] [CrossRef]
- Nikiforov, M.A.; Riblett, M.; Tang, W.-H.; Gratchouck, V.; Zhuang, D.; Fernandez, Y.; Verhaegen, M.; Varambally, S.; Chinnaiyan, A.M.; Jakubowiak, A.J. Tumor Cell-Selective Regulation of NOXA by c-MYC in Response to Proteasome Inhibition. Proc. Natl. Acad. Sci. USA 2007, 104, 19488–19493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Núñez-Vázquez, S.; Sánchez-Vera, I.; Saura-Esteller, J.; Cosialls, A.M.; Noisier, A.F.; Albericio, F.; Lavilla, R.; Pons, G.; Iglesias-Serret, D.; Gil, J. NOXA Upregulation by the Prohibitin-Binding Compound Fluorizoline Is Transcriptionally Regulated by Integrated Stress Response-Induced ATF3 and ATF4. FEBS J. 2021, 288, 1271–1285. [Google Scholar] [CrossRef]
- Sosa Seda, I.M.; Mott, J.L.; Akazawa, Y.; Barreyro, F.J.; Bronk, S.F.; Kaufmann, S.H.; Gores, G.J. Noxa Mediates Hepatic Stellate Cell Apoptosis by Proteasome Inhibition. Hepatol. Res. 2010, 40, 701–710. [Google Scholar] [CrossRef]
- Jia, L.; Yang, J.; Hao, X.; Zheng, M.; He, H.; Xiong, X.; Xu, L.; Sun, Y. Validation of SAG/RBX2/ROC2 E3 Ubiquitin Ligase as an Anticancer and Radiosensitizing Target. Clin. Cancer Res. 2010, 16, 814–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baou, M.; Kohlhaas, S.L.; Butterworth, M.; Vogler, M.; Dinsdale, D.; Walewska, R.; Majid, A.; Eldering, E.; Dyer, M.J.; Cohen, G.M. Role of NOXA and Its Ubiquitination in Proteasome Inhibitor-Induced Apoptosis in Chronic Lymphocytic Leukemia Cells. haematologica 2010, 95, 1510–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roufayel, R.; Murshid, N. CDK5: Key Regulator of Apoptosis and Cell Survival. Biomedicines 2019, 7, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowman, X.H.; McDonnell, M.A.; Kosloske, A.; Odumade, O.A.; Jenness, C.; Karim, C.B.; Jemmerson, R.; Kelekar, A. The Proapoptotic Function of Noxa in Human Leukemia Cells Is Regulated by the Kinase Cdk5 and by Glucose. Mol. Cell 2010, 40, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Narita, N.; Noda, I.; Ohtsubo, T.; Fujieda, S.; Tokuriki, M.; Saito, T.; Saito, H. Analysis of Heat-Shock Related Gene Expression in Head-and-Neck Cancer Using CDNA Arrays. Int. J. Radiat. Oncol. Biol. Phys. 2002, 53, 190–196. [Google Scholar] [CrossRef]
- Hassan, M.; Alaoui, A.; Feyen, O.; Mirmohammadsadegh, A.; Essmann, F.; Tannapfel, A.; Gulbins, E.; Schulze-Osthoff, K.; Hengge, U.R. The BH3-Only Member Noxa Causes Apoptosis in Melanoma Cells by Multiple Pathways. Oncogene 2008, 27, 4557–4568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idrus, E.; Nakashima, T.; Wang, L.; Hayashi, M.; Okamoto, K.; Kodama, T.; Tanaka, N.; Taniguchi, T.; Takayanagi, H. The Role of the BH3-Only Protein Noxa in Bone Homeostasis. Biochem. Biophys. Res. Commun. 2011, 410, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Kirejczyk, Z.; Potthoff, S.; Ploner, C.; Häcker, G. Endogenous Noxa Determines the Strong Proapoptotic Synergism of the BH3-Mimetic ABT-737 with Chemotherapeutic Agents in Human Melanoma Cells. Transl. Oncol. 2009, 2, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Hagenbuchner, J.; Ausserlechner, M.J.; Porto, V.; David, R.; Meister, B.; Bodner, M.; Villunger, A.; Geiger, K.; Obexer, P. The Anti-Apoptotic Protein BCL2L1/Bcl-XL Is Neutralized by pro-Apoptotic PMAIP1/Noxa in Neuroblastoma, Thereby Determining Bortezomib Sensitivity Independent of Prosurvival MCL1 Expression. J. Biol. Chem. 2010, 285, 6904–6912. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Takeda, K.; Oda, E.; Tanaka, H.; Murasawa, H.; Takaoka, A.; Morishita, Y.; Akira, S.; Taniguchi, T.; Tanaka, N. Integral Role of Noxa in P53-Mediated Apoptotic Response. Genes Dev. 2003, 17, 2233–2238. [Google Scholar] [CrossRef] [Green Version]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Müllauer, F.; Böck, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. P53-and Drug-Induced Apoptotic Responses Mediated by BH3-Only Proteins Puma and Noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschnek, S.; Vier, J.; Gautam, S.; Frankenberg, T.; Rangelova, S.; Eitz-Ferrer, P.; Grespi, F.; Ottina, E.; Villunger, A.; Häcker, H. Molecular Analysis of Neutrophil Spontaneous Apoptosis Reveals a Strong Role for the Pro-Apoptotic BH3-Only Protein Noxa. Cell Death Differ. 2011, 18, 1805–1814. [Google Scholar] [CrossRef]
- Muenchow, A.; Weller, S.; Hinterleitner, C.; Malenke, E.; Bugl, S.; Wirths, S.; Müller, M.R.; Schulze-Osthoff, K.; Aulitzky, W.E.; Kopp, H.-G. The BCL-2 Selective Inhibitor ABT-199 Sensitizes Soft Tissue Sarcomas to Proteasome Inhibition by a Concerted Mechanism Requiring BAX and NOXA. Cell Death Dis. 2020, 11, 701. [Google Scholar] [CrossRef] [PubMed]
- Holzerland, J.; Fénéant, L.; Banadyga, L.; Hölper, J.E.; Knittler, M.R.; Groseth, A. BH3-Only Sensors Bad, Noxa and Puma Are Key Regulators of Tacaribe Virus-Induced Apoptosis. PLoS Pathog. 2020, 16, e1008948. [Google Scholar] [CrossRef] [PubMed]
- Kurschat, C.; Metz, A.; Kirschnek, S.; Häcker, G. Importance of Bcl-2-Family Proteins in Murine Hematopoietic Progenitor and Early B Cells. Cell Death Dis. 2021, 12, 784. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.-Z.; Xin, H.; Sitailo, L.A.; Denning, M.F.; Nickoloff, B.J. Enhanced Killing of Melanoma Cells by Simultaneously Targeting Mcl-1 and NOXA. Cancer Res. 2006, 66, 9636–9645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Bougie, P.; Wuillème-Toumi, S.; Ménoret, E.; Trichet, V.; Robillard, N.; Philippe, M.; Bataille, R.; Amiot, M. Noxa Up-Regulation and Mcl-1 Cleavage Are Associated to Apoptosis Induction by Bortezomib in Multiple Myeloma. Cancer Res. 2007, 67, 5418–5424. [Google Scholar] [CrossRef] [Green Version]
- Lopez, H.; Zhang, L.; George, N.M.; Liu, X.; Pang, X.; Evans, J.J.; Targy, N.M.; Luo, X. Perturbation of the Bcl-2 Network and an Induced Noxa/Bcl-XL Interaction Trigger Mitochondrial Dysfunction after DNA Damage. J. Biol. Chem. 2010, 285, 15016–15026. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lopez, H.; George, N.M.; Liu, X.; Pang, X.; Luo, X. Selective Involvement of BH3-Only Proteins and Differential Targets of Noxa in Diverse Apoptotic Pathways. Cell Death Differ. 2011, 18, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.J.; Dai, H.; Correia, C.; Takahashi, R.; Lee, S.-H.; Schmitz, I.; Kaufmann, S.H. Noxa/Bcl-2 Protein Interactions Contribute to Bortezomib Resistance in Human Lymphoid Cells. J. Biol. Chem. 2011, 286, 17682–17692. [Google Scholar] [CrossRef] [Green Version]
- Morales, A.A.; Gutman, D.; Lee, K.P.; Boise, L.H. BH3-Only Proteins Noxa, Bmf, and Bim Are Necessary for Arsenic Trioxide–Induced Cell Death in Myeloma. Blood J. Am. Soc. Hematol. 2008, 111, 5152–5162. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Bougie, P.; Ménoret, E.; Juin, P.; Dousset, C.; Pellat-Deceunynck, C.; Amiot, M. Noxa Controls Mule-Dependent Mcl-1 Ubiquitination through the Regulation of the Mcl-1/USP9X Interaction. Biochem. Biophys. Res. Commun. 2011, 413, 460–464. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA Induces the Rapid Apoptosis of Colorectal Cancer Cells. Mol. Cell 2001, 7, 673–682. [Google Scholar] [CrossRef]
- Han, J.; Flemington, C.; Houghton, A.B.; Gu, Z.; Zambetti, G.P.; Lutz, R.J.; Zhu, L.; Chittenden, T. Expression of Bbc3, a pro-Apoptotic BH3-Only Gene, Is Regulated by Diverse Cell Death and Survival Signals. Proc. Natl. Acad. Sci. USA 2001, 98, 11318–11323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Wang, Z.; Kinzler, K.W.; Vogelstein, B.; Zhang, L. PUMA Mediates the Apoptotic Response to P53 in Colorectal Cancer Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 1931–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffers, J.R.; Parganas, E.; Lee, Y.; Yang, C.; Wang, J.; Brennan, J.; MacLean, K.H.; Han, J.; Chittenden, T.; Ihle, J.N. Puma Is an Essential Mediator of P53-Dependent and-Independent Apoptotic Pathways. Cancer Cell 2003, 4, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Fricker, M.; O’prey, J.; Tolkovsky, A.M.; Ryan, K.M. Phosphorylation of Puma Modulates Its Apoptotic Function by Regulating Protein Stability. Cell Death Dis. 2010, 1, e59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zhang, L. PUMA, a Potent Killer with or without P53. Oncogene 2008, 27, S71–S83. [Google Scholar] [CrossRef] [Green Version]
- Happo, L.; Cragg, M.S.; Phipson, B.; Haga, J.M.; Jansen, E.S.; Herold, M.J.; Dewson, G.; Michalak, E.M.; Vandenberg, C.J.; Smyth, G.K. Maximal Killing of Lymphoma Cells by DNA Damage–Inducing Therapy Requires Not Only the P53 Targets Puma and Noxa, but Also Bim. Blood J. Am. Soc. Hematol. 2010, 116, 5256–5267. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. P53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Charvet, C.; Wissler, M.; Brauns-Schubert, P.; Wang, S.-J.; Tang, Y.; Sigloch, F.C.; Mellert, H.; Brandenburg, M.; Lindner, S.E.; Breit, B. Phosphorylation of Tip60 by GSK-3 Determines the Induction of PUMA and Apoptosis by P53. Mol. Cell 2011, 42, 584–596. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Qiu, W.; Wang, P.; Yu, H.; Cheng, T.; Zambetti, G.P.; Zhang, L.; Yu, J. P53 Independent Induction of PUMA Mediates Intestinal Apoptosis in Response to Ischaemia–Reperfusion. Gut 2007, 56, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Michalak, E.M.; Villunger, A.; Adams, J.M.; Strasser, A. In Several Cell Types Tumour Suppressor P53 Induces Apoptosis Largely via Puma but Noxa Can Contribute. Cell Death Differ. 2008, 15, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Lóssio, C.F.; Verbeke, I.; Verduijn, J.; Parakhonskiy, B.; Van der Meeren, L.; Chen, P.; De Zaeytijd, J.; Skirtach, A.G.; Van Damme, E.J. The Type-1 Ribosome-Inactivating Protein OsRIP1 Triggers Caspase-Independent Apoptotic-like Death in HeLa Cells. Food Chem. Toxicol. 2021, 157, 112590. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; O’Brien, D.; Vilgelm, A.; Piazuelo, M.B.; Correa, P.; Washington, M.K.; El-Rifai, W.; Peek, R.M.; Zaika, A. Interaction of Helicobacter Pylori with Gastric Epithelial Cells Is Mediated by the P53 Protein Family. Gastroenterology 2008, 134, 1412–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, S.F.; Belz, G.T.; Strasser, A. BH3-Only Protein Puma Contributes to Death of Antigen-Specific T Cells during Shutdown of an Immune Response to Acute Viral Infection. Proc. Natl. Acad. Sci. USA 2008, 105, 3035–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunzelmann, K. Ion Channels in Regulated Cell Death. Cell. Mol. Life Sci. 2016, 73, 2387–2403. [Google Scholar] [CrossRef]
- Williams, G.T. Programmed Cell Death: Apoptosis and Oncogenesis. Cell 1991, 65, 1097–1098. [Google Scholar] [CrossRef]
- Gulbins, E.; Jekle, A.; Ferlinz, K.; Grassme, H.; Lang, F. Physiology of Apoptosis. Am. J. Physiol.-Ren. Physiol. 2000, 279, F605–F615. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M. Molecular Characterization of Mitochondrial Apoptosis-Inducing Factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Prevarskaya, N. Ion Channels in the Regulation of Apoptosis. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 2532–2546. [Google Scholar] [CrossRef] [Green Version]
- Danial, N.N. BCL-2 Family Proteins: Critical Checkpoints of Apoptotic Cell Death. Clin. Cancer Res. 2007, 13, 7254–7263. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium-a Life and Death Signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and Apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Fang, K.-M.; Chang, W.-L.; Wang, S.-M.; Su, M.-J.; Wu, M.-L. Arachidonic Acid Induces Both Na+ and Ca2+ Entry Resulting in Apoptosis. J. Neurochem. 2008, 104, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Hoffmann, E.K. Role of Ion Transport in Control of Apoptotic Cell Death. Compr. Physiol. 2012, 2, 2037–2061. [Google Scholar] [PubMed]
- Dubois, C.; Abeele, F.V.; Prevarskaya, N. Targeting Apoptosis by the Remodelling of Calcium-Transporting Proteins in Cancerogenesis. FEBS J. 2013, 280, 5500–5510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.-J.; Chen, M.; Puthiyakunnon, S.; He, C.; Xia, J.; He, C.Y.; Deng, S.-Q.; Peng, H.-J. Toxoplasma Gondii ROP18 Inhibits Human Glioblastoma Cell Apoptosis through a Mitochondrial Pathway by Targeting Host Cell P2 × 1. Parasites Vectors 2019, 12, 284. [Google Scholar] [CrossRef] [Green Version]
- Hughes, F.M., Jr.; Cidlowski, J.A. Potassium Is a Critical Regulator of Apoptotic Enzymes in Vitro and in Vivo. Adv. Enzym. Regul. 1999, 39, 157. [Google Scholar] [CrossRef]
- Bortner, C.D.; Cidlowski, J.A. The Role of Apoptotic Volume Decrease and Ionic Homeostasis in the Activation and Repression of Apoptosis. Pflügers Arch. 2004, 448, 313–318. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of Cell Volume Regulation in Vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef]
- Moreels, L.; Bhat, C.; Voráčová, M.; Peigneur, S.; Goovaerts, H.; Mäki-Lohiluoma, E.; Zahed, F.; Pardo, L.A.; Yli-Kauhaluoma, J.; Kiuru, P. Synthesis of Novel Purpurealidin Analogs and Evaluation of Their Effect on the Cancer-Relevant Potassium Channel KV10. 1. PLoS ONE 2017, 12, e0188811. [Google Scholar] [CrossRef] [PubMed]
- Oeggerli, M.; Tian, Y.; Ruiz, C.; Wijker, B.; Sauter, G.; Obermann, E.; Güth, U.; Zlobec, I.; Sausbier, M.; Kunzelmann, K. Role of KCNMA1 in Breast Cancer. PLoS ONE 2012, 7, e41664. [Google Scholar] [CrossRef] [PubMed]
- Koehl, G.E.; Spitzner, M.; Ousingsawat, J.; Schreiber, R.; Geissler, E.K.; Kunzelmann, K. Rapamycin Inhibits Oncogenic Intestinal Ion Channels and Neoplasia in APCMin/+ Mice. Oncogene 2010, 29, 1553–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, L.A.; Stühmer, W. The Roles of K+ Channels in Cancer. Nat. Rev. Cancer 2014, 14, 39–48. [Google Scholar] [CrossRef]
- Urrego, D.; Tomczak, A.P.; Zahed, F.; Stühmer, W.; Pardo, L.A. Potassium Channels in Cell Cycle and Cell Proliferation. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, L.A.; Gomez-Varela, D.; Major, F.; Sansuk, K.; Leurs, R.; Downie, B.R.; Tietze, L.F.; Stuhmer, W. Approaches Targeting Kv10. 1 Open a Novel Window for Cancer Diagnosis and Therapy. Curr. Med. Chem. 2012, 19, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Hartnett, K.A.; Nerbonne, J.M.; Levitan, E.S.; Aizenman, E. Mediation of Neuronal Apoptosis by Kv2. 1-Encoded Potassium Channels. J. Neurosci. 2003, 23, 4798–4802. [Google Scholar] [CrossRef]
- Stavrovskaya, A.A. Cellular Mechanisms of Multidrug Resistance of Tumor Cells. Biochem. C/C Biokhimiia 2000, 65, 95–106. [Google Scholar]
- Lee, E.L.; Shimizu, T.; Ise, T.; Numata, T.; Kohno, K.; Okada, Y. Impaired Activity of Volume-Sensitive Cl− Channel Is Involved in Cisplatin Resistance of Cancer Cells. J. Cell. Physiol. 2007, 211, 513–521. [Google Scholar] [CrossRef]
- Martins, J.R.; Faria, D.; Kongsuphol, P.; Reisch, B.; Schreiber, R.; Kunzelmann, K. Anoctamin 6 Is an Essential Component of the Outwardly Rectifying Chloride Channel. Proc. Natl. Acad. Sci. USA 2011, 108, 18168–18172. [Google Scholar] [CrossRef] [Green Version]
- Britschgi, A.; Bill, A.; Brinkhaus, H.; Rothwell, C.; Clay, I.; Duss, S.; Rebhan, M.; Raman, P.; Guy, C.T.; Wetzel, K. Calcium-Activated Chloride Channel ANO1 Promotes Breast Cancer Progression by Activating EGFR and CAMK Signaling. Proc. Natl. Acad. Sci. USA 2013, 110, E1026–E1034. [Google Scholar] [CrossRef] [Green Version]
- Wanitchakool, P.; Wolf, L.; Koehl, G.E.; Sirianant, L.; Schreiber, R.; Kulkarni, S.; Duvvuri, U.; Kunzelmann, K. Role of Anoctamins in Cancer and Apoptosis. Philos. Trans. R Soc. B Biol. Sci. 2014, 369, 20130096. [Google Scholar] [CrossRef]
- Qu, Z.; Yao, W.; Yao, R.; Liu, X.; Yu, K.; Hartzell, C. The Ca2+-Activated Cl− Channel, ANO1 (TMEM16A), Is a Double-Edged Sword in Cell Proliferation and Tumorigenesis. Cancer Med. 2014, 3, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.D.; Caron, A.W.; Bourget, L.; Meriin, A.B.; Sherman, M.Y.; Morimoto, R.I.; Massie, B. The Chaperone Function of Hsp70 Is Required for Protection against Stress-Induced Apoptosis. Mol. Cell. Biol. 2000, 20, 7146–7159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular Chaperones in Protein Folding and Proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Roufayel, R.; Johnston, D.S.; Mosser, D.D. The Elimination of MiR-23a in Heat-Stressed Cells Promotes NOXA-Induced Cell Death and Is Prevented by HSP70. Cell Death Dis. 2014, 5, e1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roufayel, R.; Younes, K.; Al-Sabi, A.; Murshid, N. BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis. Life 2022, 12, 256. https://doi.org/10.3390/life12020256
Roufayel R, Younes K, Al-Sabi A, Murshid N. BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis. Life. 2022; 12(2):256. https://doi.org/10.3390/life12020256
Chicago/Turabian StyleRoufayel, Rabih, Khaled Younes, Ahmed Al-Sabi, and Nimer Murshid. 2022. "BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis" Life 12, no. 2: 256. https://doi.org/10.3390/life12020256
APA StyleRoufayel, R., Younes, K., Al-Sabi, A., & Murshid, N. (2022). BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis. Life, 12(2), 256. https://doi.org/10.3390/life12020256