
Development and Biological Characterization of a Novel Selective TrkA Agonist with Neuroprotective Properties against Amyloid Toxicity

 ,
,  , ,
, ,  , , ,
, , ,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
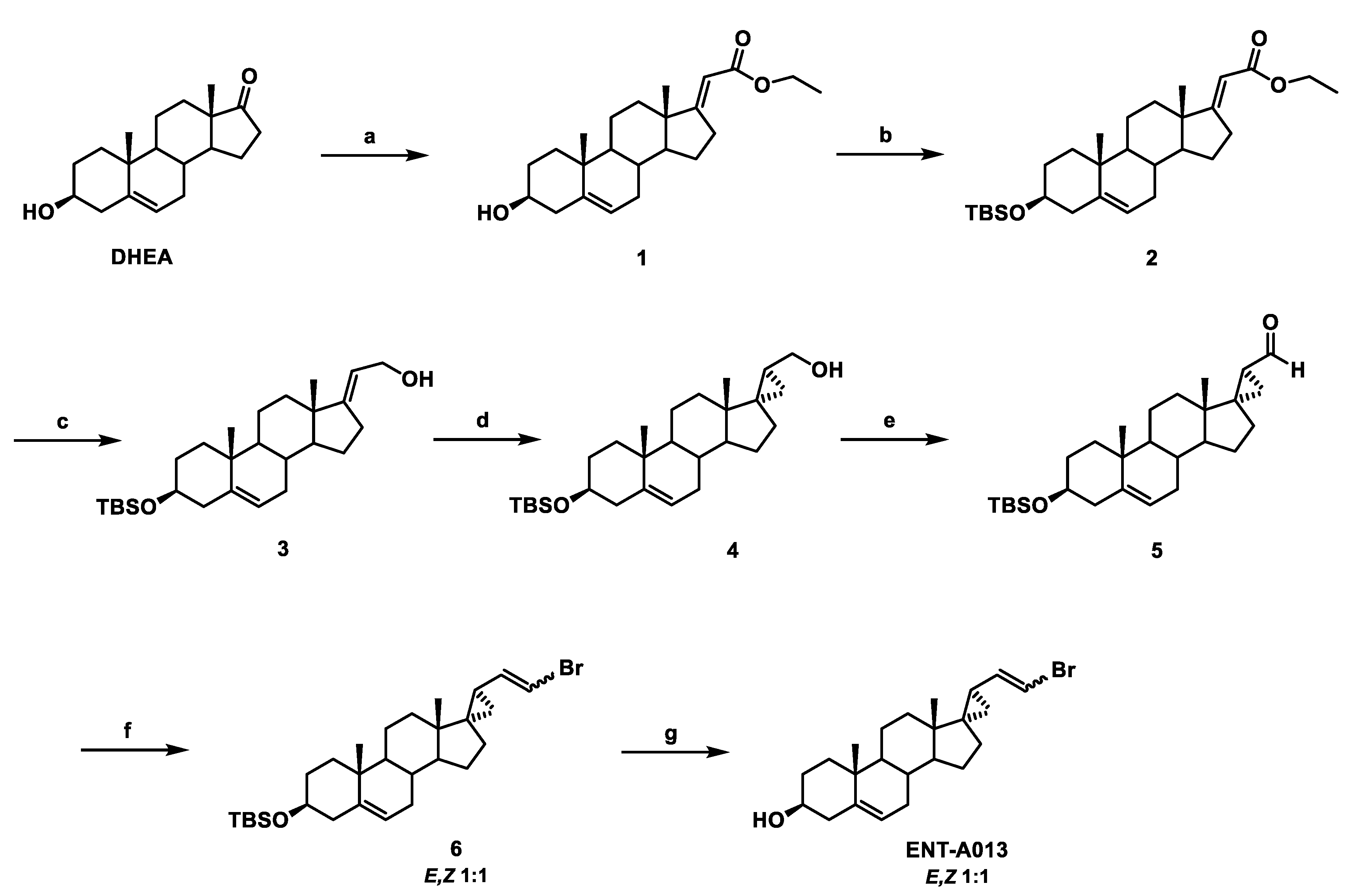
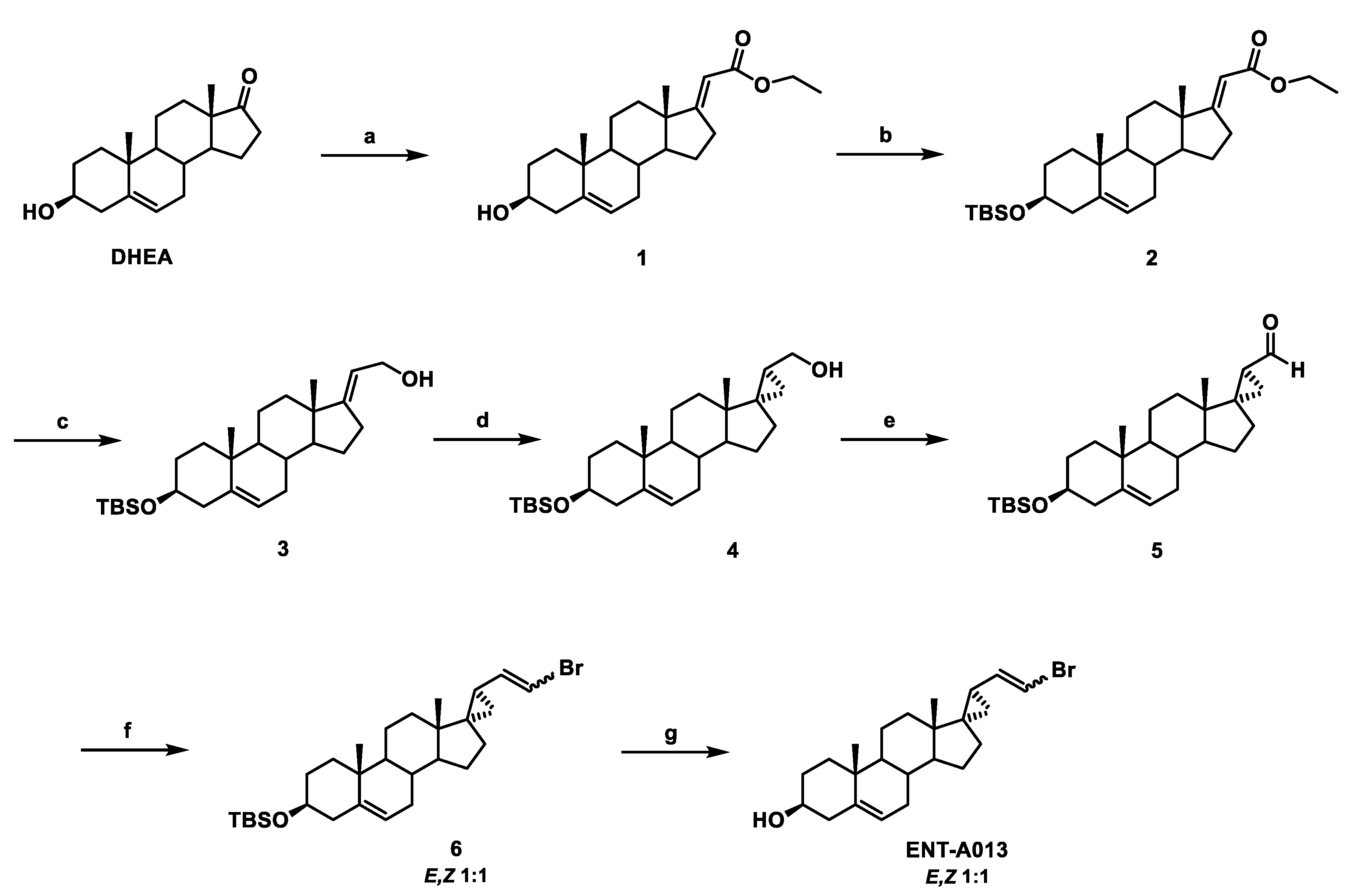
2.1. Synthesis of ENT-A013
2.2. Synthesis of (E)-(3β-Hydroxy-5-Androsten-17-Ylidene)Ethyl Ester (1)
2.3. Synthesis of (E)-[3β-(t-Butyldimethylsilyloxy)-5-Androsten-17-Ylidene]Ethyl Ester (2)
2.4. Synthesis of (E)-3β-(t-Butyldimethylsilyloxy)-Pregna-5,17(20)-Dien-21-ol (3)
2.5. Synthesis of (17S,20S)-3β-(t-Butyldimethylsilyloxy)-17α,20-Methan-5-Pregnane-21-ol (4)
2.6. Synthesis of (17S,20S)-3β-(t-Butyldimethylsilyloxy)-17α,20-Methan-5-Pregnane-21-al (5)
2.7. Synthesis of (((2′R,3S,17S)-2′-(2-Bromovinyl)-5-Androstene-17,1′-Cyclopropan)-3-yl)oxy)(Tert-Butyl)Dimethylsilane (6)
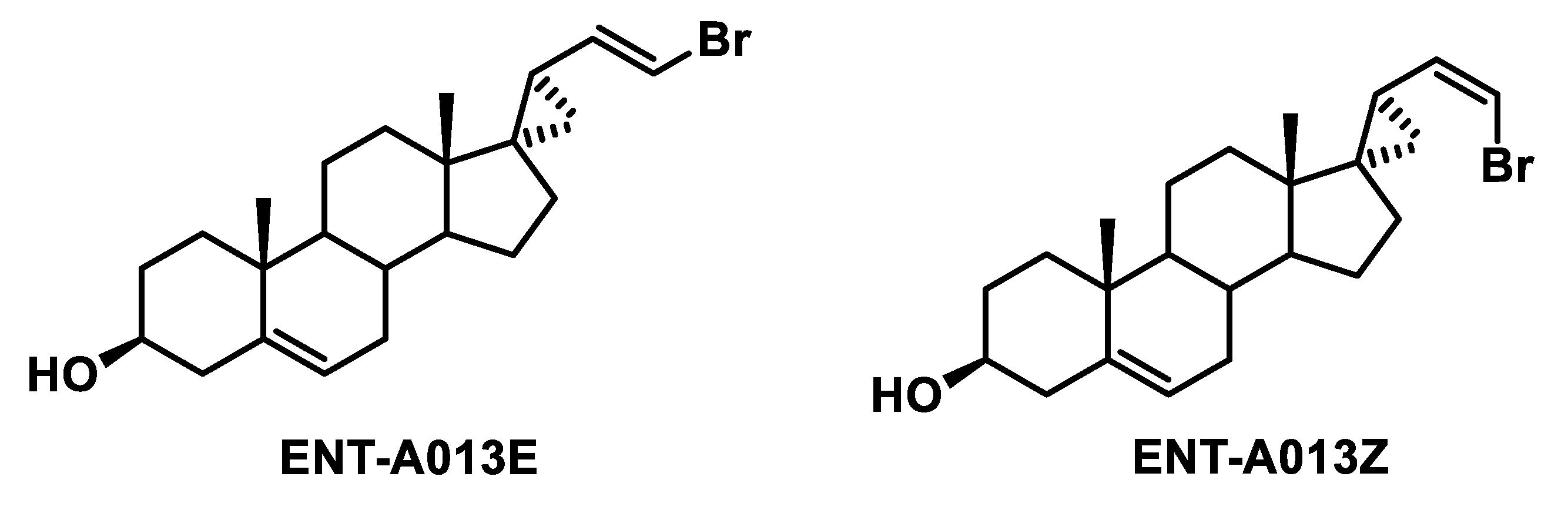
2.8. Synthesis of (2′R,3S,17S)-2′-(2-Bromovinyl)-5-Androstene-17,1′-Cyclopropan-3-ol (ENT-A013)
2.9. Cell Lines
2.10. Immunoprecipitation and Immunoblotting
2.11. CellTox Assay
2.12. Primary Dorsal Root Ganglia Neurons
2.13. Primary Hippocampal Neurons
2.14. Preparation of Aβ Oligomers
2.15. Electrophysiology
2.16. Metabolic Stability
2.17. Isozyme-Specific CYP450-Metabolism
2.18. Molecular Modelling and Docking Studies
2.19. Statistical Analysis
3. Results
3.1. Chemistry
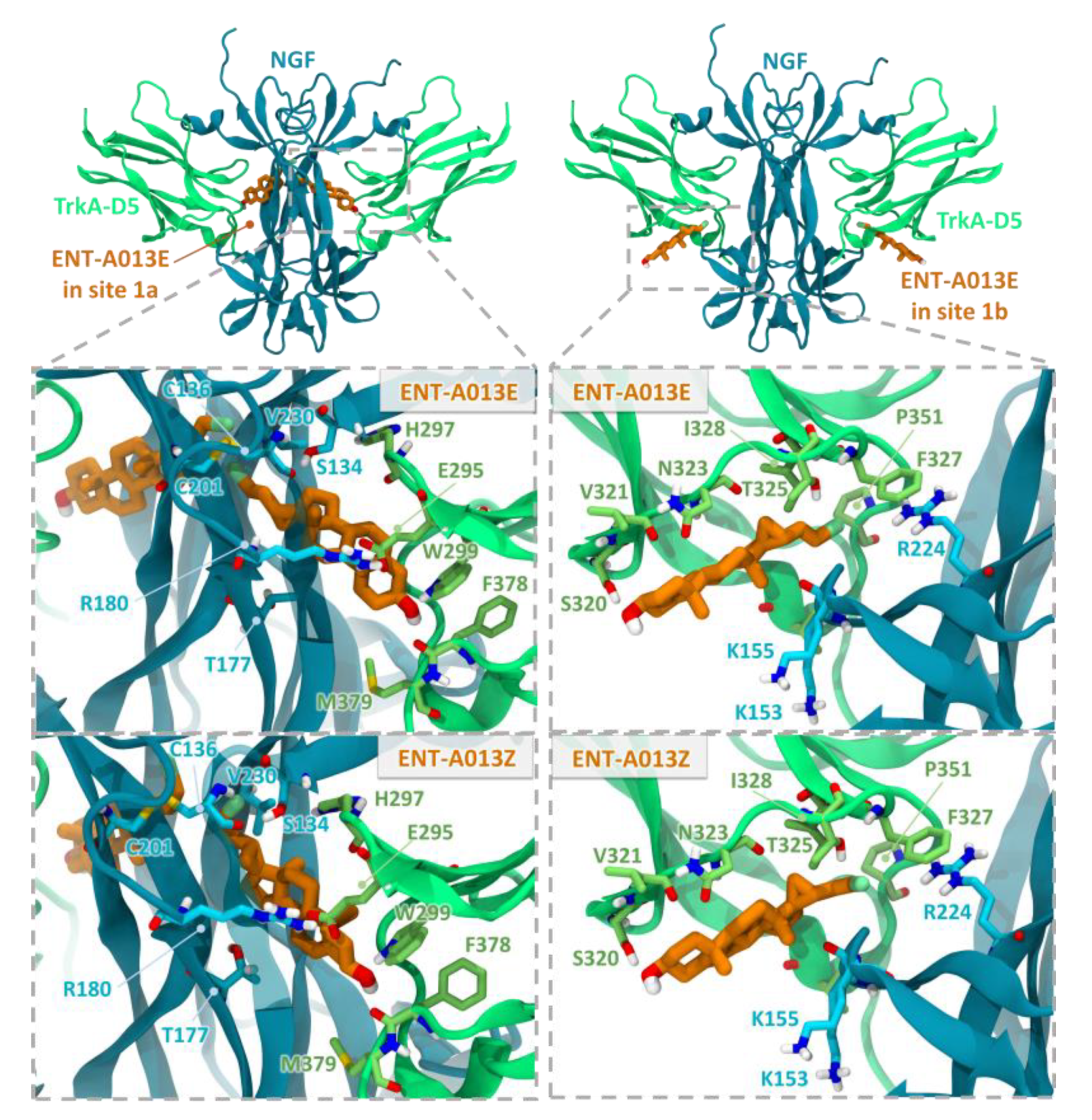
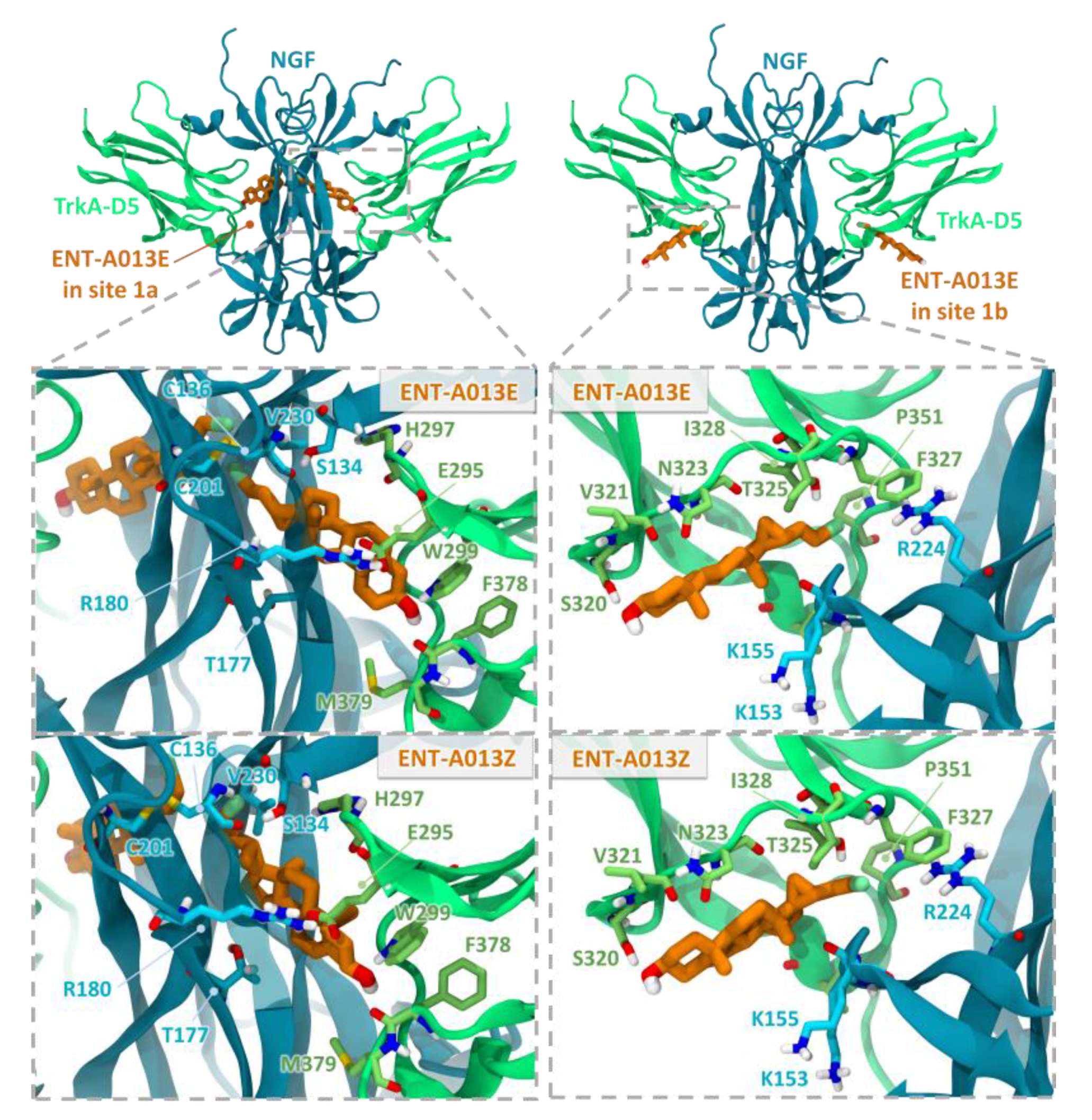
3.2. Two Possible Sites of Interaction for ENT-A013 Isomers (ENT-A013E and ENT-A013Z) with the TrkA/NGF Receptor Complex
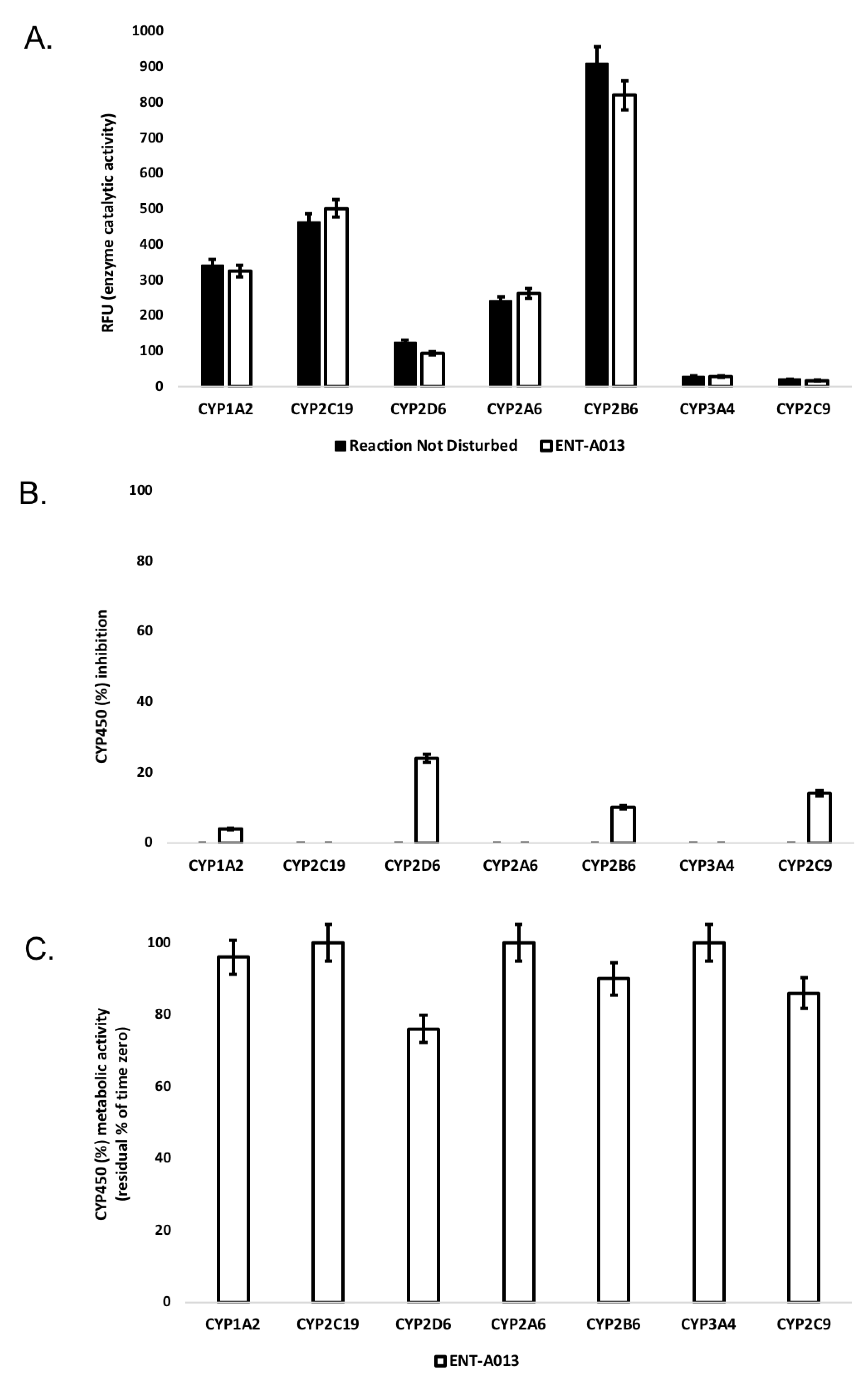
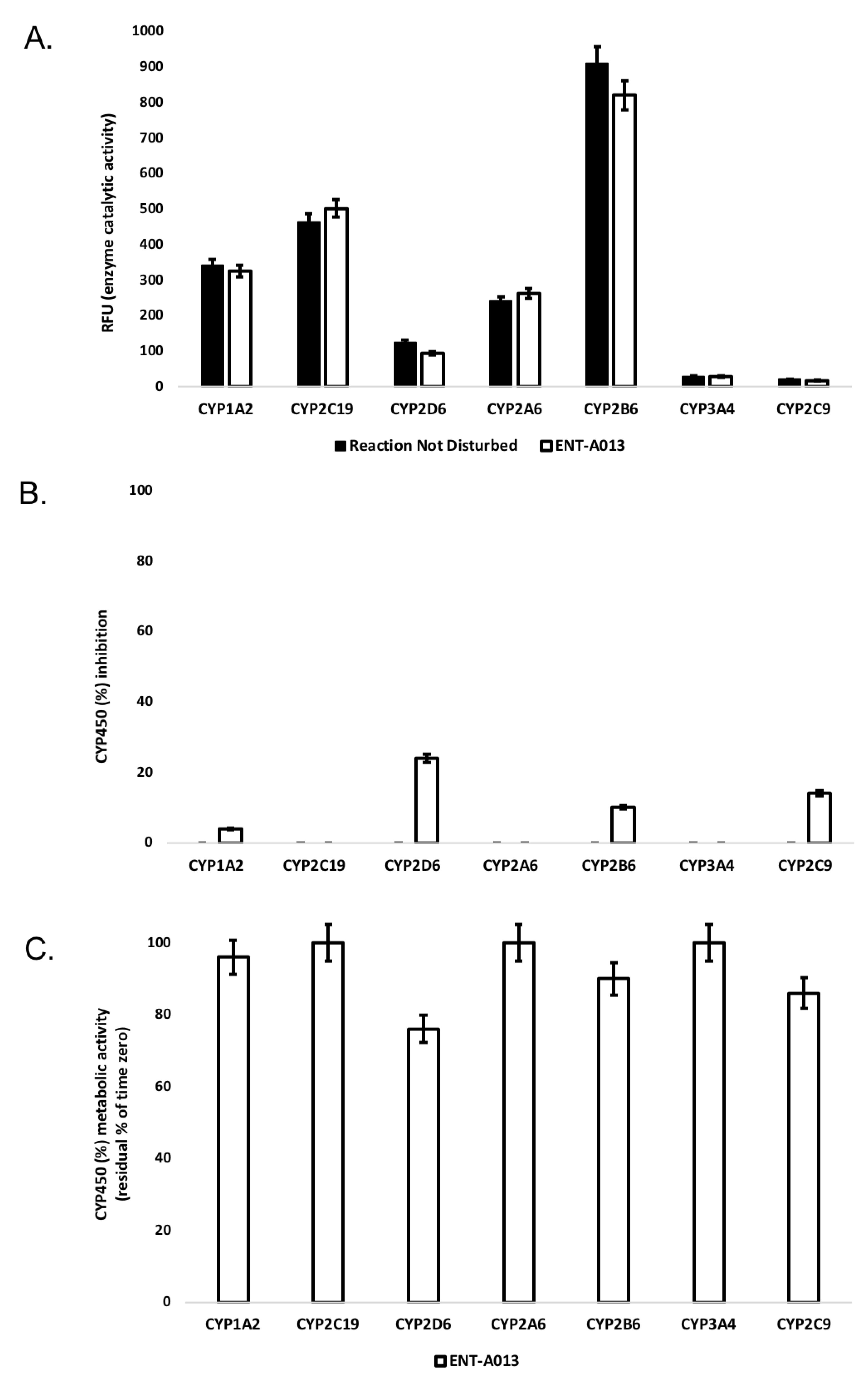
3.3. ENT-A013 Exhibits Slow Depletion in Human Liver Microsomes
3.4. ENT-A013 Shows Weak-to-Moderate CYP Inhibition
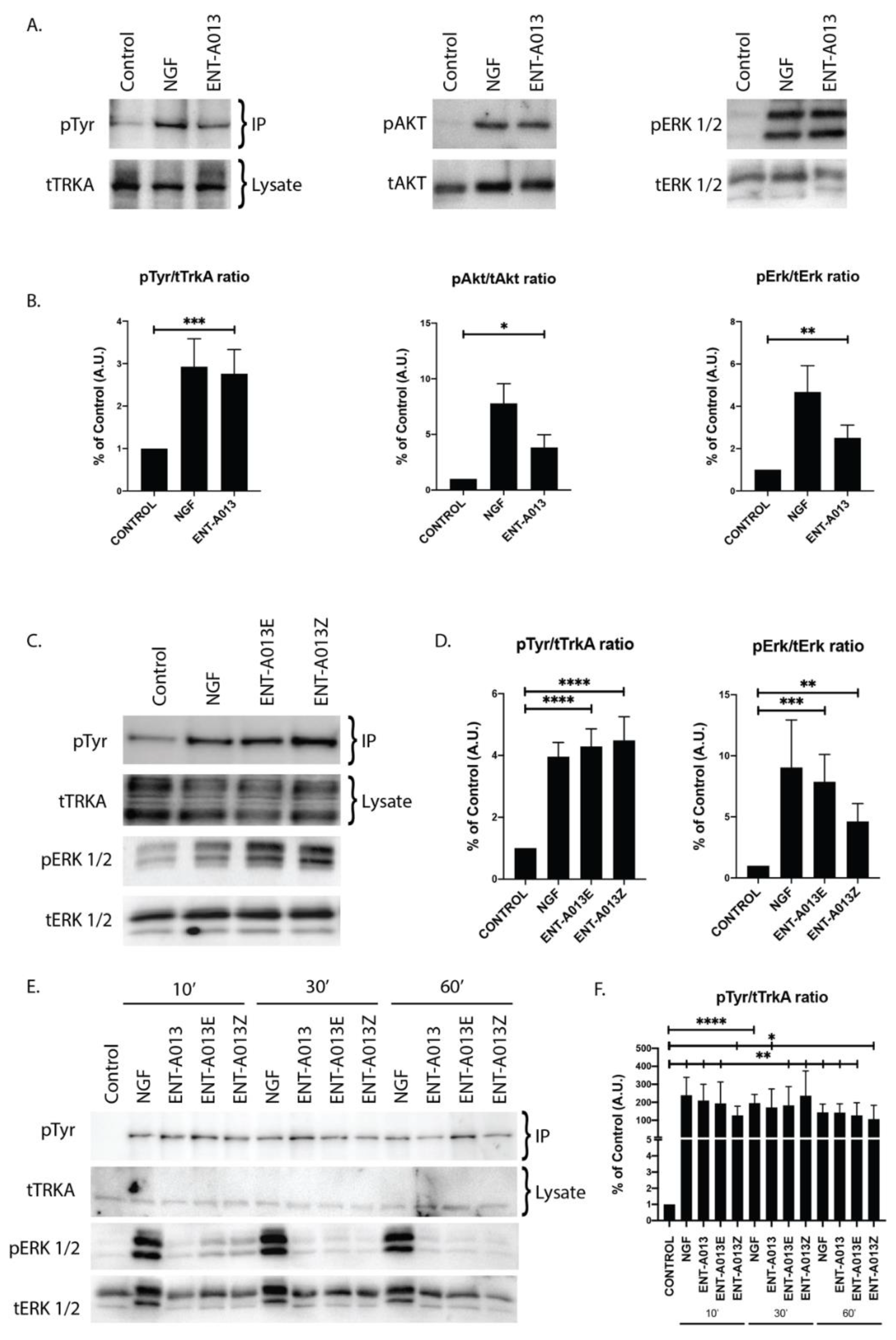
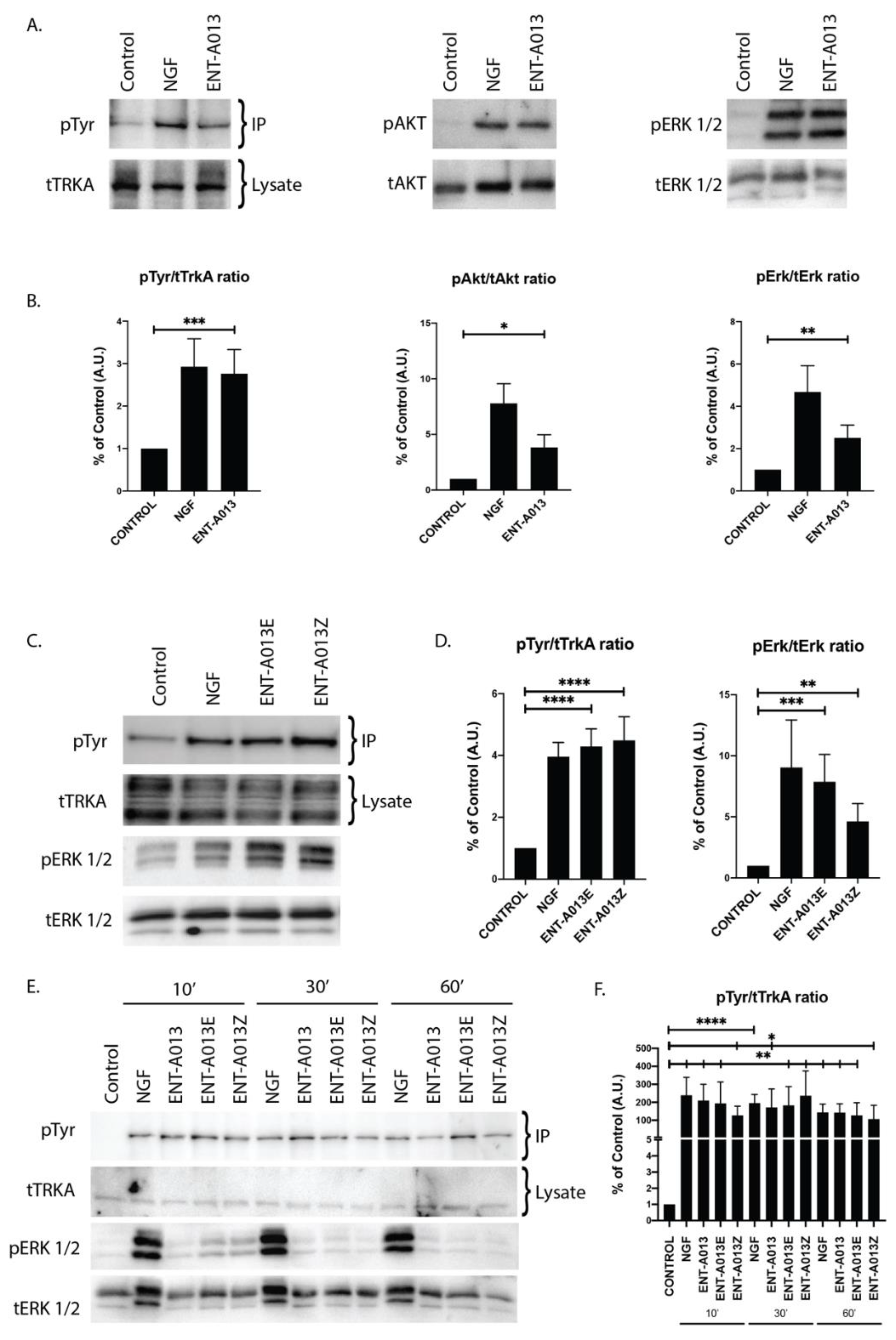
3.5. ENT-A013 Activates TrkA and Its Downstream Signaling Kinases Akt and Erk1/2 but Not TrkB, TrkC or p75NTR Neurotrophin Receptor
3.6. Both ENT-A013 Geometrical Isomers Activate TrkA Receptor and Its Downstream Signaling Kinase Erk1/2
3.7. ENT-A013, ENT-A013E and ENT-A013Z Sustain TrkA and Erk1/2 Phosphorylation Long Term and Up to 1 h after Treatment
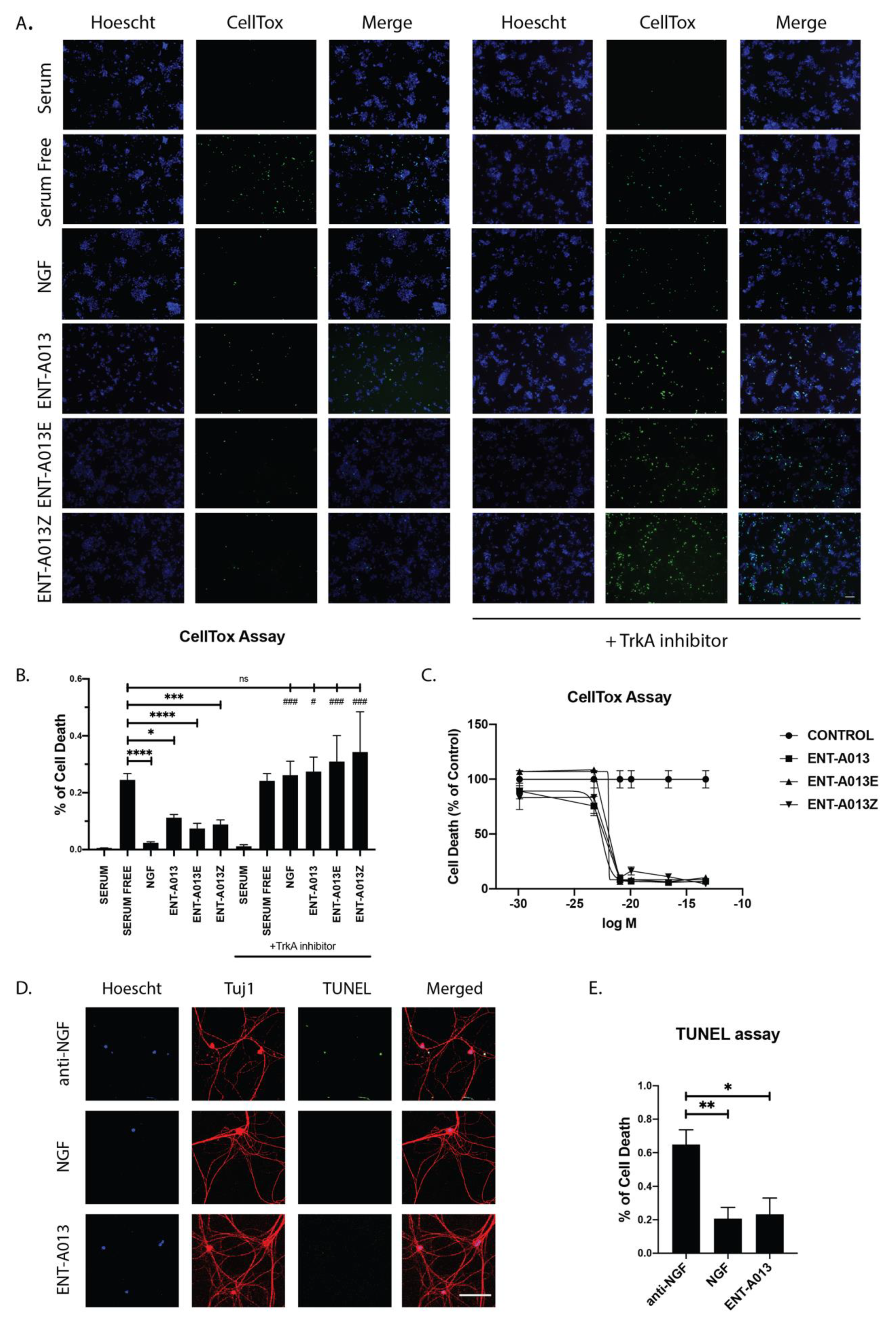
3.8. ENT-A013 Protects PC12 Cells from Serum Deprivation-Induced Cell Death through TrkA Receptor
3.9. ENT-A013 Promotes Dorsal Root Ganglia Neuron Survival in the Absence of NGF
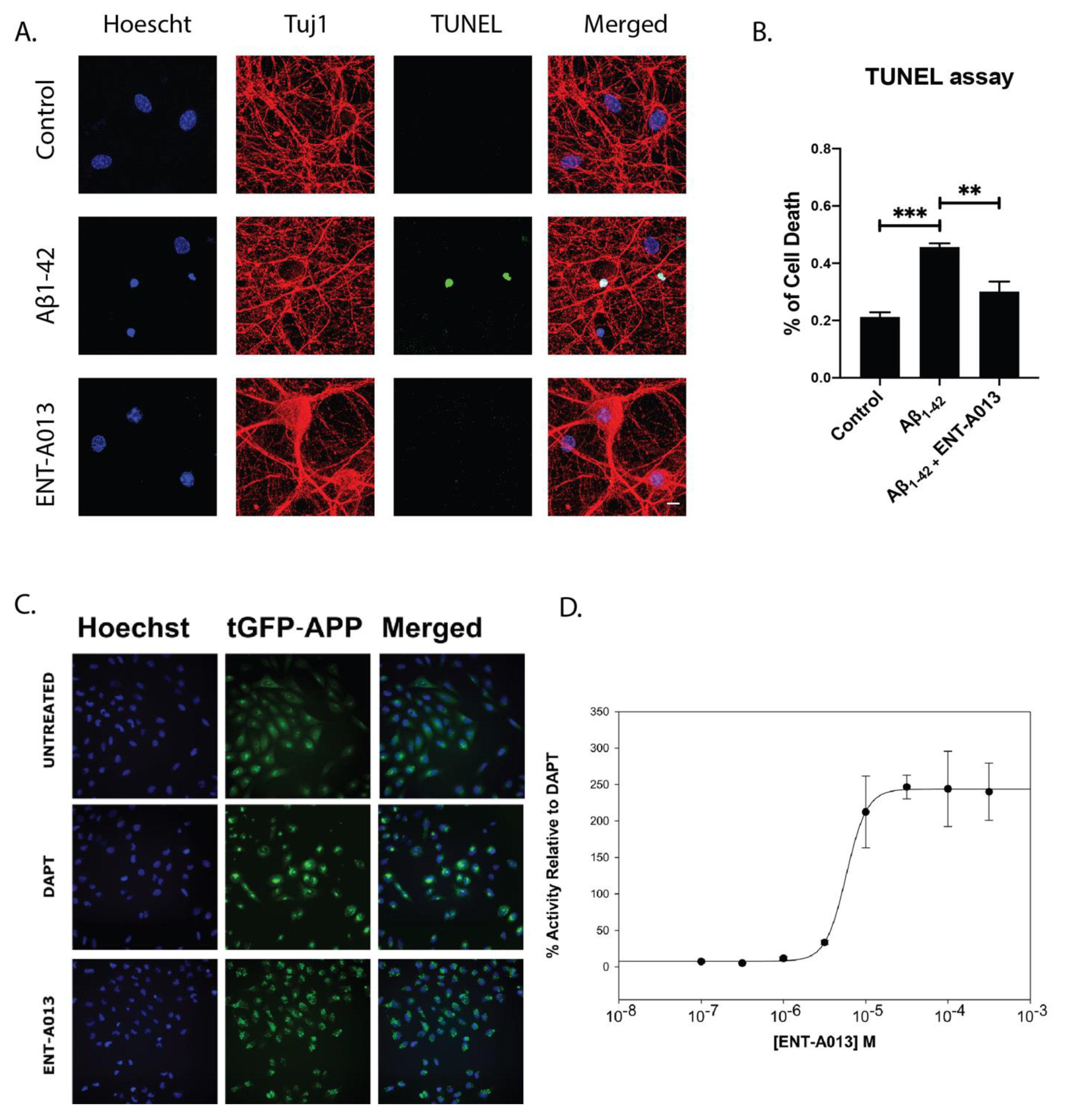
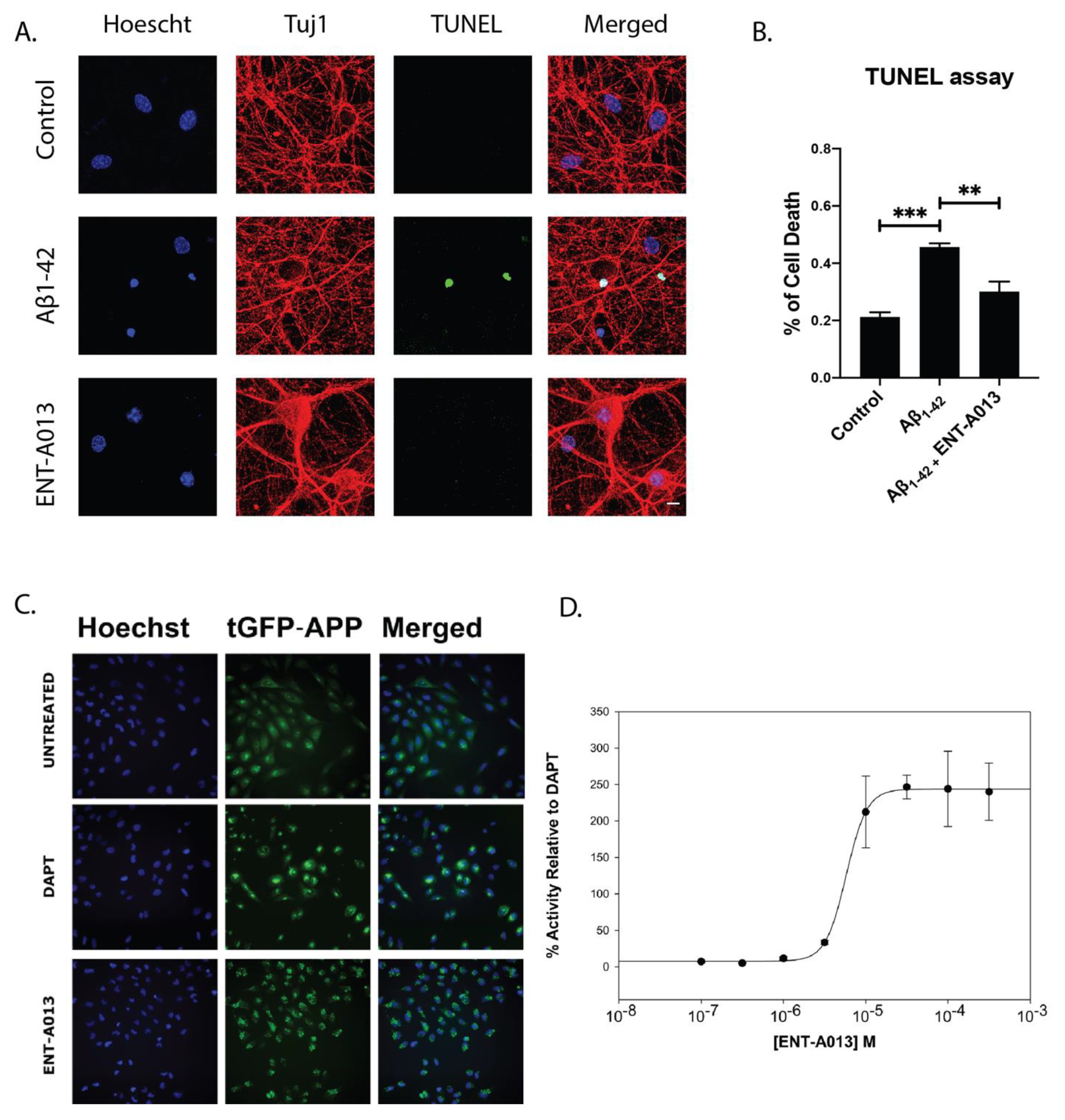
3.10. ENT-A013 Positively Modulates APP Processing and Protects Hippocampal Neurons from Aβ Toxicity
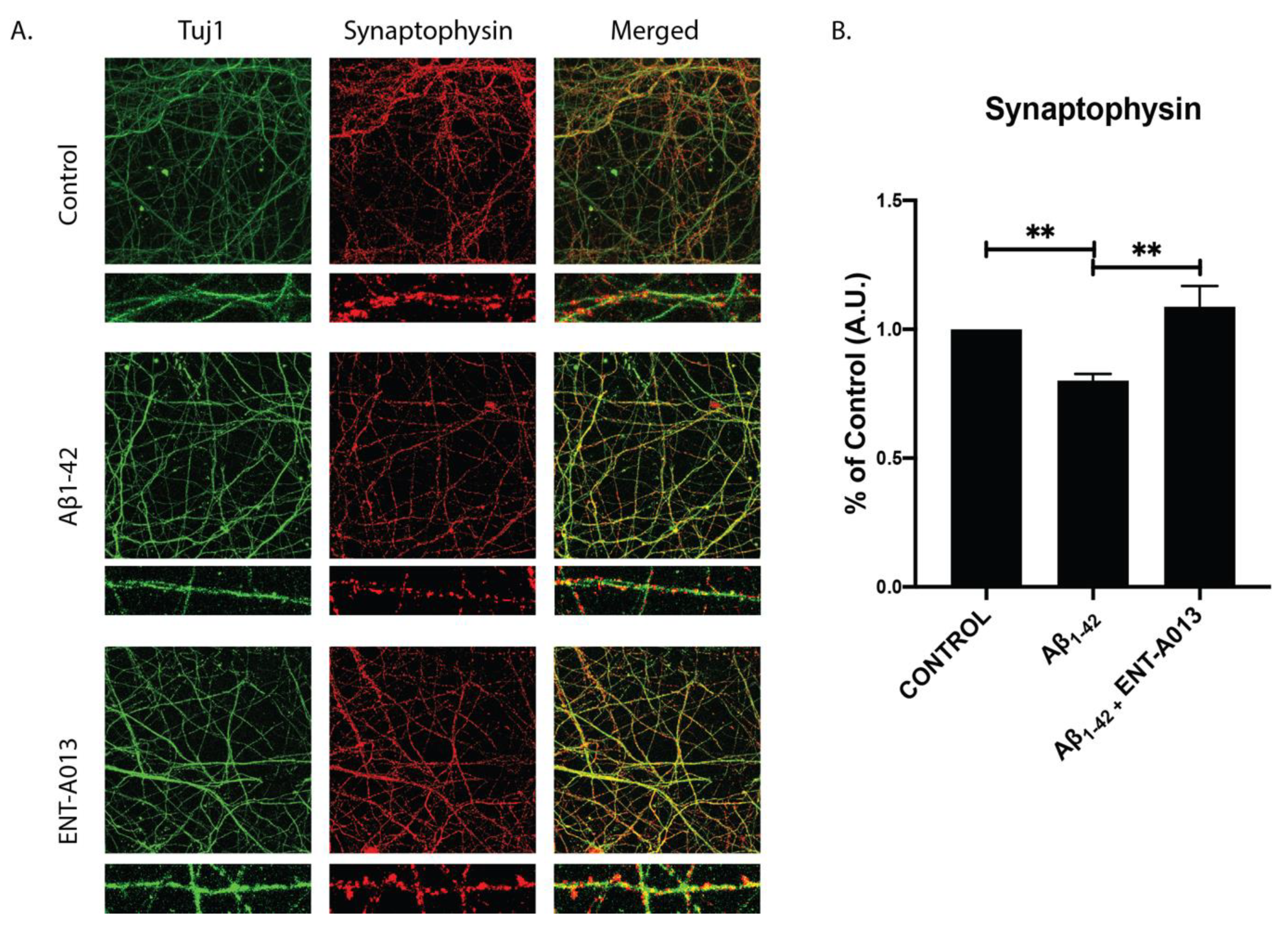
3.11. ENT-A013 Protects Hippocampal Synapses from Aβ-Induced Synapse Degeneration
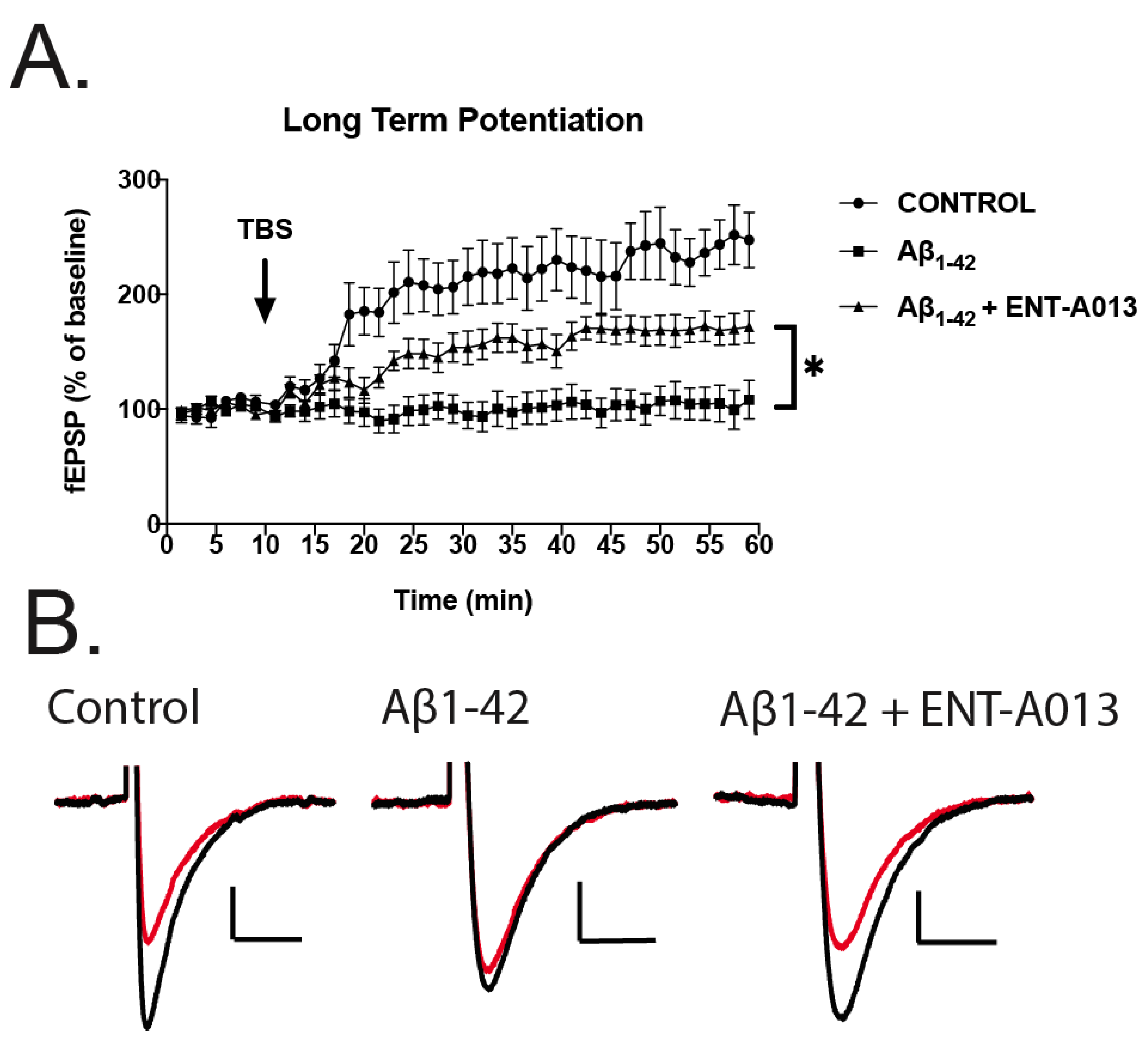
3.12. ENT-A013 Partially Reverses Long-Term Potentiation (LTP) in Brain Slices Treated with Oligomeric Aβ
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.J.; Watson, J.J.; Dawbarn, D. The Neurotrophins and Their Role in Alzheimers Disease. Curr. Neuropharmacol. 2011, 9, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Prince, M. World Alzheimer Report; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Cuello, A.; Bruno, A.; Bell, K.F. NGF-Cholinergic Dependency in Brain Aging, MCI and Alzheimers Disease. Curr. Alzheimer Res. 2007, 4, 351–358. [Google Scholar] [CrossRef]
- Calissano, P.; Amadoro, G.; Matrone, C.; Ciafre, S.; Marolda, R.; Corsetti, V.; Ciotti, M.T.; Mercanti, D.; Di Luzio, A.; Severini, C.; et al. Does the term trophic actually mean anti-amyloidogenic the case of NGF. Cell Death Differ. 2010, 17, 1126–1133. [Google Scholar] [CrossRef]
- Chen, X.Q.; Mobley, W.C. Exploring the pathogenesis of Alzheimer disease in basal forebrain cholinergic neurons: Converging insights from alternative hypotheses. Front. Neurosci. 2019, 13, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Mufson, E.J.; Counts, S.E.; Ginsberg, S.D.; Mahady, L.; Perez, S.E.; Massa, S.M.; Longo, F.M.; Ikonomovic, M.D. Nerve growth factor pathobiology during the progression of Alzheimer’s disease. Front. Neurosci. 2019, 13, 533. [Google Scholar] [CrossRef]
- Capsoni, S.; Ugolini, G.; Comparini, A.; Ruberti, F.; Berardi, N.; Cattaneo, A. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 6826–6831. [Google Scholar] [CrossRef] [Green Version]
- Capsoni, S.; Giannotta, S.; Cattaneo, A. Nerve growth factor and galantamine ameliorate early signs of neurodegeneration in anti-nerve growth factor mice. Proc. Natl. Acad. Sci. USA 2002, 99, 12432–12437. [Google Scholar] [CrossRef] [Green Version]
- Jönhagen, M.E.; Nordberg, A.; Amberla, K.; Bäckman, L.; Ebendal, T.; Meyerson, B.; Olson, L.; Seiger, Å.; Shigeta, M.; Theodorsson, E.; et al. Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 1998, 9, 246–257. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; Tong, G.; et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 2005, 11, 551–555. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Yang, J.H.; Barba, D.; Hoi-Sang, U.; Bakay, R.A.; Pay, M.M.; Masliah, E.; Conner, J.M.; Kobalka, P.; Roy, S.; et al. Nerve growth factor gene therapy activation of neuronal responses in Alzheimer disease. JAMA Neurol. 2015, 72, 1139–1147. [Google Scholar] [CrossRef]
- Rafii, M.S.; Tuszynski, M.H.; Thomas, R.G.; Barba, D.; Brewer, J.B.; Rissman, R.A.; Siffert, J.; Aisen, P.S.; AAV2-NGF Study Team. Adeno-associated viral vector (serotype 2)-nerve growth factor for patients with Alzheimer disease a randomized clinical trial. JAMA Neurol. 2018, 75, 834–841. [Google Scholar] [CrossRef]
- Lazaridis, I.; Charalampopoulos, I.; Alexaki, V.I.; Avlonitis, N.; Pediaditakis, I.; Efstathopoulos, P.; Calogeropoulou, T.; Castanas, E.; Gravanis, A. Neurosteroid dehydroepiandrosterone interacts with nerve growth factor (NGF) receptors, preventing neuronal apoptosis. PLoS Biol. 2011, 9, e1001051. [Google Scholar] [CrossRef]
- Calogeropoulou, T.; Avlonitis, N.; Minas, V.; Alexi, X.; Pantzou, A.; Charalampopoulos, I.; Zervou, M.; Vergou, V.; Katsanou, E.S.; Lazaridis, I.; et al. Novel dehydroepiandrosterone derivatives with antiapoptotic, neuroprotective activity. J. Med. Chem. 2009, 52, 6569–6587. [Google Scholar] [CrossRef]
- Pediaditakis, I.; Efstathopoulos, P.; Prousis, K.C.; Zervou, M.; Arévalo, J.C.; Alexaki, V.I.; Nikoletopoulou, V.; Karagianni, E.; Potamitis, C.; Tavernarakis, N.; et al. Selective and differential interactions of BNN27, a novel C17-spiroepoxy steroid derivative, with TrkA receptors, regulating neuronal survival and differentiation. Neuropharmacology 2016, 111, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Albuquerque, C.; Joseph, D.J.; Choudhury, P.; MacDermott, A.B. Dissection, plating, and maintenance of dorsal root ganglion neurons for monoculture and for coculture with dorsal horn neurons. Cold Spring Harb. Protoc. 2009, 4, pdb.prot5275. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Wooten, M.W. Isolation and culture of hippocampal neurons from prenatal mice. J. Vis. Exp. 2012, 65, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Poteet, E.; Xie, L.; Liu, R.; Wen, Y.; Yang, S.H. Regulation of matrix metalloproteinase 2 by oligomeric amyloid β protein. Brain Res. 2011, 1387, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Wiesmann, C.; Ultsch, M.H.; Bass, S.H.; de Vos, A.M. Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 1999, 401, 184–188. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.L.; Devine, P.W.; Phillips, J.J.; Higazi, D.R.; Lloyd, C.; Popovic, B.; Arnold, J.; Buchanan, A.; Lewis, A.; Goodman, J.; et al. Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci. Rep. 2016, 6, 38644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-2: Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA; Impact, Schrödinger, LLC: New York, NY, USA; Prime, Schrödinger, LLC: New York, NY, USA, 2020.
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-2: SiteMap; Schrödinger LLC: New York, NY, USA, 2020.
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T. New Method for Fast and Accurate Binding-site Identification and Analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-2: Maestro; Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-2: LigPrep; Schrödinger, LLC: New York, NY, USA, 2020.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2020-2: Glide; Schrödinger, LLC: New York, NY, USA, 2020.
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an Induced Fit Receptor Structure in Virtual Screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-2: Induced Fit Docking Protocol; Glide, Schrödinger, LLC: New York, NY, USA; Prime, Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-2: Prime; Schrödinger, LLC: New York, NY, USA, 2020.
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kim, H.Y.; Salvi, L.; Carroll, P.J.; Walsh, P.J. Highly enantio- and diastereoselective one-pot methods for the synthesis of halocyclopropyl alcohols. J. Am. Chem. Soc. 2009, 131, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ma, S.; Yuan, Z.; Chen, P.; Xie, X.; Wang, X.; She, X. Palladium-Promoted Neutral 1,4-Brook Rearrangement/Intramolecular Allylic Cyclization Cascade Reaction: A Strategy for the Construction of Vinyl Cyclobutanols. Org. Lett. 2017, 19, 3478–3481. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Chan, C.B.; Weinshenker, D.; Hall, R.A.; Xiao, G.; Ye, K. Amitriptyline is a TrkA and TrkB Receptor Agonist that Promotes TrkA/TrkB Heterodimerization and Has Potent Neurotrophic Activity. Chem. Biol. 2009, 16, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Shoemark, D.K.; Williams, C.; Fahey, M.S.; Watson, J.J.; Tyler, S.J.; Scoltock, S.J.; Ellis, R.Z.; Wickenden, E.; Burton, A.J.; Hemmings, J.L.; et al. Design and nuclear magnetic resonance (NMR) structure determination of the second extracellular immunoglobulin tyrosine kinase A (TrkAIg2) domain construct for binding site elucidation in drug discovery. J. Med. Chem. 2015, 58, 767–777. [Google Scholar] [CrossRef]
- Scarpi, D.; Cirelli, D.; Matrone, C.; Castronovo, G.; Rosini, P.; Occhiato, E.G.; Romano, F.; Bartali, L.; Clemente, A.M.; Bottegoni, G.; et al. Low molecular weight, non-peptidic agonists of TrkA receptor with NGF-mimetic activity. Cell Death Dis. 2012, 3, e339. [Google Scholar] [CrossRef] [PubMed]
- Barter, Z.E.; Bayliss, M.K.; Beaune, P.H.; Boobis, A.R.; Carlile, D.J.; Edwards, R.J.; Brian Houston, J.; Lake, B.G.; Lipscomb, J.C.; Pelkonen, O.R.; et al. Scaling Factors for the Extrapolation of In Vivo Metabolic Drug Clearance From In Vitro Data: Reaching a Consensus on Values of Human Micro-somal Protein and Hepatocellularity Per Gram of Liver. Curr. Drug Metab. 2006, 8, 33–45. [Google Scholar] [CrossRef]
- Masimirembwa, C.M.; Bredberg, U.; Andersson, T.B. Metabolic stability for drug discovery and development: Pharmacokinetic and biochemical challenges. Clin. Pharmacokinet. 2003, 42, 515–528. [Google Scholar] [CrossRef]
- EMA. Guideline on the Investigation of Drug Interactions; Guid Doc: London, UK, 2013; Available online: https://www.ema.europa.eu/en/documents/other/overview-comments-received-guideline-investigation-drug-interactions_en.pdf (accessed on 1 January 2022).
- EMA. Guideline on the Investigation of Drug Interactions; Guid Doc: London, UK, 2012; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf (accessed on 1 January 2022).
- FDA. Drug Development and Drug Interactions; Guid Doc: London, UK, 2020. [Google Scholar]
- Houston, J.B. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol. 1994, 47, 1469–1479. [Google Scholar] [CrossRef]
- Jusko, W.J.; Li, X. Assessment of the Kochak-Benet Equation for Hepatic Clearance for the Parallel-Tube Model: Relevance of Classic Clearance Concepts in PK and PBPK. AAPS J. 2022, 24, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Kosugi, Y.; Hosea, N. Direct Comparison of Total Clearance Prediction: Computational Machine Learning Model versus Bottom-Up Approach Using in Vitro Assay. Mol. Pharm. 2020, 17, 2299–2309. [Google Scholar] [CrossRef] [PubMed]
- Yim, D.S.; Bae, S.H.; Choi, S. Predicting human pharmacokinetics from preclinical data: Clearance. Transl. Clin. Pharmacol. 2021, 29, 78–87. [Google Scholar] [CrossRef] [PubMed]
- McGinnity, D.F.; Parker, A.J.; Soars, M.; Riley, R.J. Automated Definition of the Enzymology of Drug Oxidation by the Major Human Drug Metabolizing Cytochrome P450s. Drug Metab. Dispos. 2000, 28, 1327–1334. [Google Scholar]
- Pelkonen, O.; Turpeinen, M. In vitro-in vivo extrapolation of hepatic clearance: Biological tools, scaling factors, model assumptions and correct concentrations. Xenobiotica 2007, 37, 1066–1089. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Neltner, J.H.; Nelson, P.T. Is synaptic loss a unique hallmark of Alzheimer’s disease? Biochem. Pharmacol. 2014, 88, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wilde, M.C.; Overk, C.R.; Sijben, J.W.; Masliah, E. Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimer’s Dement. 2016, 12, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arancio, O.; Chao, M.V. Neurotrophins, synaptic plasticity and dementia. Curr. Opin. Neurobiol. 2007, 17, 325–330. [Google Scholar] [CrossRef]
- Nicoll, R.A. A Brief History of Long-Term Potentiation. Neuron 2017, 93, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.P.; O’Brien, L.C.; Brohawn, D.G. Pharmacological properties of microneurotrophin drugs developed for treatment of amyotrophic lateral sclerosis. Biochem. Pharmacol. 2016, 117, 68–77. [Google Scholar] [CrossRef]
- Tsika, C.; Tzatzarakis, M.N.; Antimisiaris, S.G.; Tsoka, P.; Efstathopoulos, P.; Charalampopoulos, I.; Gravanis, A.; Tsilimbaris, M.K. Quantification of BNN27, a novel neuroprotective 17-spiroepoxy dehydroepiandrosterone derivative in the blood and retina of rodents, after single intraperitoneal administration. Pharmacol. Res. Perspect. 2021, 9, 1–8. [Google Scholar] [CrossRef]
- Danenberg, H.D.; Haring, R.; Fisher, A.; Pittel, Z.; Gurwitz, D.; Heldman, E. Dehydroepiandrosterone (DHEA) increases production and release of Alzheimer’s amyloid precursor protein. Life Sci. 1996, 59, 1651–1657. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Bardini, P.; Santoro, G.; Davit, A.; Di Simone, D.; Danni, O.; Tabaton, M. Dehydroepiandrosterone reduces expression and activity of BACE in NT 2 neurons exposed to oxidative stress. Neurobiol. Dis. 2003, 14, 291–301. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in Neuronal Development and Function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [Green Version]
- Kilimann, I.; Grothe, M.; Heinsen, H.; Alho, E.J.; Grinberg, L.; Amaro, E., Jr.; Dos Santos, G.A.; Da Silva, R.E.; Mitchell, A.J.; Frisoni, G.B.; et al. Subregional basal forebrain atrophy in alzheimer’s disease: A multicenter study. J. Alzheimer’s Dis. 2014, 40, 687–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teipel, S.J.; Meindl, T.; Grinberg, L.; Grothe, M.; Cantero, J.L.; Reiser, M.F.; Möller, H.J.; Heinsen, H.; Hampel, H. The cholinergic system in mild cognitive impairment and Alzheimer’s disease: An in Vivo MRI and DTI study. Hum. Brain Mapp. 2011, 32, 1349–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triaca, V.; Sposato, V.; Bolasco, G.; Ciotti, M.T.; Pelicci, P.; Bruni, A.C.; Cupidi, C.; Maletta, R.; Feligioni, M.; Nisticò, R.; et al. NGF controls APP cleavage by downregulating APP phosphorylation at Thr668: Relevance for Alzheimer’s disease. Aging Cell 2016, 15, 661–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triaca, V.; Coccurello, R.; Giacovazzo, G. The neuronal Shc adaptor in Alzheimer’s Disease. Aging 2018, 10, 5–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, M.S.; Fahnestock, M. ProNGF, but not NGF, switches from neurotrophic to apoptotic activity in response to reductions in TrKA receptor levels. Int. J. Mol. Sci. 2017, 18, 599. [Google Scholar] [CrossRef] [Green Version]
- Roussarie, J.P.; Yao, V.; Rodriguez-Rodriguez, P.; Oughtred, R.; Rust, J.; Plautz, Z.; Kasturia, S.; Albornoz, C.; Wang, W.; Schmidt, E.F.; et al. Selective Neuronal Vulnerability in Alzheimer’s Disease: A Network-Based Analysis. Neuron 2020, 107, 821–835.e12. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J. Selective Loss Of Central Cholinergic Neurons In Alzheimer’s Disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Appel, S.H. A unifying hypothesis for the cause of amyotrophic lateral sclerosis, parkinsonism, and alzheimer disease. Ann. Neurol. 1981, 10, 499–505. [Google Scholar] [CrossRef]
- Birks, J.; Harvey, R. Donepezil for dementia due to Alzheimer ’ s disease (Review) summary of findings for the main comparison. Cochrane Database Syst. Rev. 2018, 6, CD001190. [Google Scholar]
- Zhu, C.W.; Livote, E.E.; Scarmeas, N.; Albert, M.; Brandt, J.; Blacker, D.; Sano, M.; Stern, Y. Long-term associations between cholinesterase inhibitors and memantine use and health outcomes among patients with Alzheimer’s disease. Alzheimer’s Dement. 2013, 9, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Secnik, J.; Schwertner, E.; Alvarsson, M.; Hammar, N.; Fastbom, J.; Winblad, B.; Garcia-Ptacek, S.; Religa, D.; Eriksdotter, M. Cholinesterase inhibitors in patients with diabetes mellitus and dementia: An open-cohort study of ∼23 000 patients from the Swedish Dementia Registry. BMJ Open Diabetes Res. Care 2020, 8, e000833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimer’s Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020, 19, 111–112. [Google Scholar] [CrossRef] [Green Version]
- Howard, R.; Liu, K.Y. Questions EMERGE as Biogen claims aducanumab turnaround. Nat. Rev. Neurol. 2020, 16, 63–64. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Feldman, H.H.; Scheltens, P. The ‘rights’ of precision drug development for Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 1–22. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, 1–24. [Google Scholar] [CrossRef]
- Longo, F.M.; Massa, S.M. Neurotrophin Receptor-Based Strategies for Alzheimer’s Disease. Curr. Alzheimer Res. 2005, 2, 167–169. [Google Scholar] [CrossRef]
- Longo, F.M.; Massa, S.M. Small-molecule modulation of neurotrophin receptors: A strategy for the treatment of neurological disease. Nat. Rev. Drug Discov. 2013, 12, 507–525. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogdakis, T.; Charou, D.; Latorrata, A.; Papadimitriou, E.; Tsengenes, A.; Athanasiou, C.; Papadopoulou, M.; Chalikiopoulou, C.; Katsila, T.; Ramos, I.; et al. Development and Biological Characterization of a Novel Selective TrkA Agonist with Neuroprotective Properties against Amyloid Toxicity. Biomedicines 2022, 10, 614. https://doi.org/10.3390/biomedicines10030614
Rogdakis T, Charou D, Latorrata A, Papadimitriou E, Tsengenes A, Athanasiou C, Papadopoulou M, Chalikiopoulou C, Katsila T, Ramos I, et al. Development and Biological Characterization of a Novel Selective TrkA Agonist with Neuroprotective Properties against Amyloid Toxicity. Biomedicines. 2022; 10(3):614. https://doi.org/10.3390/biomedicines10030614
Chicago/Turabian StyleRogdakis, Thanasis, Despoina Charou, Alessia Latorrata, Eleni Papadimitriou, Alexandros Tsengenes, Christina Athanasiou, Marianna Papadopoulou, Constantina Chalikiopoulou, Theodora Katsila, Isbaal Ramos, and et al. 2022. "Development and Biological Characterization of a Novel Selective TrkA Agonist with Neuroprotective Properties against Amyloid Toxicity" Biomedicines 10, no. 3: 614. https://doi.org/10.3390/biomedicines10030614
APA StyleRogdakis, T., Charou, D., Latorrata, A., Papadimitriou, E., Tsengenes, A., Athanasiou, C., Papadopoulou, M., Chalikiopoulou, C., Katsila, T., Ramos, I., Prousis, K. C., Wade, R. C., Sidiropoulou, K., Calogeropoulou, T., Gravanis, A., & Charalampopoulos, I. (2022). Development and Biological Characterization of a Novel Selective TrkA Agonist with Neuroprotective Properties against Amyloid Toxicity. Biomedicines, 10(3), 614. https://doi.org/10.3390/biomedicines10030614