Computational Study of Ferrocene-Based Molecular Frameworks with 2,5-Diethynylpyridine as a Chemical Bridge

Abstract
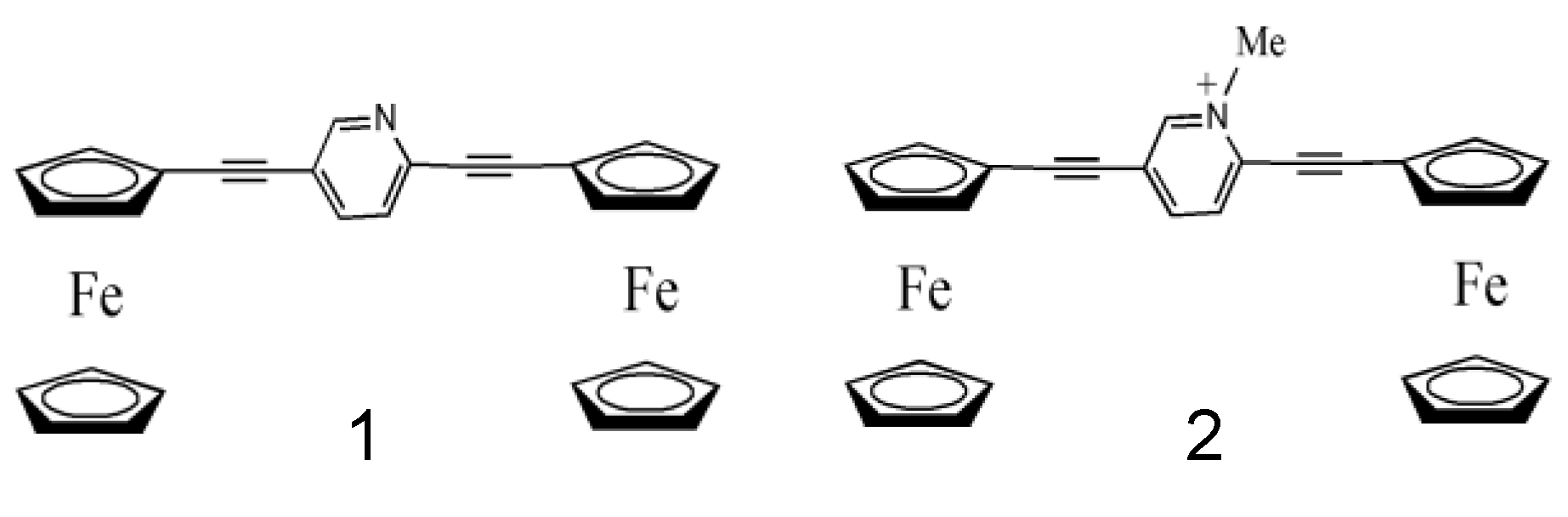
:1. Introduction


2. Computational Methods
3. Results and Discussion
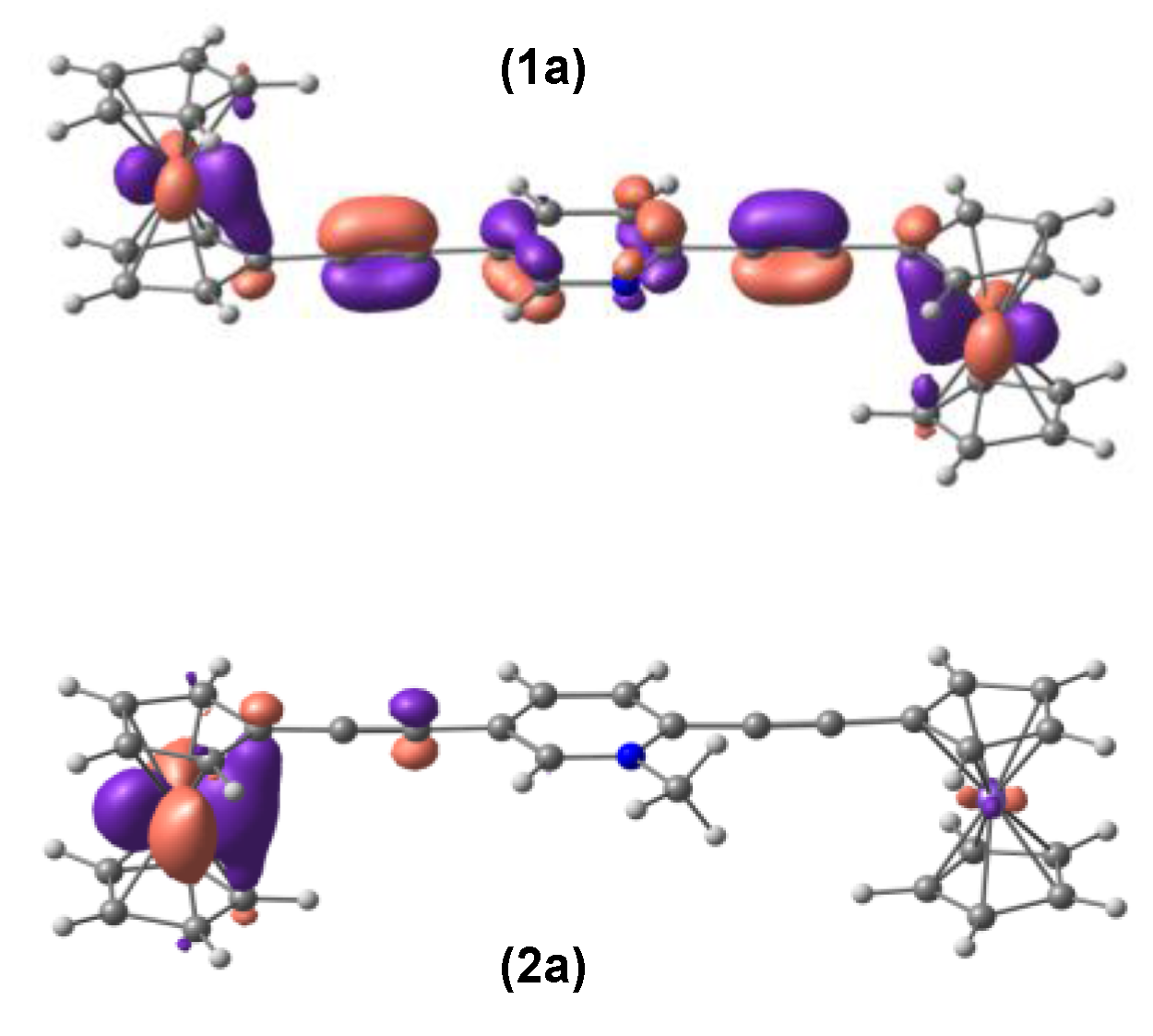
3.1. Geometries and Molecular Orbitals.


{kind=link}
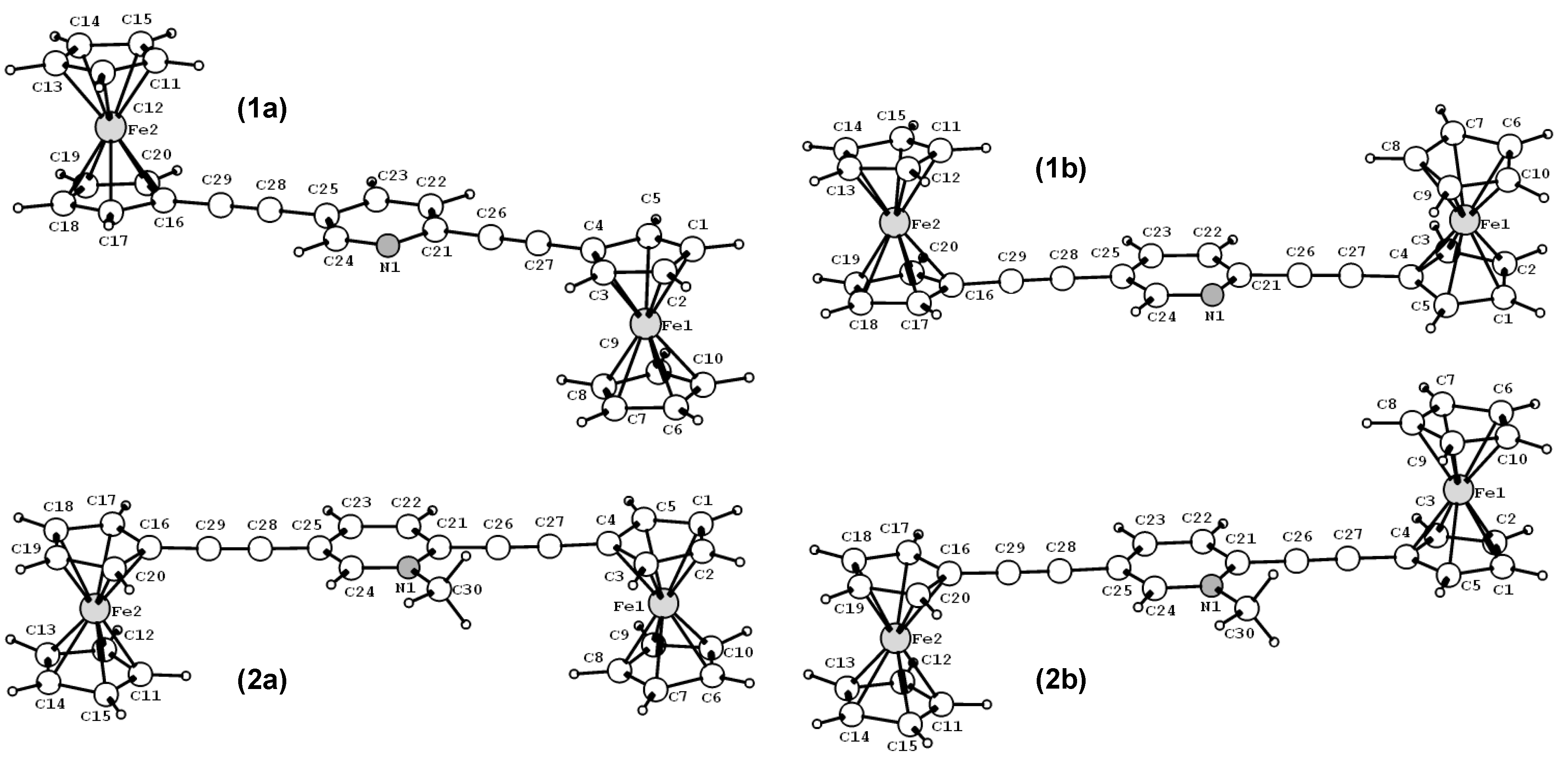{kind=link}
{kind=link}
{kind=link}
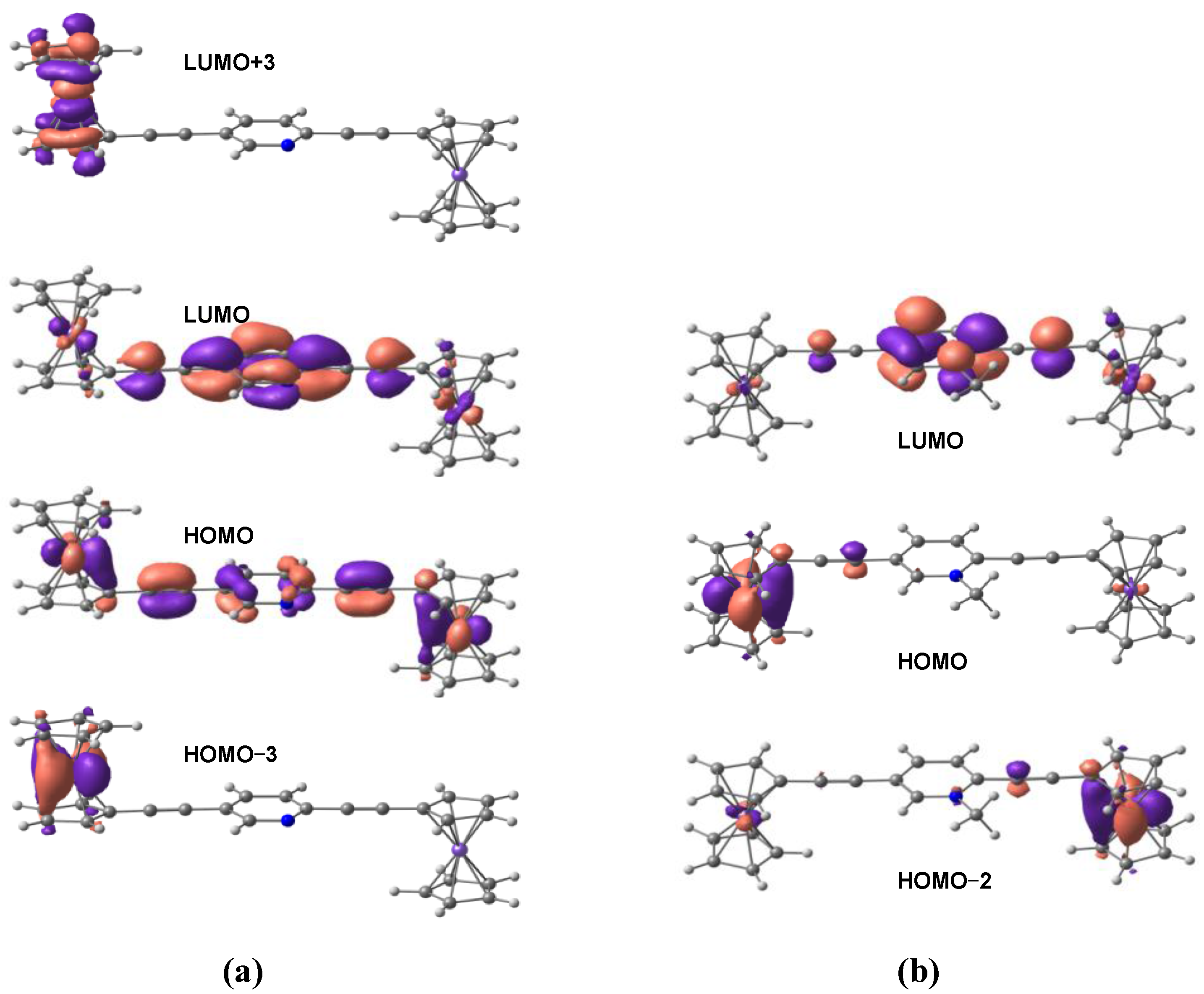{kind=link}
{kind=link}
| Bond Lengths (Ǻ) | |||
|---|---|---|---|
| Compound 1a (Gas-phase Electronic Energy: ‒1419.261490) | |||
| Fe1‒Cnt1 | 1.679 (1.658) | C16‒C29 | 1.417 (1.432) |
| Fe1‒Cnt2 | 1.674 (1.650) | C28‒C29 | 1.217 (1.186) |
| C25‒C28 | 1.419 (1.444) | N1‒C24 | 1.327 (1.356) |
| C24‒C25 | 1.414 (1.385) | N1‒C21 | 1.354 (1.375) |
| Compound 1b (Gas-phase Electronic Energy: ‒1419.261491) | |||
| Fe1‒Cnt1 | 1.679 | C16‒C29 | 1.417 |
| Fe1‒Cnt2 | 1.673 | C28‒C29 | 1.217 |
| C25‒C28 | 1.420 | N1‒C24 | 1.328 |
| C24‒C25 | 1.414 | N1‒C21 | 1.354 |
| Compound 2a (Gas-phase Electronic Energy: ‒1458.978122) | |||
| Fe1‒Cnt1 | 1.699 (1.653) | Fe2‒Cnt3 | 1.678 (1.638) |
| Fe1‒Cnt2 | 1.669 (1.641) | Fe2‒Cnt4 | 1.689 (1.655) |
| C16‒C29 | 1.408 (1.441) | N1‒C21 | 1.378 (1.390) |
| C28‒C29 | 1.220 (1.159) | C22‒C23 | 1.376 (1.376) |
| C25‒C28 | 1.408 (1.418) | C21‒C22 | 1.413 (1.373) |
| C24‒C25 | 1.396 (1.379) | C21‒C26 | 1.399 (1.457) |
| C23‒C25 | 1.423 (1.394) | C26‒C27 | 1.224 (1.180) |
| C24‒N1 | 1.354 (1.345) | C4‒C27 | 1.403 (1.425) |
| N1‒C30 | 1.477 (1.437) | ||
| Compound 2b (Gas-phase Electronic Energy: ‒1458.978163) | |||
| Fe1‒Cnt1 | 1.698 | Fe2‒Cnt3 | 1.678 |
| Fe1‒Cnt2 | 1.669 | Fe2‒Cnt4 | 1.689 |
| C16‒C29 | 1.408 | N1‒C21 | 1.378 |
| C28‒C29 | 1.220 | C22‒C23 | 1.376 |
| C25‒C28 | 1.408 | C21‒C22 | 1.413 |
| C24‒C25 | 1.396 | C21‒C26 | 1.399 |
| C23‒C25 | 1.423 | C26‒C27 | 1.224 |
| C24‒N1 | 1.355 | C4‒C27 | 1.403 |
| N1‒C30 | 1.477 | ||
| Torsion Angles | |||
| Compound 1a | |||
| C17‒C16‒C25‒C23 | ‒175.5 (‒176.2) | C3‒C4‒C21‒C22 | 4.7 (4.0) |
| C20‒C16‒C25‒C23 | 5.9 (2.7) | C20‒C16‒C21‒C23 | ‒173.8 (‒177.1) |
| Compound 2a | |||
| C20‒C16‒C25‒C24 | ‒2.6 (10.0) | C20‒C16‒C25‒C23 | 178.9 (‒171.4) |
| C17‒C16‒C25‒C24 | 175.1 (‒171.1) | C17‒C16‒C25‒C23 | ‒3.4 (7.5) |
| C22‒C21‒C4‒C5 | 3.1 (‒11.5) | N1‒C21‒C4‒C5 | ‒175.4 (165.2) |
| C22‒C21‒C4‒C3 | ‒178.4 (174.4) | N1‒C21‒C4‒C3 | 3.1 (‒8.8) |

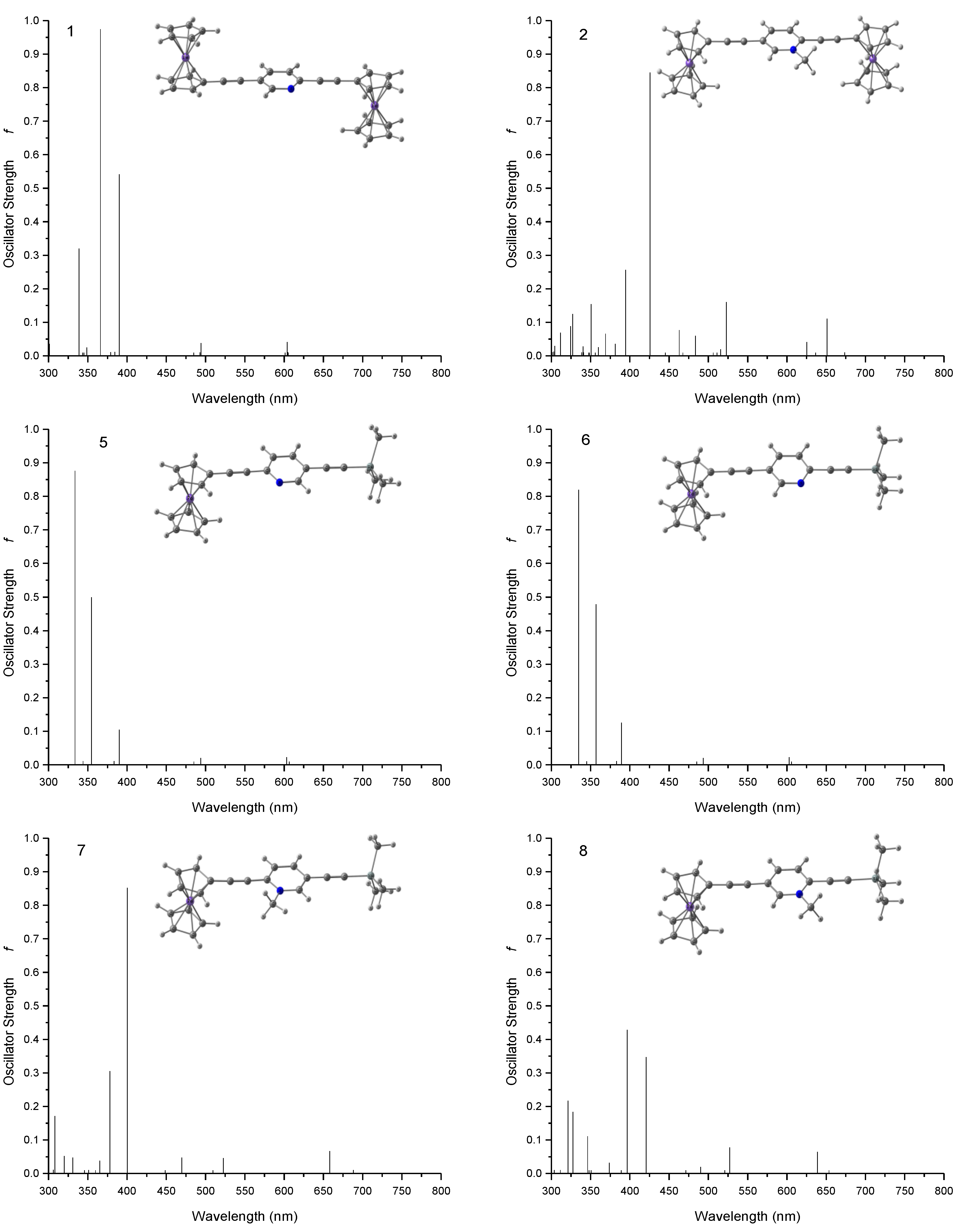
3.2. Electronic Transitions and Optical Spectra

| CH3CN solution | ||||
|---|---|---|---|---|
| Compound | Theory | Experiment | ||
| λmax (nm) | f | λmax (nm) | ε | |
| 1a | 494 | 0.032 (3.2) | 454 | 3696 (2.0) |
| 2a | 523 | 0.150 (15) | 540 | 10233 (5.4) |
| 5 | 494 | 0.010 (1.0) | 452 | 1892 (1.0) |
| 6 | 493 | 0.010 (1.0) | 446 | 2026 (1.0) |
| 7 | 522 | 0.036 (3.6) | 546 | 5810 (3.1) |
| 8 | 527 | 0.068 (6.8) | 518 | 4650 (2.5) |
| CH2Cl2 solution | ||||
| Compound | Theory | Experiment | ||
| λmax (nm) | f | λmax (nm) | ε | |
| 1a | 494 | 0.029 (2.9) | 456 | 5696 (3.0) |
| 2a | 528 | 0.159 (16) | 582 | 18376 (9.7) |
| 5 | 493 | 0.010 (1.0) | 452 | 1993 (1.1) |
| 6 | 493 | 0.010 (1.0) | 448 | 1889 (1.0) |
| 7 | 524 | 0.038 (3.8) | 586 | 9135 (4.8) |
| 8 | 535 | 0.069 (6.9) | 564 | 9656 (5.1) |

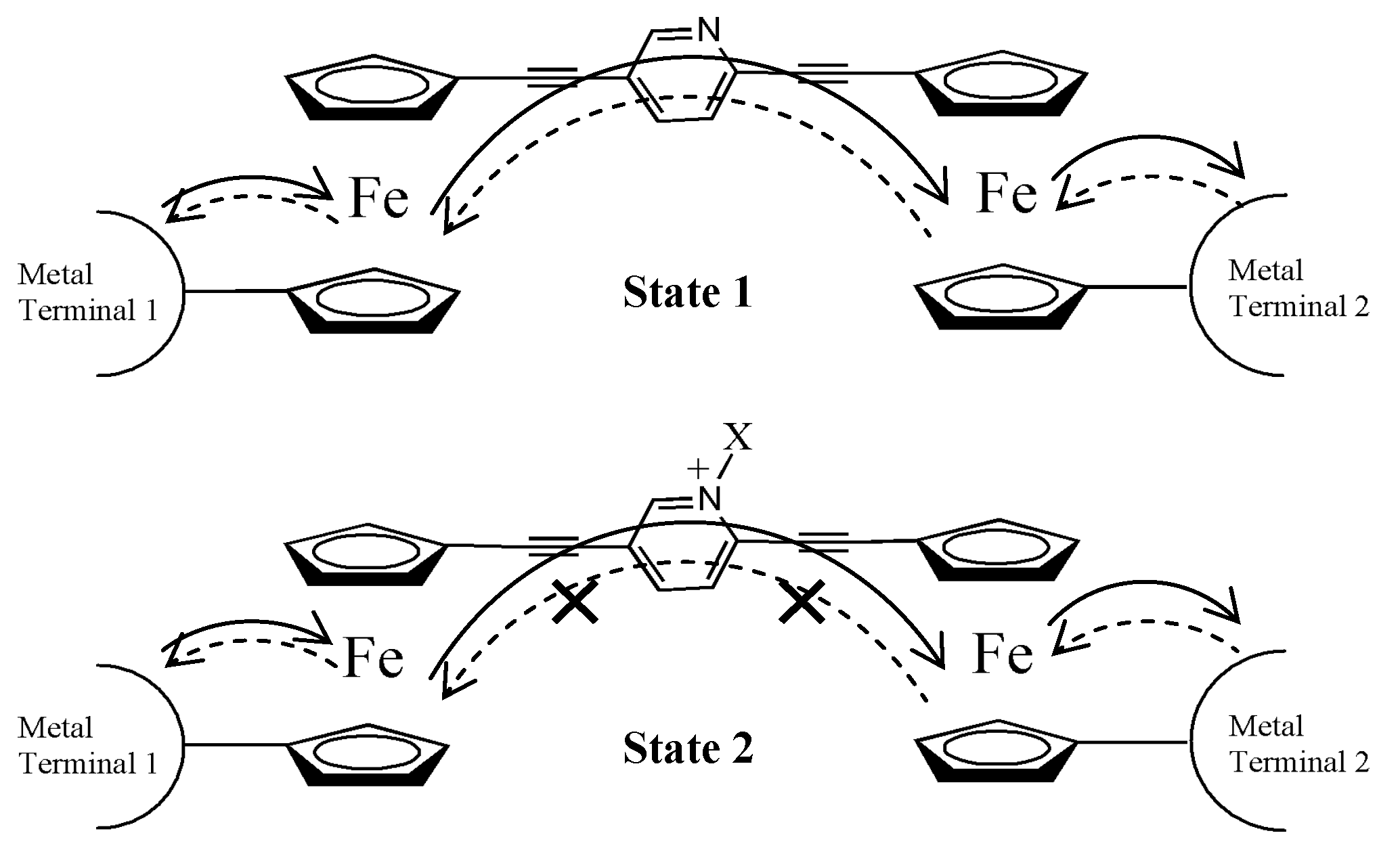
3.3. Electronic Couplings
| Compound | Hab (kcal/mol) | |
|---|---|---|
| Gas Phase | CH2Cl2 solution | |
| 1 | 1.37 | 0.56 |
| 2 | 1.36 | 0.57 |
| Fc−C≡C−Fc | 2.82 | 1.69 |
| Fc−(C≡C)3−Fc | 2.00 | 0.97 |
| Fc−(C≡C)6−Fc | 1.39 | 0.51 |
| Compound | ∆Ediabatic (kcal/mol) |
|---|---|
| 1 | 1.3 |
| 2 | 5.7 |
| Fc-oxazole-Fc state | Hab (kcal/mol) | |
|---|---|---|
| Gas Phase | CH2Cl2 solution | |
| Neutral | 1.81 | 1.04 |
| Protonated | 1.57 | 0.80 |
4. Conclusions
Acknowledgements
References and Notes
- Aviram, A.; Ratner, M.A. Molecular rectifiers. Chem. Phys. Lett. 1974, 29, 277–283. [Google Scholar]
- Eigler, D.M.; Schweizer, E.K. Positioning single atoms with a scanning tunneling microscope. Nature 1990, 344, 524–526. [Google Scholar]
- Ohnishi, H.; Kondo, Y.; Takayanagi, K. Quantized conductance through individual rows of suspended gold atoms. Nature 1998, 395, 780–783. [Google Scholar]
- Joachim, C.; Gimzewski, J.K.; Aviram, A. Electronics using hybrid-molecular and mono-molecular devices. Nature 2000, 408, 541–548. [Google Scholar]
- Joachim, C.; Gimzewski, J.K.; Schlittler, R.R.; Chavy, C. Electronic transparency of a single C-60 molecule. Phys. Rev. Lett. 1995, 74, 2102–2105. [Google Scholar]
- Reed, M.A.; Zhou, C.; Muller, C.J.; Burgin, T.P.; Tour, J.M. Conductance of a molecular junction. Science 1997, 278, 252–254. [Google Scholar]
- Reichert, J.; Ochs, R.; Beckmann, D.; Weber, H.B.; Mayor, M.; von Lohneysen, H. Driving current through single organic molecules. Phys. Rev. Lett. 2002, 88, 176804. [Google Scholar]
- Smit, R.H.M.; Noat, Y.; Untiedt, C.; Lang, N.D.; van Hemert, M.C.; van Ruitenbeek, J.M. Measurement of the conductance of a hydrogen molecule. Nature 2002, 419, 906–909. [Google Scholar]
- Xue, Y.Q.; Datta, S.; Ratner, M.A. Charge transfer and "band lineup" in molecular electronic devices: A chemical and numerical interpretation. J. Chem. Phys. 2001, 115, 4292–4299. [Google Scholar]
- Damle, P.S.; Ghosh, A.W.; Datta, S. Unified description of molecular conduction: From molecules to metallic wires. Phys. Rev. B 2001, 6420, 201403. [Google Scholar]
- Xue, Y.Q.; Datta, S.; Ratner, M.A. First-principles based matrix Green's function approach to molecular electronic devices: general formalism. Chem. Phys. 2002, 281, 151–170. [Google Scholar]
- Damle, P.; Ghosh, A.W.; Datta, S. First-principles analysis of molecular conduction using quantum chemistry software. Chem. Phys. 2002, 281, 171–187. [Google Scholar]
- Brandbyge, M.; Mozos, J.L.; Ordejon, P.; Taylor, J.; Stokbro, K. Density-functional method for nonequilibrium electron transport. Phys. Rev. B 2002, 65, 165401. [Google Scholar]
- Xue, Y.Q.; Ratner, M.A. Microscopic study of electrical transport through individual molecules with metallic contacts. I. Band lineup, voltage drop, and high-field transport. Phys. Rev. B 2003, 68, 115406. [Google Scholar]
- Xue, Y.Q.; Ratner, M.A. Microscopic study of electrical transport through individual molecules with metallic contacts. II. Effect of the interface structure. Phys. Rev. B 2003, 68, 115407. [Google Scholar]
- Fujimoto, Y.; Hirose, K. First-principles treatments of electron transport properties for nanoscale junctions. Phys. Rev. B 2003, 67, 195315. [Google Scholar]
- Calzolari, A.; Marzari, N.; Souza, I.; Nardelli, M.B. Ab initio transport properties of nanostructures from maximally localized Wannier functions. Phys. Rev. B 2004, 69, 035108. [Google Scholar]
- Rocha, A.R.; Garcia-Suarez, V.M.; Bailey, S.W.; Lambert, C.J.; Ferrer, J.; Sanvito, S. Towards molecular spintronics. Nat. Mater. 2005, 4, 335–339. [Google Scholar]
- Lee, S.U.; Belosludov, R.V.; Mizuseki, H.; Kawazoe, Y. Control of electron transport by manipulating the conjugated framework. J. Phys. Chem. C 2007, 111, 15397–15403. [Google Scholar]
- Tagami, K.; Tsukada, M. Spintronic transport through polyphenoxyl radical molecules. J. Phys. Chem. B 2004, 108, 6441–6444. [Google Scholar]
- Jalili, S.; Rafii-Tabar, H. Electronic conductance through organic nanowires. Phys. Rev. B 2005, 71, 165410. [Google Scholar]
- Xiao, X.Y.; Nagahara, L.A.; Rawlett, A.M.; Tao, N.J. Electrochemical gate-controlled conductance of single oligo(phenylene ethynylene)s. J. Am. Chem. Soc. 2005, 127, 9235–9240. [Google Scholar]
- Jiang, F.; Zhou, Y.X.; Chen, H.; Note, R.; Mizuseki, H.; Kawazoe, Y. Ab initio study of molecule transport characteristics based on nonequilibrium Green's function theory. Phys. Rev. B 2005, 72, 155408. [Google Scholar]
- Jiang, F.; Zhou, Y.X.; Chen, H.; Note, R.; Mizuseki, H.; Kawazoe, Y. Self-consistent study of single molecular transistor modulated by transverse field. J. Chem. Phys. 2006, 125, 084710. [Google Scholar]
- Jiang, F.; Zhou, Y.X.; Chen, H.; Note, R.; Mizuseki, H.; Kawazoe, Y. First-principles study of phenyl ethylene oligomers as current-switch. Phys. Lett. A 2006, 359, 487–493. [Google Scholar]
- Venkataraman, L.; Park, Y.S.; Whalley, A.C.; Nuckolls, C.; Hybertsen, M.S.; Steigerwald, M.L. Electronics and chemistry: Varying single-molecule junction conductance using chemical substituents. Nano Lett. 2007, 7, 502–506. [Google Scholar]
- Chen, H.; Lu, J. Q.; Wu, J.; Note, R.; Mizuseki, H.; Kawazoe, Y. Control of substituent ligand over current through molecular devices: An ab initio molecular orbital theory. Phys. Rev. B 2003, 67, 113408. [Google Scholar]
- Cheng, W.W.; Liao, X.Y.; Chen, H.; Note, R.; Mizuseki, H.; Kawazoe, Y. Electron transport through heterocyclic molecule: ab initio molecular orbital theory. Phys. Lett. A 2004, 326, 412–416. [Google Scholar]
- Cheng, W.W.; Chen, H.; Note, R.; Mizuseki, H.; Kawazoe, Y. Electron transport through molecular wire: effect of isomery. Phys. E 2005, 25, 643–646. [Google Scholar]
- Nguyen, T.; Sutton, A.D.; Brynda, M.; Fettinger, J.C.; Long, G.J.; Power, P.P. Synthesis of a stable compound with fivefold bonding between two chromium(I) centers. Science 2005, 310, 844–847. [Google Scholar]
- Huang, J.; Li, Q.X.; Ren, H.; Su, H.B.; Yang, J.L. Single quintuple bond [PhCrCrPh] molecule as a possible molecular switch. J. Chem. Phys. 2006, 125, 184713. [Google Scholar]
- Engtrakul, C.; Sita, L.R. Ferrocene-based nanoelectronics: 2,5-diethynylpyridine as a reversible switching element. Nano Lett. 2001, 1, 541–549. [Google Scholar]
- Getty, S.A.; Engtrakul, C.; Wang, L.; Liu, R.; Ke, S.H.; Baranger, H.U.; Yang, W.; Fuhrer, M.S.; Sita, L.R. Near-perfect conduction through a ferrocene-based molecular wire. Phys. Rev. B 2005, 71, 241401. [Google Scholar]
- Liu, R.; Ke, S.H.; Baranger, H.U.; Yang, W.T. Organometallic spintronics: Dicobaltocene switch. Nano Lett. 2005, 5, 1959–1962. [Google Scholar]
- Liu, R.; Ke, S.H.; Yang, W.T.; Baranger, H.U. Organometallic molecular rectification. J. Chem. Phys. 2006, 124, 024718. [Google Scholar]
- Wu, Q.; Van Voorhis, T. Extracting electron transfer coupling elements from constrained density functional theory. J. Chem. Phys. 2006, 125, 164105. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03; revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Bylaska, E.J.; de Jong, W.A.; Kowalski, K.; Straatsma, T.P.; Valiev, M.; Wang, D.; Aprà, E.; Windus, T.L.; Hirata, S.; Hackler, M.T.; Zhao, Y.; Fan, P.-D.; Harrison, R.J.; Dupuis, M.; Smith, D.M.A.; Nieplocha, J.; Tipparaju, V.; Krishnan, M.; Auer, A.A.; Nooijen, M.; Brown, E.; Cisneros, G.; Fann, G.I.; Früchtl, H.; Garza, J.; Hirao, K.; Kendall, R.; Nichols, J.; Tsemekhman, K.; Wolinski, K.; Anchell, J.; Bernholdt, D.; Borowski, P.; Clark, T.; Clerc, D.; Dachsel, H.; Deegan, M.; Dyall, K.; Elwood, D.; Glendening, E.; Gutowski, M.; Hess, A.; Jaffe, J.; Johnson, B.; Ju, J.; Kobayashi, R.; Kutteh, R.; Lin, Z.; Littlefield, R.; Long, X.; Meng, B.; Nakajima, T.; Niu, S.; Rosing, M.; Sandrone, G.; Stave, M.; Taylor, H.; Thomas, G.; van Lenthe, J.; Wong, A.; Zhang, Z. NWChem, A Computational Chemistry Package for Parallel Computers; Version 5.0; Pacific Northwest National Laboratory: Richland, Washington, USA, 2006. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. 3. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Abinitio effective core potentials for molecular calculations−Potentials for the transition−Metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; Defrees, D.J.; Pople, J.A. Self-consistent molecular-orbital methods. 23. A polarization-type basis set for 2Nd-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar]
- Klamt, A.; Schuurmann, G. Cosmo—A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar]
- Mochida, T.; Yamazaki, S. Mono- and diferrocenyl complexes with electron-accepting moieties formed by the reaction of ferrocenylalkynes with tetra-cyanoethylene. J. Chem. Soc. Dalton Trans. 2002, 3559–3564. [Google Scholar]
- McAdam, C.J.; Brunton, J.J.; Robinson, B.H.; Simpson, J. Ferrocenylethynylnaphthalenes and acenaphthylenes; Communication between ferrocenyl and cluster redox centres. J. Chem. Soc.-Dalton Trans. 1999, 2487–2495. [Google Scholar]
- Levanda, C.; Cowan, D.O.; Leitch, C.; Bechgaar, K. Mixed-valence diferrocenylacetylene cation. J. Am. Chem. Soc. 1974, 96, 6788–6789. [Google Scholar]
- Tarraga, A.; Molina, P.; Curiel, D.; Velasco, M.D. Homotrimetallic oxazolo-ferrocene complexes displaying tunable cooperative interactions between metal centers and redox-switchable character. Organometallics 2001, 20, 2145–2152. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ding, F.; Chen, S.; Wang, H. Computational Study of Ferrocene-Based Molecular Frameworks with 2,5-Diethynylpyridine as a Chemical Bridge. Materials 2010, 3, 2668-2683. https://doi.org/10.3390/ma3042668
Ding F, Chen S, Wang H. Computational Study of Ferrocene-Based Molecular Frameworks with 2,5-Diethynylpyridine as a Chemical Bridge. Materials. 2010; 3(4):2668-2683. https://doi.org/10.3390/ma3042668
Chicago/Turabian StyleDing, Feizhi, Shaowei Chen, and Haobin Wang. 2010. "Computational Study of Ferrocene-Based Molecular Frameworks with 2,5-Diethynylpyridine as a Chemical Bridge" Materials 3, no. 4: 2668-2683. https://doi.org/10.3390/ma3042668
APA StyleDing, F., Chen, S., & Wang, H. (2010). Computational Study of Ferrocene-Based Molecular Frameworks with 2,5-Diethynylpyridine as a Chemical Bridge. Materials, 3(4), 2668-2683. https://doi.org/10.3390/ma3042668