
Inositol Polyphosphate Kinases, Fungal Virulence and Drug Discovery

 , ,
, , 
Abstract
:1. Invasive Fungal Infections: The Clinical Burden
2. Molecular Targets of Antifungal Drugs and Drug Limitations
3. Cryptococcus neoformans: A Study Model for Virulence and Drug Development
4. Signaling via Plc1 in C. neoformans: The Role of Inositol 1,4,5-Trisphosphate (IP3)
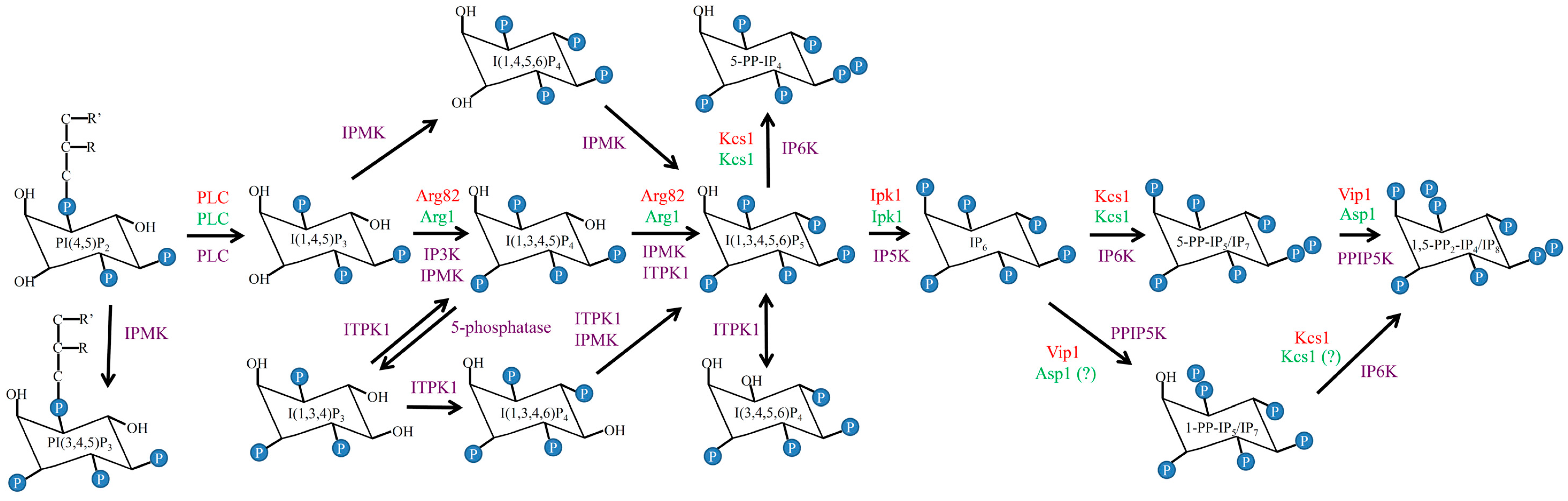
5. Delineating the IP Biosynthesis Pathway in C. neoformans
6. The Contribution of IP/PP-IP Species to Cryptococcal Cellular Function
7. 5-PP-IP5 Plays a Critical Role in Cryptococcal Virulence
8. The Plc1/IPK Pathway: A Signaling or a Metabolic Pathway?
8.1. Features of a Metabolic Pathway
8.2. Features of a Signaling Pathway
9. How IP/PP-IP Regulate Cellular Function
10. The Plc1/IPK Pathway as a Target for Antifungal Drug Development
11. Structural Studies of IPKs
12. Advances in Identifying IPK Inhibitor Specificity
13. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 113–165. [Google Scholar] [CrossRef] [PubMed]
- Vallabhaneni, S.; Mody, R.K.; Walker, T.; Chiller, T. The global burden of fungal diseases. Infect. Dis. Clin. N. Am. 2016, 30, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Low, C.Y.; Rotstein, C. Emerging fungal infections in immunocompromised patients. F1000 Med. Rep. 2011, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Miceli, M.H.; Diaz, J.A.; Lee, S.A. Emerging opportunistic yeast infections. Lancet Infect. Dis. 2011, 11, 142–151. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Diekema, D.J. Rare and emerging opportunistic fungal pathogens: Concern for resistance beyond Candida albicans and Aspergillus fumigatus. J. Clin. Microbiol. 2004, 42, 4419–4431. [Google Scholar] [CrossRef] [PubMed]
- Armstrong-James, D.; Meintjes, G.; Brown, G.D. A neglected epidemic: Fungal infections in HIV/AIDS. Trends Microbiol. 2014, 22, 120–127. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS. Global AIDS Update. Available online: http://www.unaids.org/en/resources/documents/2016/Global-AIDS-update-2016. (Accessed on 1 July 2016).
- CDC. Opportunistic Infections. Available online: http://www.cdc.gov/hiv/basics/livingwithhiv/opportunisticinfections.html (accessed on 10 June 2016).
- Menzin, J.; Meyers, J.L.; Friedman, M.; Korn, J.R.; Perfect, J.R.; Langston, A.A.; Danna, R.P.; Papadopoulos, G. The economic costs to United States hospitals of invasive fungal infections in transplant patients. Am. J. Infect. Control 2011, 39, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Abruzzo, G.K.; Flattery, A.M.; Gill, C.J.; Kong, L.; Smith, J.G.; Pikounis, V.B.; Balkovec, J.M.; Bouffard, A.F.; Dropinski, J.F.; Rosen, H.; et al. Evaluation of the echinocandin antifungal MK-0991 (L-743,872): Efficacies in mouse models of disseminated aspergillosis, candidiasis, and cryptococcosis. Antimicrob. Agents Chemother. 1997, 41, 2333–2338. [Google Scholar] [PubMed]
- Bartizal, K.; Gill, C.J.; Abruzzo, G.K.; Flattery, A.M.; Kong, L.; Scott, P.M.; Smith, J.G.; Leighton, C.E.; Bouffard, A.; Dropinski, J.F.; et al. In vitro preclinical evaluation studies with the echinocandin antifungal MK-0991 (L-743,872). Antimicrob. Agents Chemother. 1997, 41, 2326–2332. [Google Scholar] [PubMed]
- Kartsonis, N.A.; Nielsen, J.; Douglas, C.M. Caspofungin: The first in a new class of antifungal agents. Drug Resist. Updates 2003, 6, 197–218. [Google Scholar] [CrossRef]
- Krishnarao, T.V.; Galgiani, J.N. Comparison of the in vitro activities of the echinocandin LY303366, the pneumocandin MK-0991, and fluconazole against Candida species and Cryptococcus neoformans. Antimicrob. Agents Chemother. 1997, 41, 1957–1960. [Google Scholar] [PubMed]
- Howard, S.J.; Cerar, D.; Anderson, M.J.; Albarrag, A.; Fisher, M.C.; Pasqualotto, A.C.; Laverdiere, M.; Arendrup, M.C.; Perlin, D.S.; Denning, D.W. Frequency and evolution of Azole resistance in Aspergillus fumigatus associated with treatment failure. Emerg. Infect. Dis. 2009, 15, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.J.; Webster, I.; Moore, C.B.; Gardiner, R.E.; Park, S.; Perlin, D.S.; Denning, D.W. Multi-azole resistance in Aspergillus fumigatus. Int. J. Antimicrob. Agents 2006, 28, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, A.; Kathuria, S.; Randhawa, H.S.; Gaur, S.N.; Klaassen, C.H.; Meis, J.F. Isolation of multiple-triazole-resistant Aspergillus fumigatus strains carrying the TR/L98H mutations in the cyp51A gene in India. J. Antimicrob. Chemother. 2012, 67, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Bueid, A.; Howard, S.J.; Moore, C.B.; Richardson, M.D.; Harrison, E.; Bowyer, P.; Denning, D.W. Azole antifungal resistance in Aspergillus fumigatus: 2008 and 2009. J. Antimicrob. Chemother. 2010, 65, 2116–2118. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.D.; Johnson, M.D.; Pfeiffer, C.D.; Jimenez-Ortigosa, C.; Catania, J.; Booker, R.; Castanheira, M.; Messer, S.A.; Perlin, D.S.; Pfaller, M.A. Increasing echinocandin resistance in Candida glabrata: Clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin. Infect. Dis. 2013, 56, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, A.A.; Farley, M.M.; Harrison, L.H.; Stein, B.; Hollick, R.; Lockhart, S.R.; Magill, S.S.; Derado, G.; Park, B.J.; Chiller, T.M. Changes in incidence and antifungal drug resistance in candidemia: Results from population-based laboratory surveillance in Atlanta and Baltimore, 2008–2011. Clin. Infect. Dis. 2012, 55, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A.; Rhomberg, P.R.; Messer, S.A.; Jones, R.N.; Castanheira, M. Isavuconazole, micafungin, and 8 comparator antifungal agents’ susceptibility profiles for common and uncommon opportunistic fungi collected in 2013: Temporal analysis of antifungal drug resistance using CLSI species-specific clinical breakpoints and proposed epidemiological cutoff values. Diagn. Microbiol. Infect. Dis. 2015, 82, 303–313. [Google Scholar] [PubMed]
- WHO. Antimicrobial Resistance: Global Report on Surveillance; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- WHO. Rapid Advice: Diagnosis, Prevention and Management of Cryptococcal Disease in HIV-Infected Adults, Adolescents and Children; WHO: Geneva, Switzerland, 2011. [Google Scholar]
- Perfect, J.R.; Dismukes, W.E.; Dromer, F.; Goldman, D.L.; Graybill, J.R.; Hamill, R.J.; Harrison, T.S.; Larsen, R.A.; Lortholary, O.; Nguyen, M.H.; et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the infectious diseases society of America. Clin. Infect. Dis. 2010, 50, 291–322. [Google Scholar] [CrossRef] [PubMed]
- Day, J.N.; Chau, T.T.; Wolbers, M.; Mai, P.P.; Dung, N.T.; Mai, N.H.; Phu, N.H.; Nghia, H.D.; Phong, N.D.; Thai, C.Q.; et al. Combination antifungal therapy for cryptococcal meningitis. N. Engl. J. Med. 2013, 368, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Loyse, A.; Dromer, F.; Day, J.; Lortholary, O.; Harrison, T.S. Flucytosine and cryptococcosis: Time to urgently address the worldwide accessibility of a 50-year-old antifungal. J. Antimicrob. Chemother. 2013, 68, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Govender, N.P.; Meintjes, G.; Banoo, S. Access to flucytosine for HIV-infected patients with cryptococcal meningitis—An urgent need. S. Afr. Med. J. 2014, 104, 594–595. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.; Dlamini, S.; Paul, N.; Dedicoat, M. Treatment of acute cryptococcal meningitis in HIV infected adults, with an emphasis on resource-limited settings. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef] [Green Version]
- Powderly, W.G. Therapy for cryptococcal meningitis in patients with AIDS. Clin. Infect. Dis. 1992, 14, 54–59. [Google Scholar] [CrossRef]
- Momoff, N.; Parrish, A. Fluconazole-resistant cryptococcal meningitis. S. Afr. Med. J. 2003, 93, 444. [Google Scholar] [PubMed]
- Smith, K.D.; Achan, B.; Hullsiek, K.H.; McDonald, T.R.; Okagaki, L.H.; Alhadab, A.A.; Akampurira, A.; Rhein, J.R.; Meya, D.B.; Boulware, D.R.; et al. Increased antifungal drug resistance in clinical isolates of Cryptococcus neoformans in Uganda. Antimicrob. Agents Chemother. 2015, 59, 7197–7204. [Google Scholar] [CrossRef] [PubMed]
- Bii, C.C.; Makimura, K.; Abe, S.; Taguchi, H.; Mugasia, O.M.; Revathi, G.; Wamae, N.C.; Kamiya, S. Antifungal drug susceptibility of Cryptococcus neoformans from clinical sources in Nairobi, Kenya. Mycoses 2007, 50, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A.; Diekema, D.J.; Gibbs, D.L.; Newell, V.A.; Bijie, H.; Dzierzanowska, D.; Klimko, N.N.; Letscher-Bru, V.; Lisalova, M.; Muehlethaler, K.; et al. Results from the ARTEMIS DISK Global Antifungal Surveillance Study, 1997 to 2007: 10.5-year analysis of susceptibilities of noncandidal yeast species to fluconazole and voriconazole determined by CLSI standardized disk diffusion testing. J. Clin. Microbiol. 2009, 47, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Sar, B.; Monchy, D.; Vann, M.; Keo, C.; Sarthou, J.L.; Buisson, Y. Increasing in vitro resistance to fluconazole in Cryptococcus neoformans Cambodian isolates: April 2000 to March 2002. J. Antimicrob. Chemother. 2004, 54, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Park, B.J.; Wannemuehler, K.A.; Marston, B.J.; Govender, N.; Pappas, P.G.; Chiller, T.M. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 2009, 23, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Lightowler, J.V.; Cooke, G.S.; Mutevedzi, P.; Lessells, R.J.; Newell, M.L.; Dedicoat, M. Treatment of cryptococcal meningitis in KwaZulu-Natal, South Africa. PLoS ONE 2010, 5, e8630. [Google Scholar] [CrossRef] [PubMed]
- Lomes, N.R.; Melhem, M.S.; Szeszs, M.W.; Martins, M.D.; Buccheri, R. Cryptococcosis in non-HIV/non-transplant patients: A Brazilian case series. Med. Mycol. 2016, 54, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Kambugu, A.; Meya, D.B.; Rhein, J.; O‘Brien, M.; Janoff, E.N.; Ronald, A.R.; Kamya, M.R.; Mayanja-Kizza, H.; Sande, M.A.; Bohjanen, P.R.; et al. Outcomes of cryptococcal meningitis in Uganda before and after the availability of highly active antiretroviral therapy. Clin. Infect. Dis. 2008, 46, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Brizendine, K.D.; Baddley, J.W.; Pappas, P.G. Predictors of mortality and differences in clinical features among patients with Cryptococcosis according to immune status. PLoS ONE 2013, 8, e60431. [Google Scholar] [CrossRef] [PubMed]
- Bruatto, M.; Vidotto, V.; Maina, A.M. Growth of Cryptococcus neoformans in a thiamine-free medium. Mycopathologia 1992, 119, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Vidotto, V.; Aoki, S.; Campanini, G. A vitamin-free minimal synthetic medium for Cryptococcus neoformans. Mycopathologia 1996, 133, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Hull, C.M.; Heitman, J. Sexual reproduction between partners of the same mating type in Cryptococcus neoformans. Nature 2005, 434, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Wickes, B.L.; Mayorga, M.E.; Edman, U.; Edman, J.C. Dimorphism and haploid fruiting in Cryptococcus neoformans: Association with the alpha-mating type. Proc. Natl. Acad. Sci. USA 1996, 93, 7327–7331. [Google Scholar] [CrossRef] [PubMed]
- Toffaletti, D.L.; Rude, T.H.; Johnston, S.A.; Durack, D.T.; Perfect, J.R. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J. Bacteriol. 1993, 175, 1405–1411. [Google Scholar] [PubMed]
- Hull, C.M.; Heitman, J. Genetics of Cryptococcus neoformans. Annu. Rev. Genet. 2002, 36, 557–615. [Google Scholar] [CrossRef] [PubMed]
- Tenor, J.L.; Oehlers, S.H.; Yang, J.L.; Tobin, D.M.; Perfect, J.R. Live Imaging of Host-Parasite Interactions in a zebrafish infection model reveals cryptococcal determinants of virulence and central nervous system invasion. mBio 2015, 6, e01425–e01415. [Google Scholar] [CrossRef] [PubMed]
- Perfect, J.R.; Lang, S.D.; Durack, D.T. Chronic cryptococcal meningitis: A new experimental model in rabbits. Am. J. Pathol. 1980, 101, 177–194. [Google Scholar] [PubMed]
- Mylonakis, E.; Ausubel, F.M.; Perfect, J.R.; Heitman, J.; Calderwood, S.B. Killing of Caenorhabditis elegans by Cryptococcus neoformans as a model of yeast pathogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 15675–15680. [Google Scholar] [CrossRef] [PubMed]
- Mylonakis, E.; Moreno, R.; El Khoury, J.B.; Idnurm, A.; Heitman, J.; Calderwood, S.B.; Ausubel, F.M.; Diener, A. Galleria mellonella as a model system to study Cryptococcus neoformans pathogenesis. Infect. Immun. 2005, 73, 3842–3850. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, N.P.; Najvar, L.K.; Bocanegra, R.; Kirkpatrick, W.R.; Sorrell, T.C.; Patterson, T.F. Limited activity of miltefosine in murine models of cryptococcal meningoencephalitis and disseminated cryptococcosis. Antimicrob. Agents Chemother. 2013, 57, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.F.; Guillot, L.; Qureshi, S.T. Mammalian model hosts of cryptococcal infection. Comp. Med. 2007, 57, 9–17. [Google Scholar] [PubMed]
- Sabiiti, W.; May, R.C.; Pursall, E.R. Experimental models of cryptococcosis. Int. J. Microbiol. 2012, 2012, 626745. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Kwon-Chung, K.J. Complementation of a capsule-deficient mutation of Cryptococcus neoformans restores its virulence. Mol. Cell. Biol. 1994, 14, 4912–4919. [Google Scholar] [CrossRef] [PubMed]
- Nosanchuk, J.D.; Valadon, P.; Feldmesser, M.; Casadevall, A. Melanization of Cryptococcus neoformans in murine infection. Mol. Cell. Biol. 1999, 19, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Salas, S.D.; Bennett, J.E.; Kwon-Chung, K.J.; Perfect, J.R.; Williamson, P.R. Effect of the laccase gene CNLAC1, on virulence of Cryptococcus neoformans. J. Exp. Med. 1996, 184, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Alspaugh, J.A.; Cavallo, L.M.; Perfect, J.R.; Heitman, J. RAS1 regulates filamentation, mating and growth at high temperature of Cryptococcus neoformans. Mol. Microbiol. 2000, 36, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Chayakulkeeree, M.; Johnston, S.A.; Oei, J.B.; Lev, S.; Williamson, P.R.; Wilson, C.F.; Zuo, X.; Leal, A.L.; Vainstein, M.H.; Meyer, W.; et al. SEC14 is a specific requirement for secretion of phospholipase B1 and pathogenicity of Cryptococcus neoformans. Mol. Microbiol. 2011, 80, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Okagaki, L.H.; Strain, A.K.; Nielsen, J.N.; Charlier, C.; Baltes, N.J.; Chretien, F.; Heitman, J.; Dromer, F.; Nielsen, K. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog. 2010, 6, e1000953. [Google Scholar] [CrossRef]
- Crabtree, J.N.; Okagaki, L.H.; Wiesner, D.L.; Strain, A.K.; Nielsen, J.N.; Nielsen, K. Titan cell production enhances the virulence of Cryptococcus neoformans. Infect. Immun. 2012, 80, 3776–3785. [Google Scholar] [CrossRef] [PubMed]
- Olszewski, M.A.; Noverr, M.C.; Chen, G.H.; Toews, G.B.; Cox, G.M.; Perfect, J.R.; Huffnagle, G.B. Urease expression by Cryptococcus neoformans promotes microvascular sequestration, thereby enhancing central nervous system invasion. Am. J. Pathol. 2004, 164, 1761–1771. [Google Scholar] [CrossRef]
- Idnurm, A.; Reedy, J.L.; Nussbaum, J.C.; Heitman, J. Cryptococcus neoformans virulence gene discovery through insertional mutagenesis. Eukaryot. Cell 2004, 3, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Liu, O.W.; Chun, C.D.; Chow, E.D.; Chen, C.; Madhani, H.D.; Noble, S.M. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell 2008, 135, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.W.; Yang, D.H.; Maeng, S.; Lee, K.T.; So, Y.S.; Hong, J.; Choi, J.; Byun, H.J.; Kim, H.; Bang, S.; et al. Systematic functional profiling of transcription factor networks in Cryptococcus neoformans. Nat. Commun. 2015, 6, 6757. [Google Scholar] [CrossRef] [PubMed]
- Leidich, S.D.; Ibrahim, A.S.; Fu, Y.; Koul, A.; Jessup, C.; Vitullo, J.; Fonzi, W.; Mirbod, F.; Nakashima, S.; Nozawa, Y.; et al. Cloning and disruption of caPLB1, a phospholipase B gene involved in the pathogenicity of Candida albicans. J. Biol. Chem. 1998, 273, 26078–26086. [Google Scholar] [CrossRef] [PubMed]
- Theiss, S.; Ishdorj, G.; Brenot, A.; Kretschmar, M.; Lan, C.Y.; Nichterlein, T.; Hacker, J.; Nigam, S.; Agabian, N.; Kohler, G.A. Inactivation of the phospholipase B gene PLB5 in wild-type Candida albicans reduces cell-associated phospholipase A2 activity and attenuates virulence. Int. J. Med. Microbiol. 2006, 296, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Mirbod-Donovan, F.; Schaller, R.; Hung, C.Y.; Xue, J.; Reichard, U.; Cole, G.T. Urease produced by Coccidioides posadasii contributes to the virulence of this respiratory pathogen. Infect. Immun. 2006, 74, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Gresnigt, M.S.; Bozza, S.; Becker, K.L.; Joosten, L.A.; Abdollahi-Roodsaz, S.; van der Berg, W.B.; Dinarello, C.A.; Netea, M.G.; Fontaine, T.; De Luca, A.; et al. A polysaccharide virulence factor from Aspergillus fumigatus elicits anti-inflammatory effects through induction of Interleukin-1 receptor antagonist. PLoS Pathog. 2014, 10, e1003936. [Google Scholar] [CrossRef] [PubMed]
- Jahn, B.; Koch, A.; Schmidt, A.; Wanner, G.; Gehringer, H.; Bhakdi, S.; Brakhage, A.A. Isolation and characterization of a pigmentless-conidium mutant of Aspergillus fumigatus with altered conidial surface and reduced virulence. Infect. Immun. 1997, 65, 5110–5117. [Google Scholar] [PubMed]
- Odom, A.; Muir, S.; Lim, E.; Toffaletti, D.L.; Perfect, J.; Heitman, J. Calcineurin is required for virulence of Cryptococcus neoformans. EMBO J. 1997, 16, 2576–2589. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, C.A.; Alspaugh, J.A.; Yue, C.; Harashima, T.; Cox, G.M.; Perfect, J.R.; Heitman, J. Cyclic AMP-dependent protein kinase controls virulence of the fungal pathogen Cryptococcus neoformans. Mol. Cell. Biol. 2001, 21, 3179–3191. [Google Scholar] [CrossRef] [PubMed]
- Bahn, Y.S.; Kojima, K.; Cox, G.M.; Heitman, J. Specialization of the HOG pathway and its impact on differentiation and virulence of Cryptococcus neoformans. Mol. Biol. Cell 2005, 16, 2285–2300. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Kim, S.; Snyder, S.H. Inositol pyrophosphates as mammalian cell signals. Sci. Signal. 2011, 4, re1. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, T.R.; Norton, D.; Price, M.S.; Hay, C.; Clements, M.F.; Nichols, C.B.; Alspaugh, J.A. Interaction of Cryptococcus neoformans Rim101 and protein kinase A regulates capsule. PLoS Pathog. 2010, 6, e1000776. [Google Scholar] [CrossRef] [PubMed]
- Kozubowski, L.; Lee, S.C.; Heitman, J. Signalling pathways in the pathogenesis of Cryptococcus. Cell. Microbiol. 2009, 11, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Gerik, K.J.; Bhimireddy, S.R.; Ryerse, J.S.; Specht, C.A.; Lodge, J.K. PKC1 is essential for protection against both oxidative and nitrosative stresses, cell integrity, and normal manifestation of virulence factors in the pathogenic fungus Cryptococcus neoformans. Eukaryot. Cell 2008, 7, 1685–1698. [Google Scholar] [CrossRef] [PubMed]
- Chayakulkeeree, M.; Sorrell, T.C.; Siafakas, A.R.; Wilson, C.F.; Pantarat, N.; Gerik, K.J.; Boadle, R.; Djordjevic, J.T. Role and mechanism of phosphatidylinositol-specific phospholipase C in survival and virulence of Cryptococcus neoformans. Mol. Microbiol. 2008, 69, 809–826. [Google Scholar] [PubMed]
- Flick, J.S.; Thorner, J. Genetic and biochemical characterization of a phosphatidylinositol-specific phospholipase C in Saccharomyces cerevisiae. Mol. Cell. Biol. 1993, 13, 5861–5876. [Google Scholar] [CrossRef] [PubMed]
- Yoko-o, T.; Matsui, Y.; Yagisawa, H.; Nojima, H.; Uno, I.; Toh-e, A. The putative phosphoinositide-specific phospholipase C gene, PLC1, of the yeast Saccharomyces cerevisiae is important for cell growth. Proc. Natl. Acad. Sci. USA 1993, 90, 1804–1808. [Google Scholar] [CrossRef] [PubMed]
- Payne, W.E.; Fitzgerald-Hayes, M. A mutation in PLC1, a candidate phosphoinositide-specific phospholipase C gene from Saccharomyces cerevisiae, causes aberrant mitotic chromosome segregation. Mol. Cell. Biol. 1993, 13, 4351–4364. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.L.; Majerus, P.W. Identification and properties of two distinct phosphatidylinositol-specific phospholipase C enzymes from sheep seminal vesicular glands. J. Biol. Chem. 1982, 257, 6461–6469. [Google Scholar] [PubMed]
- Lev, S.; Desmarini, D.; Li, C.; Chayakulkeeree, M.; Traven, A.; Sorrell, T.C.; Djordjevic, J.T. Phospholipase C of Cryptococcus neoformans regulates homeostasis and virulence by providing inositol trisphosphate as a substrate for Arg1 kinase. Infect. Immun. 2013, 81, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Heung, L.J.; Luberto, C.; Plowden, A.; Hannun, Y.A.; Del Poeta, M. The sphingolipid pathway regulates Pkc1 through the formation of diacylglycerol in Cryptococcus neoformans. J. Biol. Chem. 2004, 279, 21144–21153. [Google Scholar] [CrossRef] [PubMed]
- Gerik, K.J.; Donlin, M.J.; Soto, C.E.; Banks, A.M.; Banks, I.R.; Maligie, M.A.; Selitrennikoff, C.P.; Lodge, J.K. Cell wall integrity is dependent on the PKC1 signal transduction pathway in Cryptococcus neoformans. Mol. Microbiol. 2005, 58, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W., Jr. Formation and actions of calcium-mobilizing messenger, inositol 1,4,5-trisphosphate. Am. J. Physiol. 1987, 252, 149–157. [Google Scholar]
- Putney, J.W., Jr.; Aub, D.L.; Taylor, C.W.; Merritt, J.E. Formation and biological action of inositol 1,4,5-trisphosphate. Fed. Proc. 1986, 45, 2634–2638. [Google Scholar] [PubMed]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, T.; Marks, A.R. Calcineurin is downstream of the inositol 1,4,5-trisphosphate receptor in the apoptotic and cell growth pathways. J. Biol. Chem. 2000, 275, 6417–6420. [Google Scholar] [CrossRef] [PubMed]
- Silverman-Gavrila, L.B.; Lew, R.R. An IP3-activated Ca2+ channel regulates fungal tip growth. J. Cell Sci. 2002, 115, 5013–5025. [Google Scholar] [CrossRef] [PubMed]
- Menniti, F.S.; Miller, R.N.; Putney, J.W., Jr.; Shears, S.B. Turnover of inositol polyphosphate pyrophosphates in pancreatoma cells. J. Biol. Chem. 1993, 268, 3850–3856. [Google Scholar] [PubMed]
- Tsui, M.M.; York, J.D. Roles of inositol phosphates and inositol pyrophosphates in development, cell signaling and nuclear processes. Adv. Enzyme Regul. 2010, 50, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Livermore, T.M.; Saiardi, A. Inositol pyrophosphates: Between signalling and metabolism. Biochem. J. 2013, 452, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Roman, A.R.; Wente, S.R. Inositol polyphosphates: A new frontier for regulating gene expression. Chromosoma 2008, 117, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lev, S.; Li, C.; Desmarini, D.; Saiardi, A.; Fewings, N.L.; Schibeci, S.D.; Sharma, R.; Sorrell, T.C.; Djordjevic, J.T. Fungal inositol pyrophosphate IP7 is crucial for metabolic adaptation to the host environment and pathogenicity. mBio 2015, 6, e00531–e00515. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lev, S.; Saiardi, A.; Desmarini, D.; Sorrell, T.C.; Djordjevic, J.T. Identification of a major IP5 kinase in Cryptococcus neoformans confirms that PP-IP5/IP7, not IP6, is essential for virulence. Sci. Rep. 2016, 6, 23927. [Google Scholar] [CrossRef] [PubMed]
- Saiardi, A.; Erdjument-Bromage, H.; Snowman, A.M.; Tempst, P.; Snyder, S.H. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr. Biol. 1999, 9, 1323–1326. [Google Scholar] [CrossRef]
- Odom, A.R.; Stahlberg, A.; Wente, S.R.; York, J.D. A role for nuclear inositol 1,4,5-trisphosphate kinase in transcriptional control. Science 2000, 287, 2026–2029. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, J.; Fleig, U. Asp1, a conserved 1/3 inositol polyphosphate kinase, regulates the dimorphic switch in Schizosaccharomyces pombe. Mol. Cell. Biol. 2010, 30, 4535–4547. [Google Scholar] [CrossRef] [PubMed]
- Fridy, P.C.; Otto, J.C.; Dollins, D.E.; York, J.D. Cloning and characterization of two human VIP1-like inositol hexakisphosphate and diphosphoinositol pentakisphosphate kinases. J. Biol. Chem. 2007, 282, 30754–30762. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, J.; Risse, C.; Seidel, C.; Pohlmann, T.; Jakopec, V.; Walla, E.; Ramrath, P.; Takeshita, N.; Baumann, S.; Feldbrugge, M.; et al. The Vip1 inositol polyphosphate kinase family regulates polarized growth and modulates the microtubule cytoskeleton in fungi. PLoS Genet. 2014, 10, e1004586. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Nair, V.S.; Holland, A.A.; Capolicchio, S.; Jessen, H.J.; Johnson, M.K.; Shears, S.B. Asp1 from Schizosaccharomyces pombe binds a [2Fe-2S](2+) cluster which inhibits inositol pyrophosphate 1-phosphatase activity. Biochemistry 2015, 54, 6462–6474. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.A.; Zaremba, A.; Shears, S.B. Receptor-dependent compartmentalization of PPIP5K1, a kinase with a cryptic polyphosphoinositide binding domain. Biochem. J. 2011, 434, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Mulugu, S.; Bai, W.; Fridy, P.C.; Bastidas, R.J.; Otto, J.C.; Dollins, D.E.; Haystead, T.A.; Ribeiro, A.A.; York, J.D. A conserved family of enzymes that phosphorylate inositol hexakisphosphate. Science 2007, 316, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Glennon, M.C.; Shears, S.B. Turnover of inositol pentakisphosphates, inositol hexakisphosphate and diphosphoinositol polyphosphates in primary cultured hepatocytes. Biochem. J. 1993, 293, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Steidle, E.A.; Chong, L.S.; Wu, M.; Crooke, E.; Fiedler, D.; Resnick, A.C.; Rolfes, R.J. A novel inositol pyrophosphate phosphatase in Saccharomyces cerevisiae: Siw14 protein selectively cleaves the beta-phosphate from 5-diphosphoinositol pentakisphosphate (5PP-IP5). J. Biol. Chem. 2016, 291, 6772–6783. [Google Scholar] [CrossRef] [PubMed]
- Pietrella, D.; Cherniak, R.; Strappini, C.; Perito, S.; Mosci, P.; Bistoni, F.; Vecchiarelli, A. Role of mannoprotein in induction and regulation of immunity to Cryptococcus neoformans. Infect. Immun. 2001, 69, 2808–2814. [Google Scholar] [CrossRef] [PubMed]
- Delfino, D.; Cianci, L.; Migliardo, M.; Mancuso, G.; Cusumano, V.; Corradini, C.; Teti, G. Tumor necrosis factor-inducing activities of Cryptococcus neoformans components. Infect. Immun. 1996, 64, 5199–5204. [Google Scholar] [PubMed]
- Levitz, S.M.; Specht, C.A. The molecular basis for the immunogenicity of Cryptococcus neoformans mannoproteins. FEMS Yeast Res. 2006, 6, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Nong, S.H.; Mansour, M.K.; Specht, C.A.; Levitz, S.M. Purification and characterization of a second immunoreactive mannoprotein from Cryptococcus neoformans that stimulates T-cell responses. Infect. Immun. 2002, 70, 5485–5493. [Google Scholar] [CrossRef] [PubMed]
- Voglmaier, S.M.; Bembenek, M.E.; Kaplin, A.I.; Dorman, G.; Olszewski, J.D.; Prestwich, G.D.; Snyder, S.H. Purified inositol hexakisphosphate kinase is an ATP synthase: Diphosphoinositol pentakisphosphate as a high-energy phosphate donor. Proc. Natl. Acad. Sci. USA 1996, 93, 4305–4310. [Google Scholar] [CrossRef] [PubMed]
- Lonetti, A.; Szijgyarto, Z.; Bosch, D.; Loss, O.; Azevedo, C.; Saiardi, A. Identification of an evolutionarily conserved family of inorganic polyphosphate endopolyphosphatases. J. Biol. Chem. 2011, 286, 31966–31974. [Google Scholar] [CrossRef] [PubMed]
- Boer, V.M.; Crutchfield, C.A.; Bradley, P.H.; Botstein, D.; Rabinowitz, J.D. Growth-limiting intracellular metabolites in yeast growing under diverse nutrient limitations. Mol. Biol. Cell 2010, 21, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Belde, P.J.; Vossen, J.H.; Borst-Pauwels, G.W.; Theuvenet, A.P. Inositol 1,4,5-trisphosphate releases Ca2+ from vacuolar membrane vesicles of Saccharomyces cerevisiae. FEBS Lett. 1993, 323, 113–118. [Google Scholar] [CrossRef]
- Cornelius, G.; Gebauer, G.; Techel, D. Inositol trisphosphate induces calcium release from Neurospora crassa vacuoles. Biochem. Biophys. Res. Commun. 1989, 162, 852–856. [Google Scholar] [CrossRef]
- Calvert, C.M.; Sanders, D. Inositol trisphosphate-dependent and -independent Ca2+ mobilization pathways at the vacuolar membrane of Candida albicans. J. Biol. Chem. 1995, 270, 7272–7280. [Google Scholar] [PubMed]
- Ho, M.W.; Yang, X.; Carew, M.A.; Zhang, T.; Hua, L.; Kwon, Y.U.; Chung, S.K.; Adelt, S.; Vogel, G.; Riley, A.M.; et al. Regulation of Ins(3,4,5,6)P(4) signaling by a reversible kinase/phosphatase. Curr. Biol. 2002, 12, 477–482. [Google Scholar] [CrossRef]
- Xie, W.; Kaetzel, M.A.; Bruzik, K.S.; Dedman, J.R.; Shears, S.B.; Nelson, D.J. Inositol 3,4,5,6-tetrakisphosphate inhibits the calmodulin-dependent protein kinase II-activated chloride conductance in T84 colonic epithelial cells. J. Biol. Chem. 1996, 271, 14092–14097. [Google Scholar] [PubMed]
- Renstrom, E.; Ivarsson, R.; Shears, S.B. Inositol 3,4,5,6-tetrakisphosphate inhibits insulin granule acidification and fusogenic potential. J. Biol. Chem. 2002, 277, 26717–26720. [Google Scholar] [CrossRef] [PubMed]
- Ismailov, I.I.; Fuller, C.M.; Berdiev, B.K.; Shlyonsky, V.G.; Benos, D.J.; Barrett, K.E. A biologic function for an “orphan” messenger: d-myo-inositol 3,4,5,6-tetrakisphosphate selectively blocks epithelial calcium-activated chloride channels. Proc. Natl. Acad. Sci. USA 1996, 93, 10505–10509. [Google Scholar] [CrossRef] [PubMed]
- Vajanaphanich, M.; Schultz, C.; Rudolf, M.T.; Wasserman, M.; Enyedi, P.; Craxton, A.; Shears, S.B.; Tsien, R.Y.; Barrett, K.E.; Traynor-Kaplan, A. Long-term uncoupling of chloride secretion from intracellular calcium levels by Ins(3,4,5,6)P4. Nature 1994, 371, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Rudolf, M.; Carew, M.A.; Yoshida, M.; Nerreter, V.; Riley, A.M.; Chung, S.K.; Bruzik, K.S.; Potter, B.V.; Schultz, C.; et al. Inositol 1,3,4-trisphosphate acts in vivo as a specific regulator of cellular signaling by inositol 3,4,5,6-tetrakisphosphate. J. Biol. Chem. 1999, 274, 18973–18980. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, P.P.; Qian, X.; Stiles, A.R.; Cho, J.; Jones, D.H.; Lesley, S.A.; Grabau, E.A.; Shears, S.B.; Spraggon, G. Integration of inositol phosphate signaling pathways via human ITPK1. J. Biol. Chem. 2007, 282, 28117–28125. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Mollapour, E.; Shears, S.B. Signal transduction during environmental stress: InsP8 operates within highly restricted contexts. Cell. Signal. 2005, 17, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Pesesse, X.; Choi, K.; Zhang, T.; Shears, S.B. Signaling by higher inositol polyphosphates. Synthesis of bisdiphosphoinositol tetrakisphosphate (“InsP8”) is selectively activated by hyperosmotic stress. J. Biol. Chem. 2004, 279, 43378–43381. [Google Scholar] [CrossRef] [PubMed]
- Ongusaha, P.P.; Hughes, P.J.; Davey, J.; Michell, R.H. Inositol hexakisphosphate in Schizosaccharomyces pombe: Synthesis from Ins(1,4,5)P3 and osmotic regulation. Biochem. J. 1998, 335, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Worley, J.; Luo, X.; Capaldi, A.P. Inositol pyrophosphates regulate cell growth and the environmental stress response by activating the HDAC Rpd3L. Cell Rep. 2013, 3, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- York, J.D. Regulation of nuclear processes by inositol polyphosphates. Biochim. Biophys. Acta 2006, 1761, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Wild, R.; Gerasimaite, R.; Jung, J.Y.; Truffault, V.; Pavlovic, I.; Schmidt, A.; Saiardi, A.; Jessen, H.J.; Poirier, Y.; Hothorn, M.; et al. Control of eukaryotic phosphate homeostasis by inositol polyphosphate sensor domains. Science 2016, 352, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Secco, D.; Wang, C.; Shou, H.; Whelan, J. Phosphate homeostasis in the yeast Saccharomyces cerevisiae, the key role of the SPX domain-containing proteins. FEBS Lett. 2012, 586, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Mulugu, S.; York, J.D.; O’Shea, E.K. Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Science 2007, 316, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.J.; Watson, P.J.; Celardo, I.; Gordiyenko, Y.; Cowley, S.M.; Robinson, C.V.; Fairall, L.; Schwabe, J.W. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol. Cell. 2013, 51, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012, 481, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.; Radenberg, T.; Thiel, U.; Vogel, G.; Khoo, K.H.; Dell, A.; Jackson, T.R.; Hawkins, P.T.; Mayr, G.W. The detection, purification, structural characterization, and metabolism of diphosphoinositol pentakisphosphate(s) and bisdiphosphoinositol tetrakisphosphate(s). J. Biol. Chem. 1993, 268, 4009–4015. [Google Scholar] [PubMed]
- Laussmann, T.; Eujen, R.; Weisshuhn, C.M.; Thiel, U.; Vogel, G. Structures of diphospho-myo-inositol pentakisphosphate and bisdiphospho-myo-inositol tetrakisphosphate from Dictyostelium resolved by NMR analysis. Biochem. J. 1996, 315, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Saiardi, A.; Bhandari, R.; Resnick, A.C.; Snowman, A.M.; Snyder, S.H. Phosphorylation of proteins by inositol pyrophosphates. Science 2004, 306, 2101–2105. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Saiardi, A.; Ahmadibeni, Y.; Snowman, A.M.; Resnick, A.C.; Kristiansen, T.Z.; Molina, H.; Pandey, A.; Werner, J.K., Jr.; Juluri, K.R.; et al. Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc. Natl. Acad. Sci. USA 2007, 104, 15305–15310. [Google Scholar] [CrossRef] [PubMed]
- Szijgyarto, Z.; Garedew, A.; Azevedo, C.; Saiardi, A. Influence of inositol pyrophosphates on cellular energy dynamics. Science 2011, 334, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Saiardi, A.; Cockcroft, S. Human ITPK1: A reversible inositol phosphate kinase/phosphatase that links receptor-dependent phospholipase C to Ca2+-activated chloride channels. Sci. Signal. 2008, 1, pe5. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.J.; Wilson, M.P.; Majerus, P.W.; Hurley, J.H. Specificity determinants in inositol polyphosphate synthesis: Crystal structure of inositol 1,3,4-trisphosphate 5/6-kinase. Mol. Cell 2005, 18, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Holmes, W.; Jogl, G. Crystal structure of inositol phosphate multikinase 2 and implications for substrate specificity. J. Biol. Chem. 2006, 281, 38109–38116. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; DeRose, E.F.; London, R.E.; Shears, S.B. IP6K structure and the molecular determinants of catalytic specificity in an inositol phosphate kinase family. Nat. Commun. 2014, 5, 4178. [Google Scholar] [CrossRef] [PubMed]
- Endo-Streeter, S.; Tsui, M.K.; Odom, A.R.; Block, J.; York, J.D. Structural studies and protein engineering of inositol phosphate multikinase. J. Biol. Chem. 2012, 287, 35360–35369. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.J.; Hurley, J.H. Crystal structure of the catalytic core of inositol 1,4,5-trisphosphate 3-kinase. Mol. Cell 2004, 15, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, B.; Schell, M.J.; Letcher, A.J.; Veprintsev, D.B.; Irvine, R.F.; Williams, R.L. Structure of a human inositol 1,4,5-trisphosphate 3-kinase: Substrate binding reveals why it is not a phosphoinositide 3-kinase. Mol. Cell 2004, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- El Bakkoury, M.; Dubois, E.; Messenguy, F. Recruitment of the yeast MADS-box proteins, ArgRI and Mcm1 by the pleiotropic factor ArgRIII is required for their stability. Mol. Microbiol. 2000, 35, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Messenguy, F.; Dubois, E. Genetic evidence for a role for MCM1 in the regulation of arginine metabolism in Saccharomyces cerevisiae. Mol. Cell. Biol. 1993, 13, 2586–2592. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Godage, H.Y.; Riley, A.M.; Weaver, J.D.; Shears, S.B.; Potter, B.V. Synthetic inositol phosphate analogs reveal that PPIP5K2 has a surface-mounted substrate capture site that is a target for drug discovery. Chem. Biol. 2014, 21, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Choi, G.; Bae, Y.S.; Burdett, M.; Moon, H.S.; Lee, J.W.; Gray, N.S.; Schultz, P.G.; Meijer, L.; Chung, S.K.; et al. Purine-based inhibitors of inositol-1,4,5-trisphosphate-3-kinase. Chembiochem 2002, 3, 897–901. [Google Scholar] [CrossRef]
- Padmanabhan, U.; Dollins, D.E.; Fridy, P.C.; York, J.D.; Downes, C.P. Characterization of a selective inhibitor of inositol hexakisphosphate kinases: Use in defining biological roles and metabolic relationships of inositol pyrophosphates. J. Biol. Chem. 2009, 284, 10571–10582. [Google Scholar] [CrossRef] [PubMed]
- Dubois, E.; Scherens, B.; Vierendeels, F.; Ho, M.M.; Messenguy, F.; Shears, S.B. In Saccharomyces cerevisiae, the inositol polyphosphate kinase activity of Kcs1p is required for resistance to salt stress, cell wall integrity, and vacuolar morphogenesis. J. Biol. Chem. 2002, 277, 23755–23763. [Google Scholar] [CrossRef] [PubMed]
- Mayr, G.W.; Windhorst, S.; Hillemeier, K. Antiproliferative plant and synthetic polyphenolics are specific inhibitors of vertebrate inositol-1,4,5-trisphosphate 3-kinases and inositol polyphosphate multikinase. J. Biol. Chem. 2005, 280, 13229–13240. [Google Scholar] [CrossRef] [PubMed]
- Fraifeld, V.; Seidman, R.; Sagi, O.; Muradian, K.; Wolfson, M. Aurintricarboxylic acid decreases proliferative potential of SKOV3 and MCF7 human carcinoma cells. Anticancer Res. 2001, 21, 1975–1978. [Google Scholar] [PubMed]
- Mori, H.; Kawabata, K.; Matsunaga, K.; Ushida, J.; Fujii, K.; Hara, A.; Tanaka, T.; Murai, H. Chemopreventive effects of coffee bean and rice constituents on colorectal carcinogenesis. Biofactors 2000, 12, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Benezra, M.; Ben-Sasson, S.A.; Regan, J.; Chang, M.; Bar-Shavit, R.; Vlodavsky, I. Antiproliferative activity to vascular smooth muscle cells and receptor binding of heparin-mimicking polyaromatic anionic compounds. Arterioscler. Thromb. 1994, 14, 1992–1999. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.M.; Kully, M.; Khan, J.K.; Hattori, M.; Daneshtalab, M. Synthesis of chlorogenic acid derivatives with promising antifungal activity. Bioorg. Med. Chem. 2007, 15, 6830–6833. [Google Scholar] [CrossRef] [PubMed]
- Mellon, J.E.; Dowd, M.K.; Beltz, S.B.; Moore, G.G. Growth inhibitory effects of gossypol and related compounds on fungal cotton root pathogens. Lett. Appl. Microbiol. 2014, 59, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.W.; Lee, D.G. Antifungal action of chlorogenic acid against pathogenic fungi, mediated by membrane disruption. Pure Appl. Chem. 2010, 82, 219–226. [Google Scholar] [CrossRef]
- Da Silva, C.R.; de Andrade Neto, J.B.; de Sousa Campos, R.; Figueiredo, N.S.; Sampaio, L.S.; Magalhaes, H.I.; Cavalcanti, B.C.; Gaspar, D.M.; de Andrade, G.M.; Lima, I.S.; et al. Synergistic effect of the flavonoid catechin, quercetin, or epigallocatechin gallate with fluconazole induces apoptosis in Candida tropicalis resistant to fluconazole. Antimicrob. Agents Chemother. 2014, 58, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Guo, X.; Dawuti, G.; Aibai, S. Antifungal activity of ellagic acid in vitro and in vivo. Phytother. Res. 2015, 29, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Rezusta, A.; Lopez-Chicon, P.; Paz-Cristobal, M.P.; Alemany-Ribes, M.; Royo-Diez, D.; Agut, M.; Semino, C.; Nonell, S.; Revillo, M.J.; Aspiroz, C.; et al. In vitro fungicidal photodynamic effect of hypericin on Candida species. Photochem. Photobiol. 2012, 88, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Paz-Cristobal, M.P.; Gilaberte, Y.; Alejandre, C.; Pardo, J.; Revillo, M.J.; Rezusta, A. In vitro fungicidal photodynamic effect of hypericin on Trichophyton spp. Mycopathologia 2014, 178, 221–225. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
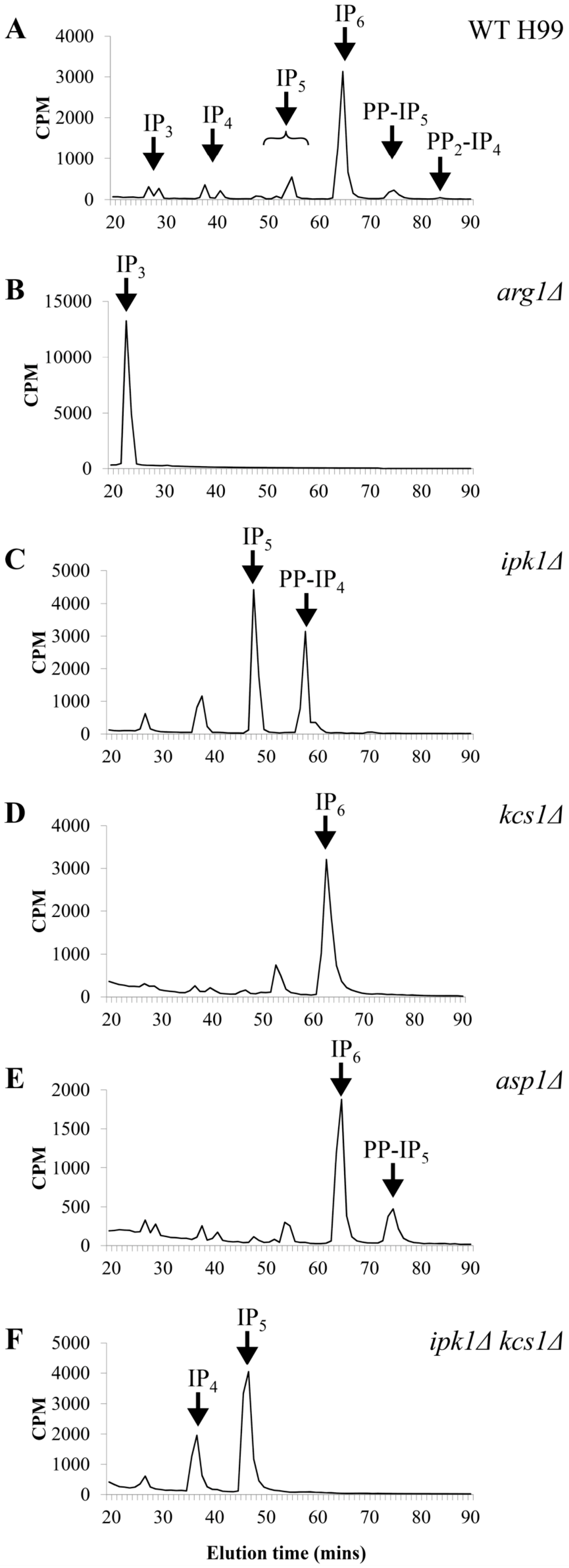
| Strains | IP Species | ||||||
|---|---|---|---|---|---|---|---|
| IP3 | IP4 | IP5 | PP-IP4 | IP6 | PP-IP5 | PP2-IP4 | |
| WT H99 | − | + | + | − | + | + | + |
| arg1Δ | +++ | − | − | − | − | − | − |
| ipk1Δ | + | + | +++ | +++ | − | − | − |
| kcs1Δ | + | + | + | − | + | − | − |
| asp1Δ | + | + | + | − | + | + | − |
| ipk1Δ kcs1Δ | + | + | +++ | − | − | − | − |
| Mutant | plc1Δ | arg1Δ | ipk1Δ | kcs1Δ | ipk1Δ kcs1Δ | asp1Δ |
|---|---|---|---|---|---|---|
| Virulence in mice | Avirulent | N/A | Hypovirulent | Avirulent | Avirulent | Fully virulent |
| Virulence in invertebrate models | Hypovirulent in C. elegans (25 °C) and G. mellonella (30 °C) | Hypovirulent in G. mellonella (30 °C) | N/A | Hypovirulent in G. mellonella (30 °C) | N/A | N/A |
| Cell wall integrity | Compromised | Compromised | Compromised | Compromised | Compromised | Normal |
| Capsule production | Normal/reduced epending on growth conditions | Reduced | Normal * | Increased | Normal * | Normal |
| Urease production | Reduced | Reduced | Reduced | Reduced | Reduced | Normal |
| Mutant-specific features | Abnormally layered cell wall; large vacuoles; no septal dissolution | Abnormally thick cell wall; enlarged vacuoles; cell separation defect; accelerated endocytosis | Mucoid | Enlarged cell size; mucoid colony appearance | Mucoid | None |
| Carbon source utilization | N/A | N/A | Partially compromised | Defective | Defective | N/A |
| 37 °C growth | Reduced | Reduced | Slightly reduced | Slightly reduced | Slightly reduced | Normal |
| Mating | Defective | Defective | Normal * | Defective | N/A | Normal |
| Melanization | Reduced | Reduced | Normal * | Reduced | Normal * | Normal |
| Laccase activity | Reduced * | Reduced * | Reduced | Reduced | Reduced | Normal * |
| Compound | Target Enzymes | Fungal Species/Inhibitory Concentration | Reference |
|---|---|---|---|
| Gossypol | Mammalian IP3K/IPMK | Pythium irregulare ED50 = 4.0 μg/mL Rhizoctonia solani ED50 = 34.6 μg/mL | [154] |
| Chlorogenic acid | Mammalian IPMK | Candida albicans MIC = 80 μg/mL Trichosporon beigelii MIC = 40 μg/mL Malassezia furfur MIC = 40 μg/mL | [155] |
| Quercetin | Mammalian IP3K | Synergistic effect with fluconazole (16 μg/mL) in fluconazole-resistant Candida tropicalis MIC50 ≤ 0.5 μg/mL | [156] |
| Ellagic acid | Mammalian IP3K/IPMK | Trichophyton rubrum MIC = 18.75 μg/mL Trichophyton mentagrophytes MIC = 32.29 μg/mL Microsporum canis MIC = 58.33 μg/mL Candida albicans MIC = 25 μg/mL Candida tropicalis MIC = 75 μg/mL | [157] |
| Hypericin | Mammalian IP3K | Natural photosensitizer, 3 log10 fungicidal effect at fluence of 37 J/cm2 Candida albicans 0.625 µM Candida parapsilosis 1.25 µM Candida krusei 20 μM Trichophyton rubrum 10–20 µM Trichophyton mentagrophytes 20–50 µM | [158] [159] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Lev, S.; Saiardi, A.; Desmarini, D.; Sorrell, T.C.; Djordjevic, J.T. Inositol Polyphosphate Kinases, Fungal Virulence and Drug Discovery. J. Fungi 2016, 2, 24. https://doi.org/10.3390/jof2030024
Li C, Lev S, Saiardi A, Desmarini D, Sorrell TC, Djordjevic JT. Inositol Polyphosphate Kinases, Fungal Virulence and Drug Discovery. Journal of Fungi. 2016; 2(3):24. https://doi.org/10.3390/jof2030024
Chicago/Turabian StyleLi, Cecilia, Sophie Lev, Adolfo Saiardi, Desmarini Desmarini, Tania C. Sorrell, and Julianne T. Djordjevic. 2016. "Inositol Polyphosphate Kinases, Fungal Virulence and Drug Discovery" Journal of Fungi 2, no. 3: 24. https://doi.org/10.3390/jof2030024
APA StyleLi, C., Lev, S., Saiardi, A., Desmarini, D., Sorrell, T. C., & Djordjevic, J. T. (2016). Inositol Polyphosphate Kinases, Fungal Virulence and Drug Discovery. Journal of Fungi, 2(3), 24. https://doi.org/10.3390/jof2030024