1. Introduction
The molecule with the formula C
H
O has six structural isomers: vinyl ether, 2-butenal, cis-2-butenal, cyclobutanone, furan, 2, 3-dihydro, and furan, 2, 5-dihydro. Vinyl ether is an important molecule for biomedical applications. It is used as an anesthetic in surgical procedures in children for short durations with fast recovery time [
1]. Mass spectrometry is one of the techniques to monitor gas concentration for the patient undergoing an anaesthetic procedure [
2]. Vinyl ether is also used as an antioxidant to reduce the side effect of free radicals (oxidants) in the human body that can cause genetic disorders [
3,
4]. There is a possibility of having free electrons and oxidants (having unpaired electrons in the outermost shell) in the human body for further reaction to damage DNA [
3,
4]. Plasmalogens (endogenous antioxidants) containing vinyl ether bonds are found in high concentrations in brain and heart [
5]. Plasmalogens deficiency can cause neurodegenerative diseases, cancer, stroke, etc. [
6,
7]. Hence, to understand the chemistry of this biological environment, one needs to model the system with various cross sections and reaction rates as input. Another important application of C
H
O molecule is in the military, where it is considered as a range compound [
8], and its cross section and rate coefficients are useful in gas transport modeling. The presence of C
H
O on the surface of Martian soil was detected as early as 1977 [
9]. Natural processes make it possible to create this molecule in the atmosphere of Mars [
10]. Furan, 2, 5-dihydro is used in pharmaceuticals and commodity chemicals [
11] as well.
The wide range of applications for C
H
O molecule from biological sciences to atmospheric physics has encouraged us to perform the present calculations. To the best of our knowledge, there is no previous data available to compare our results in the energy range chosen here (from ionization potential of the target molecule to 5 keV). The only single data point available is from Tureček et al. [
12], who evaluated relative ionization cross section value for cis-2-butenal, cyclobutanone, and furan, 2, 5-dihydro at 75 eV. Due to the unavailability of previously reported data, our results are compared with another molecule with similar structure, i.e., tetrahydrofuran (THF). THF and furan, 2, 5-dihydro have a similar structure “(with THF having 2 additional electrons)” and very close ionization energy and electric dipole polarizability, thus making it a good candidate for comparison. Theoretical and experimental data for THF are available in the literature [
13,
14,
15,
16,
17,
18,
19].
The present study of electron scattering from C
H
O and its isomers has been performed based on spherical complex optical potential formalism (SCOP) [
20,
21] and complex scattering potential ionization contribution method (CSP-ic) [
20,
21]. The theoretical approach of SCOP evaluates integral elastic (
) and inelastic (
) cross section values. The addition of these two cross sections provides the total (
). The CSP-ic method is applied to evaluate ionization cross section (
). The electrostatic potential surface method is employed to identify the centers that scatters the incoming electron. Due to the non-spherical and complex structure of these targets, a multi-center approach called group additivity rule is used [
22], which could predict reasonable cross sections. Further fine tuning is performed by a geometrical screening correction technique to compensate for the overestimation in cross section due to overlapping of electron charge densities. The
,
, and
values for vinyl ether and its isomers are reported first. The correlation of
with (
and effective target diameter (
) and the projectile wavelength (
) for vinyl ether and its isomers produces a good understanding of ionization cross section and molecular parameters.
Furthermore, a parametric fitting of
with energy (A
) at the intermediate energy range shown. Present electron-impact total scattering cross sections are then utilized to estimate the gas-kinetic radius and van der Waals coefficients. The theoretical description is given in
Section 2, and
Section 3 explains the present results. The final section concludes with an overall summary of the work.
2. Theoretical Methodology
Present target molecules, C
H
O and its isomers, are structurally complex and large in size in terms of an electron-molecule quantum collision problem. The geometries of the present target molecules are shown in
Figure 1, while in
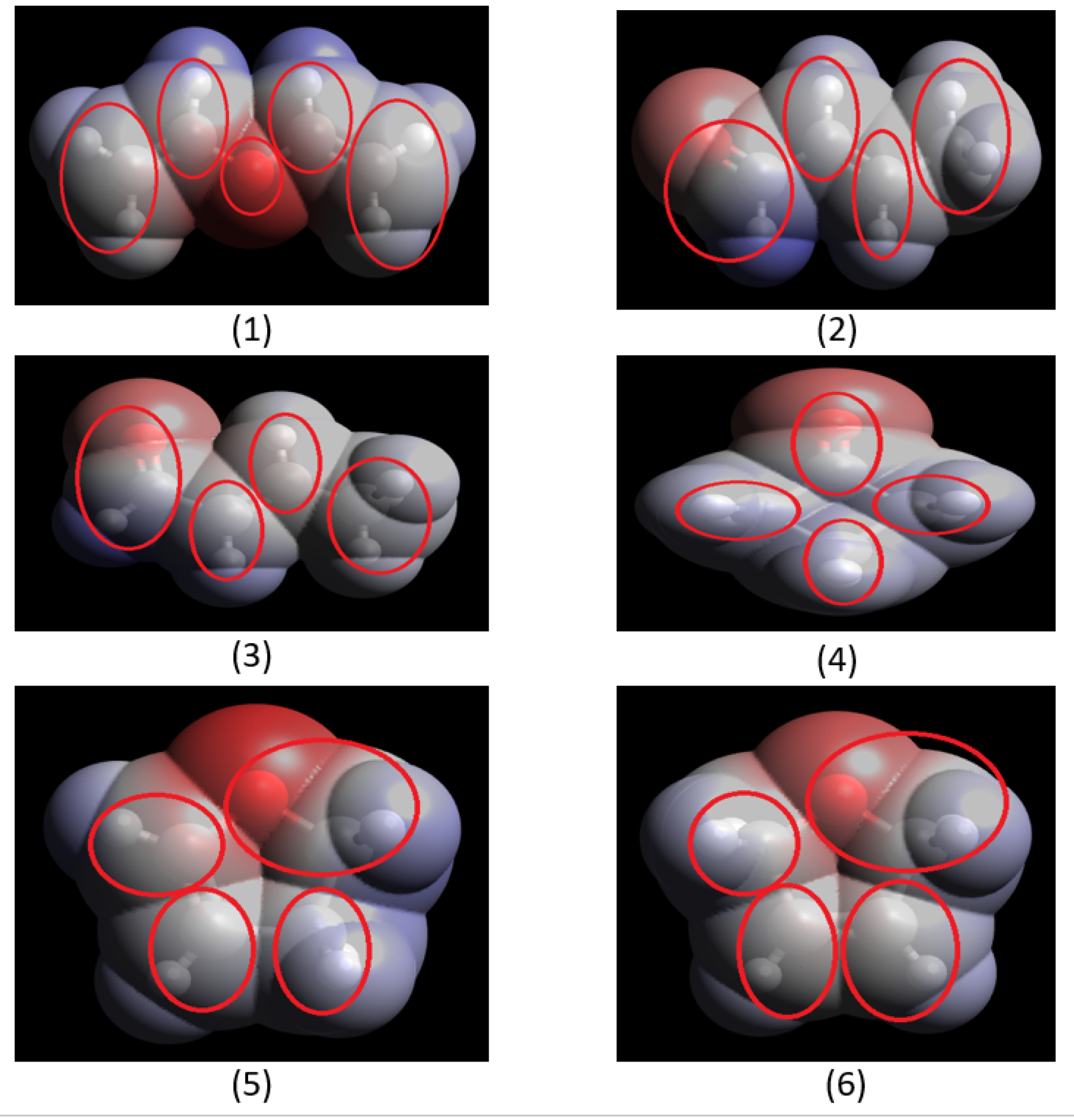
Table 1, their ionization energies and polarizabilities are illustrated. Due to this reason, a target molecule is divided into groups/scattering centres to have a viable calculation and to obtain reasonable cross-section values. These scattering centers are identified based on molecular geometrical configuration and its electron charge density distribution. For the present set of molecules, these centers are identified through Avogadro software [
23] using the electrostatic potential surface method, as shown in
Figure 2.
For each scattering center, total charge density is calculated via expansion of charge density of individual atoms from the center of mass of that particular group. In the present case, the scattering center contains more than one heavier atom along with hydrogen atoms. The expansion of charge density for heavier atoms is performed from the center of mass, and for hydrogen atoms, charge density expansion is induced by heavier atoms. This expanded charge density is re-normalized to obtain the total number of electrons in each scattering centre. The charge density
is calculated using the atomic parameters as described in Cox and Bonham [
25]. For each scattering centre, the Schrödinger equation comprises complex optical potential and solves it with the help of partial wave method. This method is called the group additivity rule (GAR) [
22]. From the contribution of each scattering center, the overall cross section of the target molecule is calculated. Further information on this methodology may be obtained from the articles [
26,
27].
The potential includes all standard possible projectile-target interactions. The optical potential (
) consists of real (
) and imaginary (
) parts expressed as follows.
Here,
is the projectile field energy in eV and
r is the projectile-target interaction distance.
comprises static potential (
), exchange potential (
), and polarization potential (
) modeled from [
25,
28,
29], respectively.
is experienced by the incoming electron due to target’s static charge cloud.
occurs due to the interaction and the exchange of incident and target electrons.
arises via the redistribution of target charge cloud by the interaction with incident field, which creates a perturbation in static charge cloud.
in Equation (
1) corresponds to the absorption of incident flux due to inelastic channels such as excitation and ionization, which is modeled using the non-empirical formula reported by Staszewska et al. [
30,
31]. The solution of Schrödinger equation is obtained as phase shifts (
), possessing the signature of all scattering channels. Using these complex phase shifts, the cross sections are estimated as follows [
20,
21]:
where
k is the wave vector, and
=
is the ‘inelasticity’ or ‘absorption factor’ for each partial wave
l. The sum of elastic and inelastic cross section provides the total cross section. Furthermore, ionization cross section is estimated through an energy-dependent ratio,
R(
) based on CSP-ic method [
20,
21]. The ratio is defined as follows:
such that
following the boundary conditions:
where
is the energy where
reaches its maximum value. The first choice in Equation (
5) confirms that the target’s ionization channel is not active below its threshold
I. The second condition represents the contribution of ionization in the inelastic cross section is
(about 70–80%) [
32] at peak energy (
). This is an empirical estimation based on benchmark experimental/theoretical results. According to the third condition,
approaches unity as projectile energy increases. In the present calculations, we have taken
for all targets. Using this
,
is predicted with an uncertainity of approximately
%, which is termed as the CSP-ic method [
20,
21].
For all these molecules, a screening correction technique is applied [
33] to obtain reliable cross sections. As the number of atoms in a molecule increases, the possibility of screening effect due to charge density overlapping of atoms within the molecule also enhances. This overlapping causes an overestimation of cross section from its actual values. Hence, a geometrical screening correction technique is applied for the present target molecules to overcome this. The present calculation considers a screening correction to the central atom of an individual group. The screening correction between the groups is not considered as the charge overlapping between the groups is assumed to be negligible. The screening correction included in the present calculation is explained below. The overlap cross section for each group is calculated with the following.
Here,
N provides the number of neighboring atoms of the central atom.
and
represent the cross section values of
ith and
jth atoms of these neighbors.
is the max
and
the distance between
ith and
jth atoms. Thus, the screening corrected cross section
for the scattering group is obtained as follows:
where
is the cross section calculated based on the group additivity rule.
3. Results and Discussion
This section presents the cross sections predicted for all the targets studied. The cross section is plotted along y-axis in Å
and energy along x-axis in eV (logarithmic scale). The present results are compared with THF, both theoretical [
13,
14] and experimental [
16,
17,
18,
19] cross sections, due to the non-availability of previous data for vinyl ether and its isomers. The structure of THF and furan, 2, 5-dihydro is quite similar, except for the difference of two hydrogen atoms. In addition, the target parameters such as the ionization threshold and polarizabilities are also close to each other, which makes the comparison justified.
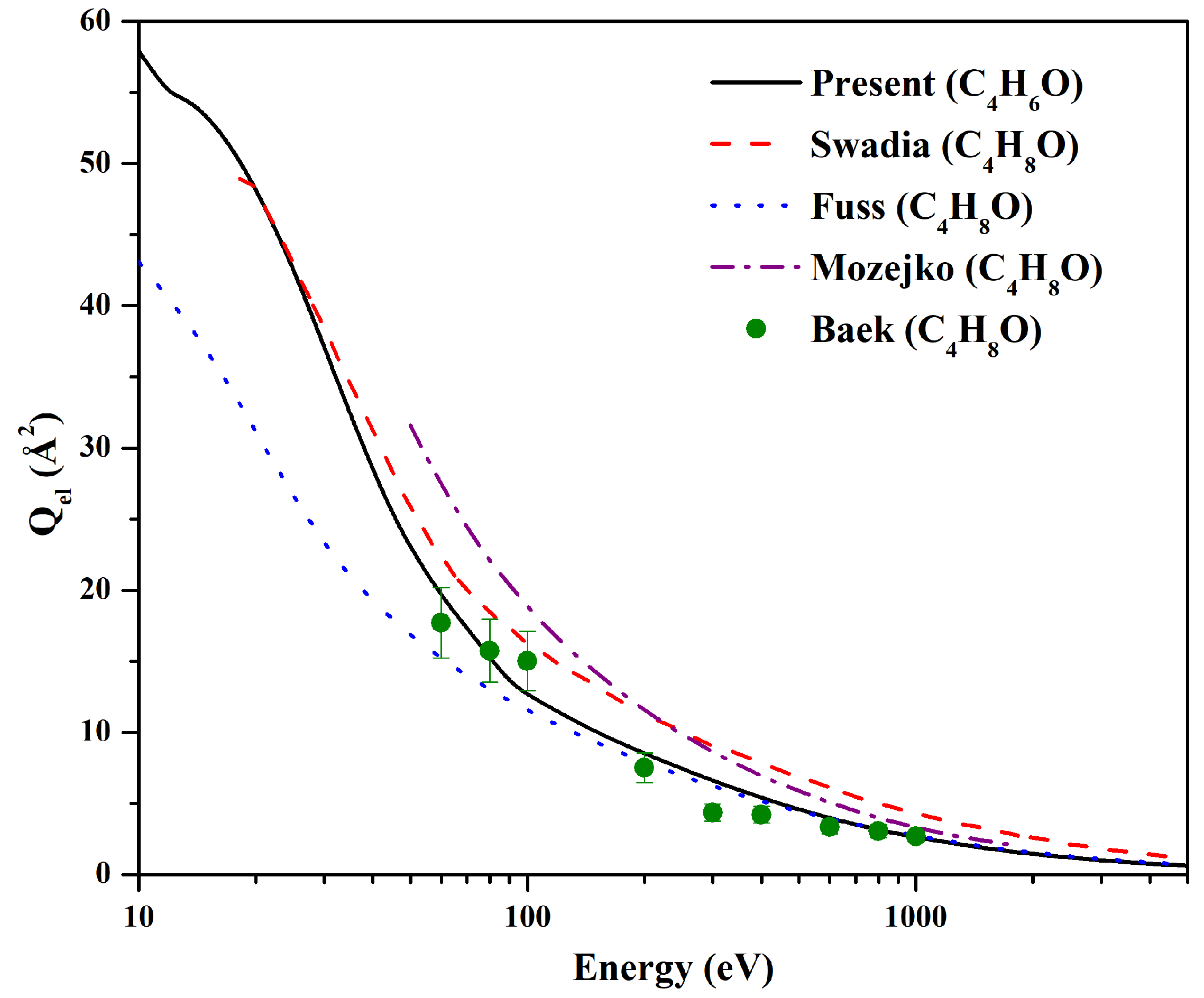
In
Figure 3, present furan, 2, 5-dihydro cross section values are presented and compared with previous data of THF computed by Swadia et al. [
13], Fuss et al. [
18], and Mozejko et al. [
14] and measured by Baek et al. [
19]. The theoretical values by Swadia et al. [
13] are comparable in the 18–30 eV energy range, but they are higher beyond that. Swadia et al. [
13] obtained
through the optical potential approach and, hence, is similar to the present calculation. The values of Fuss et al. [
18] are lower than the present result up to 50 eV, while their results are comparable above that. At higher energies, both results merge together. Fuss et al. [
18] computed
by using a screening-corrected independent atom approach using the optical potential. The difference in additivity rule and screening correction method used in present and Fuss calculations creates the deviation in the outputs. Mozejko et al. [
14] results are higher than present outcomes throughout the energy range. The method employed by Mozejko is different than the present approach. In addition, they have used an independent atom model (IAM) approach, which does not consider the redistribution of atomic electrons due to molecular bonding and, hence, produces higher values compared to the present result. However, above 400 eV, they are comparable to each other as the projectile electron obtains less time to spend with the target. The experimental data of Baek et al. [
20] are quite close to the present values throughout the energy range within the experimental error.
In
Figure 4, present
results are presented along with comparison (THF) [
13,
17,
18,
19]. The values for THF are in general lower than our cross section below 50–80 eV. Beyond that, the total cross section agrees quite well with all the available data for THF. The theoretical curves of Fuss et al. [
18] are much lower than all others up to about 200 eV. However, the measurements of Fuss et al. [
18] produce an excellent agreement with present results throughout the energy range. The experimental result of Mozejko et al. [
17] is lower than present curve around the ionization threshold region and then become comparable above 60–80 eV. The same is true for the measurements of Baek et al. [
19] as well.
Figure 3.
for furan, 2, 5-dihydro. Solid line: present
; dash line:
for THF by Swadia et al. [
13]; dotted line:
for THF by Fuss et al. [
18]; dash-dot line:
for THF by Mozejko et al. [
14]; solid circle:
for THF by Baek et al. [
19].
Figure 3.
for furan, 2, 5-dihydro. Solid line: present
; dash line:
for THF by Swadia et al. [
13]; dotted line:
for THF by Fuss et al. [
18]; dash-dot line:
for THF by Mozejko et al. [
14]; solid circle:
for THF by Baek et al. [
19].
Figure 5,
Figure 6 and
Figure 7 show present ionization cross section values of furan, 2, 5-dihydro, cis-2-butenal, and cyclobutanone, respectively. In
Figure 5, the present
of furan, 2, 5-dihydro compares with the experimental and theoretical data points of furan, 2, 5-dihydro from Tureček et al. [
12]. Present
values of furan, 2, 5-dihydro are also compared with THF values reported by Swadia et al. [
13] and Mozejko et al. [
14] and the experimental values given by Fuss et al. [
18] and Dampc et al. [
16]. Present values are higher than all other data near the intermediate energy range. However, it merges with most of them at low and high energies. Mozejko et al. [
14] computed
using the Binary-Encounter-Bethe (BEB) model, which estimates direct ionization cross-sections for the ground states of the geometrically optimized molecule. Present
values are comparable to the experimental result of Fuss et al. [
18] within their error bars. Moreover, it shows excellent agreement with the measurements of Dampc et al. [
16] up to 30 eV. The experimental data points of Tureček et al. [
12] is significantly less than the present values at 75 eV, while the theoretical data point is slightly close to present data and much closer to that of THF values. In
Figure 6, the theoretical and experimental data points of Tureček et al. [
12] is compared with the present data for cis-2-butenal. The data points at 75 eV are quite lower than our results. In
Figure 7, the cross section for cyclobutanone is plotted along with the data point at 75 eV by Tureček et al. [
12]. This is similar to the previous figure, except that they do not agree with each other.
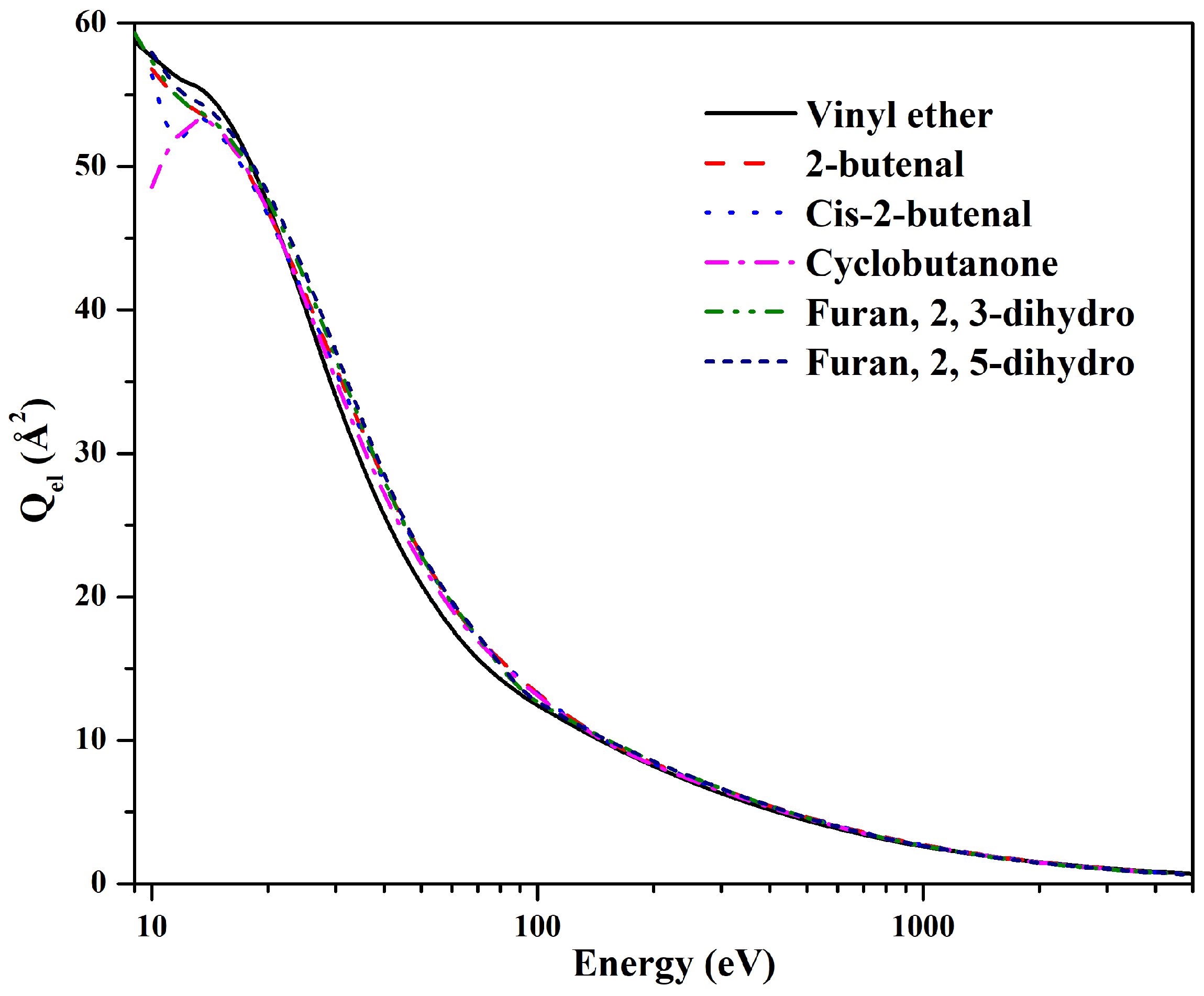
Figure 8 and
Figure 9 represent
and
for vinyl ether and its isomers, respectively. Near the ionization threshold, the difference in cross sections of isomers is enough to visualize the isomeric effect in the present target molecules. This difference decreases at the intermediate energy range, and at higher energies cross section values are close to each other. The isomeric effect is due to the difference in the structure of isomers and, hence, the distribution of charge density of individual atoms in the target molecule.
have branched and cyclic structures, which is contributing to the isomeric effect. Vinyl ether, 2-butenal, and cis-2-butenal are branched isomers. The
and
values of 2-butenal and cis-2-butenal are quite close throughout the energy range except between 10-14 eV. The hump at low energy in
is enhanced due to the inclusion (or opening) of more inelastic channels while calculating elastic cross sections. In contrast, the cross section for vinyl ether is different from 2-butenal and cis-2-butenal, reflecting the electron cloud and polarizability of the branched isomers. The cyclic isomers, furan, 2, 3-dihydro, and furan, 2, 5-dihydro have similar
and
values throughout the energy range except for the 10–17 eV and 15–30 eV energy ranges, respectively. Cyclobutanone, another cyclic isomer, has different
and
values than furan, 2, 3-dihydro and furan, 2, 5-dihydro near-ionization threshold, and intermediate energy range, which shows structural effect. However, at high energies, the isomeric effect vanishes for all targets. This is because, as the incident electron field energy increases, the de Broglie wavelength of the incident electron becomes so small that it does not recognize molecular structures any more. Thus, the structural effects and/or threshold effects become negligible, and the isomeric effect in cross section disappears.
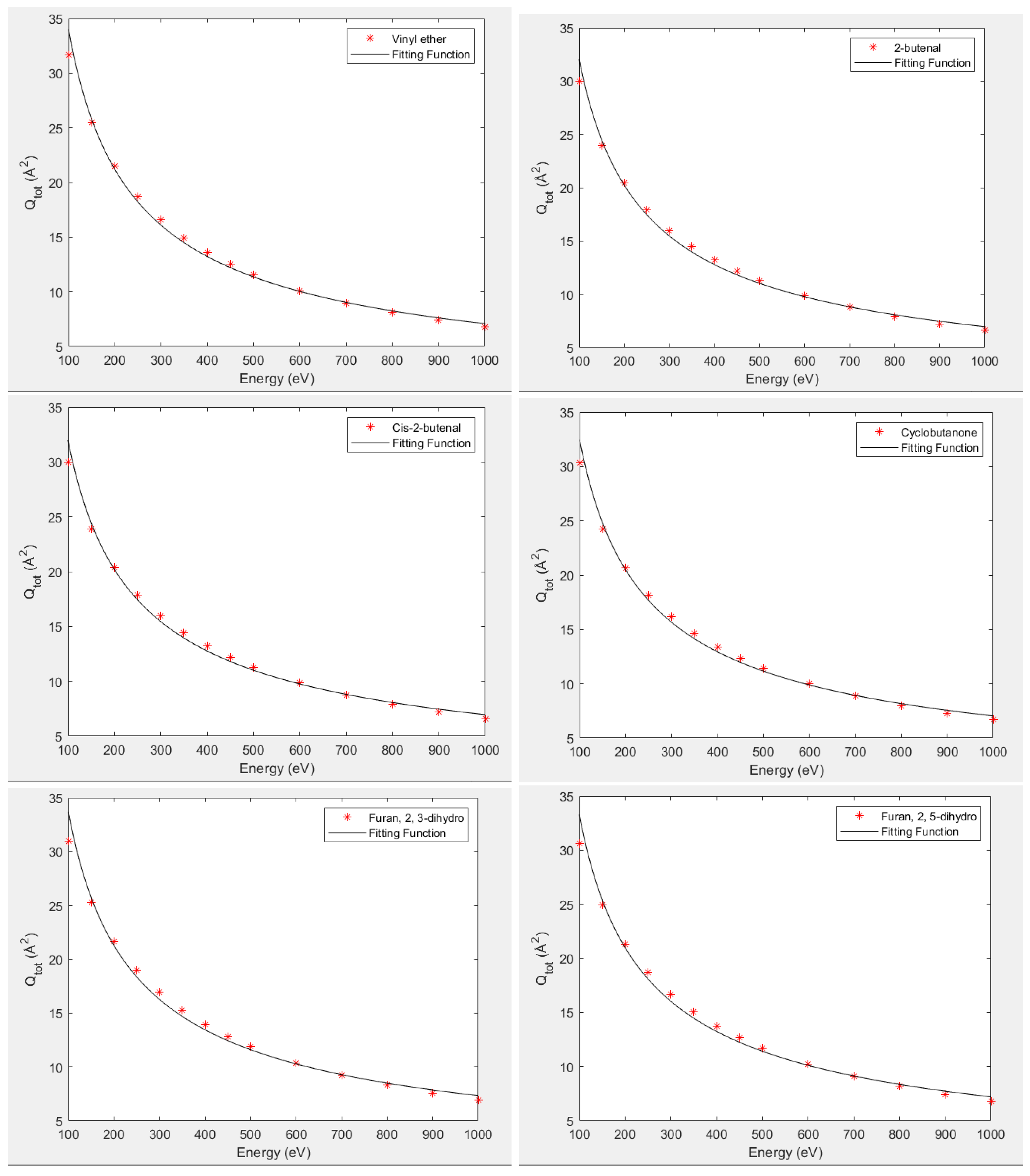
To study the consistency and reliability of the data, we have also analytically fitted the total cross section at intermediate and high energy range (100–1000 eV) using the following formulae [
34]:
where A and B are fitting parameters and
is the incident electron energy. The fitting given by Joshipura et al. [
34] is for the molecular systems having 10 electrons. Furthermore, Garcia et al. [
35] has also reported such fittings of the total cross section at intermediate energies for molecules having 10–22 electrons. The resent target molecules and its isomers have 38 electrons. The calculated and fitted cross sections are given in
Figure 10. It shows excellent corroboration and proves the validity of our data. The parameters A and B are given in
Table 2. We can see that the value of B is quite close for all the isomers, while parameter A varies quite significantly. Since the magnitude of total cross section can be considered as a size-effect, it should depend on the spread of an electron cloud, which is reflected by the polarizability of the molecule.
Furthermore, the gas-kinetic radius (
R) and van der Waals coefficient (
b) are two important quantities for characterizing the motion of any molecules in the gas phase. A formula for calculation the gas-kinetic radius (
R) and the van der Waals coefficient (
b) using total electron scattering cross-sections (TCS) was given in Hirschfelder et al. [
36]. In the present study, an analytical approach for calculations of
R and
b is used from the electron impact cross-sections data of vinyl ether and its isomers. The present estimation of
R and
b is based on the work of Szmytkowski et al. [
37]. According to the previously reported approach of Szmytkowski et al. [
37] and Goswami et al. [
38],
Q at 350 eV, the gas-kinetic radius
R and van der Waals coefficient
b of the target can be respectively expressed as follows:
and the following is the case.
Here,
N is the Avogadro number. The
R and
b calculated for the present targets are listed in
Table 2. It can be observed that only minor variations are seen in
R and
b due to the same electron number, which is slightly different due to change in geometrical distribution of the constituent atoms. For the present isomers, the maximum change in position of the center of mass is only about 9% by the rearrangement of C, H and O atoms in these molecules. A similar effect is observed in the previous calculations of Goswami et al. [
38].
In
Figure 11, the total ionization cross sections for vinyl ether and its isomers were compared. The graph shows that the cross section values for vinyl ether are more prominent in magnitude than other isomers. The curves of furan, 2, 3-dihydro and furan, 2, 5-dihydro are very close throughout the energy range. In contrast, the values of 2-butenal and cis-2-butenal cross sections merges together. The cross section for cyclobutanone is very close to furan, 2, 5-dihydro below 40 eV. The peak of present ionization cross section is at 60 eV for vinyl ether, cyclobutanone, furan, 2, 3-dihydro, and furan, 2, 5-dihydro and at 70 eV for 2-butenal and cis-2-butenal.
For a particular incident energy, the wavelength of the projectile electron matches molecular diameter and gives maximum cross section values. Harland et al. [
39] showed that the effective diameter of the molecule may be obtained using the radius of the polarizability sphere. The radius of the polarizability sphere is obtained through the polarizability volume of the target molecule. The present target molecule has six different structural formulas with 38 electrons. The electron cloud of a molecule is proportional to its polarizability volume. The polarizability volume provides an idea of molecular charge distribution and adequate sizes of the molecule.
Figure 12 represents the relation between effective molecular diameter,
and incident electron wavelength at Q
. In
Figure 12, the effective diameter of each isomer is slightly higher than the electron wavelength at which
attains its maximum value. The incident electron at the de Broglie wavelength of 1.462 Å and 1.579 Å is comparable to the effective diameter of 2-butenal and cis-2-butenal and vinyl ether, cyclobutanone, furan, 2, 3-dihydro, and furan, 2, 5-dihydro, respectively. The probability of ionization increases from the threshold region and reaches its maximum value as the de Broglie wavelength of incident electron matches to an effective diameter of the target molecule.
The maximum ionization cross section value also depicts the relation between polarizability and ionization energy of the target molecule.
Figure 13 represents the relationship of Q
with (
for vinyl ether and its isomers. In
Figure 13, the correlation of Q
with (
for vinyl ether, cyclobutanone, furan, 2, 3-dihydro, and furan, 2, 5-dihydro is close to a straight line. On the other hand, for 2-butenal and cis-2-butenal, the correlation is quite small. The polarizability value for 2-butenal is slightly higher than cis-2-butenal, making the point falls towards the right in the figure. The reliability of the present Q
is also estimated through the relation Q
= 11.92 (
, as given in Harland et al. [
39] and is shown in
Table 3.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}