
Bispecific Antibody-Based Immune-Cell Engagers and Their Emerging Therapeutic Targets in Cancer Immunotherapy


Abstract
:1. Introduction
2. Action Mechanism of bsAb-Based ICEs in Cancer
2.1. T-Cell Engagers
2.2. NK-Cell Engagers
3. Role of Known and Emerging Targets of ICEs
3.1. Single-Pass ICE Targets in Solid Cancers
3.1.1. Delta-Like Ligand 3
3.1.2. Epidermal Growth Factor Receptor
3.1.3. EpCAM
3.1.4. Glycoprotein A33
3.1.5. Human EGFR 2
3.1.6. Mucin 16
3.1.7. Mucin 17
3.1.8. Prostate-Specific Membrane Antigen
3.2. Multi-Pass Transmembrane Proteins as ICE Targets in Solid Cancers
3.2.1. Claudin-18 Isoform 2
3.2.2. Six-Transmembrane Epithelial Antigen of Prostate 1
3.2.3. Somatostatin Receptor 2
3.3. Glycosylphosphatidylinositol-Anchored Proteins as ICE Targets in Solid Cancers
3.3.1. Carcinoembryonic Antigen
3.3.2. Glypican 3
3.4. Sphingolipid as ICE Targets in Solid Cancers
GD2
3.5. Single Transmembrane Proteins as ICE Targets in Hematological Cancers
3.5.1. B-Cell Maturation Antigen
3.5.2. CD19
3.5.3. CD22
3.5.4. CD30
3.5.5. CD33
3.5.6. CD38
3.5.7. CD123
3.5.8. C-Type Lectin Domain Family 12 Member A
3.5.9. FMS-Like Tyrosine Kinase 3
3.6. Multi-Pass Transmembrane Proteins as ICE Targets in Hematological Cancers
3.6.1. CD20
3.6.2. GPCR Class C Group 5 Member D
4. Design and Structure of bsAb-Based ICEs
4.1. Fv-Based ICEs
4.1.1. BiTE
4.1.2. Dual-Affinity Retargeting Protein
4.1.3. Tandem Diabody
4.2. IgG-Based ICEs: Symmetric Format
4.2.1. Fabs-In-Tandem Ig
4.2.2. IgG–[L]–scFv2
4.2.3. IgG–[H]–scFv2
4.2.4. scFv2–Fc–scFv2
4.3. IgG-Based ICEs: Asymmetric Format
4.3.1. Mouse–Rat Hybrid IgG
4.3.2. KiH-Based IgG
4.3.3. Charge-Pair-Based IgG
4.3.4. cFAE-Based IgG
4.3.5. Common Light-Chain-Based IgG
4.3.6. CrossMab-Based IgG
4.3.7. Fab–scFv–Fc
4.3.8. Fc-Fused Fvs
5. Current Development Status of bsAb-Based ICEs
5.1. ICEs Targeting Tumor Antigens in Solid Cancers
5.1.1. CEA-Specific ICE
5.1.2. CLDN18.2-Specific ICE
5.1.3. DLL3-Specific ICE
5.1.4. EGFR-Specific ICE
5.1.5. EpCAM-Specific ICE
5.1.6. GD2-Specific ICE
5.1.7. GPA33-Specific ICE
5.1.8. GPC3-Specific ICE
5.1.9. HER2-Specific ICE
5.1.10. MUC16-Specific ICE
5.1.11. MUC17-Specific ICE
5.1.12. PSMA-Specific ICE
5.1.13. STEAP1-Specific ICE
5.1.14. SSTR2-Specific ICE
5.2. ICEs Targeting Tumor Antigens on Hematological Cancers
5.2.1. BCMA-Specific ICE
5.2.2. CD19-Specific ICE
5.2.3. CD20-Specific ICE
5.2.4. CD22-Specific ICE
5.2.5. CD30-Specific ICE
5.2.6. CD33-Specific ICE
5.2.7. CD38-Specific ICE
5.2.8. CD123-Specific ICE
5.2.9. CLEC12A-Specific ICE
5.2.10. FLT3-Specific ICE
5.2.11. GPRC5D-Specific ICE
6. Practical Considerations for the Development of Next-Generation ICEs
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Estimated Number of New Cases in 2020, Worldwide, Both Sexes, All Ages. Available online: https://gco.iarc.fr/today/online-analysispie?v=2020&mode=cancer&mode_population=continents&population=900&populations=900&key=total&sex=0&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&nb_items=7&group_cancer=1&include_nmsc=1&include_nmsc_other=1&half_pie=0&donut=0 (accessed on 21 February 2022).
- Estimated Number of Deaths in 2020, Worldwide, Both Sexes, All Ages. Available online: https://gco.iarc.fr/today/online-analysispie?v=2020&mode=cancer&mode_population=continents&population=900&populations=900&key=total&sex=0&cancer=39&type=1&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&nb_items=7&group_cancer=1&include_nmsc=1&include_nmsc_other=1&half_pie=0&donut=0 (accessed on 21 February 2022).
- Estimated Number of New Cases From 2020 to 2040, Both Sexes, Ages (0–85). Available online: https://gco.iarc.fr/tomorrow/en/dataviz/bars?types=0&sexes=0&mode=population&group_populations=1&multiple_populations=1&multiple_cancers=1&cancers=39&populations=903_904_905_908_909_935&apc=cat_ca20v1.5_ca23v1.5&group_cancers=1 (accessed on 21 February 2022).
- Arruebo, M.; Vilaboa, N.; Sáez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; González-Fernández, A. Assessment of the evolution of cancer treatment therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, D.; Arrighi, S.; Darwiche, Y.; Deb, S. Comparison of Anticancer Drug Toxicities: Paradigm Shift in Adverse Effect Profile. Life 2021, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Baldo, B.A. Immune- and Non-Immune-Mediated Adverse Effects of Monoclonal Antibody Therapy: A Survey of 110 Approved Antibodies. Antibodies 2022, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. The structure of a typical antibody molecule. In Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redman, J.M.; Hill, E.M.; AlDeghaither, D.; Weiner, L.M. Mechanisms of action of therapeutic antibodies for cancer. Mol. Immunol. 2015, 67, 28–45. [Google Scholar] [CrossRef] [Green Version]
- Van Erp, E.A.; Luytjes, W.; Ferwerda, G.; van Kasteren, P.B. Fc-Mediated Antibody Effector Functions During Respiratory Syncytial Virus Infection and Disease. Front. Immunol. 2019, 10, 548. [Google Scholar] [CrossRef] [Green Version]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Wang, S.; Chen, K.; Lei, Q.; Ma, P.; Yuan, A.Q.; Zhao, Y.; Jiang, Y.; Fang, H.; Xing, S.; Fang, Y. The state of the art of bispecific antibodies for treating human malignancies. EMBO Mol. Med. 2021, 13, 14291. [Google Scholar] [CrossRef]
- Mazor, Y.; Oganesyan, V.; Yang, C.; Hansen, A.; Wang, J.; Liu, H.; Sachsenmeier, K.; Carlson, M.; Gadre, D.V.; Borrok, M.J.; et al. Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs 2015, 7, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical development and future directions. mAbs 2010, 2, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Ruf, P.; Bauer, H.W.; Schoberth, A.; Kellermann, C.; Lindhofer, H. First time intravesically administered trifunctional antibody catumaxomab in patients with recurrent non-muscle invasive bladder cancer indicates high tolerability and local immunological activity. Cancer Immunol. Immunother. 2021, 70, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Nagorsen, D.; Kufer, P.; Baeuerle, P.A.; Bargou, R. Blinatumomab: A historical perspective. Pharmacol. Ther. 2012, 136, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Bispecific antibodies rise again: Amgen’s blinatumomab is setting the stage for a bispecific-antibody revival, enabled by new formats that may solve the field’s long-standing problems. Nat. Rev. Drug Discov. 2014, 13, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, M.; Ren, F.; Meng, X.; Yu, J. The landscape of bispecific T cell engager in cancer treatment. Biomark. Res. 2021, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Liu, M.; Zhang, Y.; Wang, X. Bispecific T cell engagers: An emerging therapy for management of hematologic malignancies. J. Hematol. Oncol. 2021, 14, 75. [Google Scholar] [CrossRef]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse: A molecular machine controlling T cell activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.; Keefe, D.; Boulant, S.; Boucrot, E.; Walch, M.; Martinvalet, D.; Goping, I.S.; Bleackley, R.C.; Kirchhausen, T.; Lieberman, J. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat. Immunol. 2011, 12, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [Green Version]
- Dain Moon, N.T.; Park, Y.; Lee, S.-W.; Kim, D.H. Development of bispecific antibody for cancer immunotherapy: Focus on T cell engaging antibody. Immune Netw. 2022, 22, 4. [Google Scholar] [CrossRef]
- Kamakura, D.; Asano, R.; Yasunaga, M. T Cell Bispecific Antibodies: An Antibody-Based Delivery System for Inducing Antitumor Immunity. Pharmaceuticals 2021, 14, 1172. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef] [PubMed]
- Aptsiauri, N.; Ruiz-Cabello, F.; Garrido, F. The transition from HLA-I positive to HLA-I negative primary tumors: The road to escape from T-cell responses. Curr. Opin. Immunol. 2018, 51, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Kim, T.; Song, S.Y.; Park, S.; Cho, H.H.; Jung, S.H.; Ahn, J.S.; Kim, H.J.; Lee, J.J.; Kim, H.O.; et al. Naive CD8(+) T cell derived tumor-specific cytotoxic effectors as a potential remedy for overcoming TGF-beta immunosuppression in the tumor microenvironment. Sci. Rep. 2016, 6, 28208. [Google Scholar] [CrossRef]
- Demaria, O.; Gauthier, L.; Debroas, G.; Vivier, E. Natural killer cell engagers in cancer immunotherapy: Next generation of immuno-oncology treatments. Eur. J. Immunol. 2021, 51, 1934–1942. [Google Scholar] [CrossRef]
- Poli, A.; Michel, T.; Thérésine, M.; Andrès, E.; Hentges, F.; Zimmer, J. CD56bright natural killer (NK) cells: An important NK cell subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef]
- Amand, M.; Iserentant, G.; Poli, A.; Sleiman, M.; Fievez, V.; Sanchez, I.P.; Sauvageot, N.; Michel, T.; Aouali, N.; Janji, B.; et al. Human CD56dimCD16dim Cells As an Individualized Natural Killer Cell Subset. Front. Immunol. 2017, 8, 699. [Google Scholar] [CrossRef]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, N.L.; Herrera, A.F.; Domingo-Domenech, E.; Mehta, A.; Forero-Torres, A.; Garcia-Sanz, R.; Armand, P.; Devata, S.; Izquierdo, A.R.; Lossos, I.S.; et al. A phase 1b study of AFM13 in combination with pembrolizumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2020, 136, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.H.; Giffin, M.J.; Bailis, J.M.; Smit, M.D.; Carbone, D.P.; He, K. DLL3: An emerging target in small cell lung cancer. J. Hematol. Oncol. 2019, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Furuta, M.; Kikuchi, H.; Shoji, T.; Takashima, Y.; Kikuchi, E.; Kikuchi, J.; Kinoshita, I.; Dosaka-Akita, H.; Sakakibara-Konishi, J. DLL 3 regulates the migration and invasion of small cell lung cancer by modulating Snail. Cancer Sci. 2019, 110, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Furuta, M.; Sakakibara, J.K.; Shoji, T.; Takashima, Y.; Kikuchi, H.; Kikuchi, E.; Kikuchi, J.; Kinoshita, I.; Akita, H.D.; Nishimura, M. Abstract 3158: DLL3 regulates migration and invasion of small cell lung cancer. Cancer Res. 2018, 78, 3158. [Google Scholar] [CrossRef]
- Vitorino, P.; Chuang, C.-H.; Iannello, A.; Zhao, X.; Anderson, W.; Ferrando, R.; Zhang, Z.; Madhavan, S.; Karsunky, H.; Saunders, L. Rova-T enhances the anti-tumor activity of anti-PD1 in a murine model of small cell lung cancer with endogenous Dll3 expression. Transl. Oncol. 2021, 14, 100883. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.A.P.; Noguti, J.; Oshima, C.T.F.; Ribeiro, D.A. Effective targeting of the epidermal growth factor receptor (EGFR) for treating oral cancer: A promising approach. Anticancer. Res. 2014, 34, 1547–1552. [Google Scholar]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Yaish, P.; Gazit, A.; Gilon, C.; Levitzki, A. Blocking of EGF-dependent cell proliferation by EGF receptor kinase inhibitors. Science 1988, 242, 933–935. [Google Scholar] [CrossRef]
- Lu, Z.; Jiang, G.; Blume-Jensen, P.; Hunter, T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol. Cell Biol. 2001, 21, 4016–4031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spano, J.P.; Lagorce, C.; Atlan, D.; Milano, G.; Domont, J.; Benamouzig, R.; Attar, A.; Benichou, J.; Martin, A.; Morere, J.F. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. 2005, 16, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.; Sainsbury, C.; Needham, G.; Farndon, J.; Malcolm, A.; Harris, A. Epidermal-growth-factor receptor status as predictor of early recurrence of and death from breast cancer. Lancet 1987, 329, 1398–1402. [Google Scholar] [CrossRef]
- Ekstrand, A.J.; James, C.D.; Cavenee, W.K.; Seliger, B.; Pettersson, R.F.; Collins, V.P. Genes for epidermal growth factor receptor, transforming growth factor α, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991, 51, 2164–2172. [Google Scholar] [PubMed]
- Hendler, F.J.; Ozanne, B.W. Human squamous cell lung cancers express increased epidermal growth factor receptors. J. Clin. Investig. 1984, 74, 647–651. [Google Scholar] [CrossRef]
- Ishitoya, J.; Toriyama, M.; Oguchi, N.; Kitamura, K.; Ohshima, M.; Asano, K.; Yamamoto, T. Gene amplification and overexpression of EGF receptor in squamous cell carcinomas of the head and neck. Brit. J. Cancer 1989, 59, 559–562. [Google Scholar] [CrossRef] [Green Version]
- You, B.; Chen, E. Anti-EGFR Monoclonal antibodies for treatment of colorectal cancers: Development of cetuximab and panitumumab. J. Clin. Pharmacol. 2012, 52, 128–155. [Google Scholar] [CrossRef]
- Huang, L.; Yang, Y.; Yang, F.; Liu, S.; Zhu, Z.; Lei, Z.; Guo, J. Functions of EpCAM in physiological processes and diseases. Int. J. Mol. Med. 2018, 42, 1771–1785. [Google Scholar] [CrossRef] [Green Version]
- Trzpis, M.; McLaughlin, P.M.J.; de Leij, L.M.F.H.; Harmsen, M.C. Epithelial cell adhesion molecule: More than a carcinoma marker and adhesion molecule. Am. J. Pathol. 2007, 171, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Münz, M.; Kieu, C.; Mack, B.; Schmitt, B.; Zeidler, R.; Gires, O. The carcinoma-associated antigen EpCAM upregulates c-myc and induces cell proliferation. Oncogene 2004, 23, 5748–5758. [Google Scholar] [CrossRef]
- Went, P.; Vasei, M.; Bubendorf, L.; Terracciano, L.; Tornillo, L.; Riede, U.; Kononen, J.; Simon, R.; Sauter, G.; Baeuerle, P.A. Frequent high-level expression of the immunotherapeutic target Ep-CAM in colon, stomach, prostate and lung cancers. Brit. J. Cancer 2006, 94, 128–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-H.; Herlyn, D.; Wong, K.-K.; Park, D.-C.; Schorge, J.O.; Lu, K.H.; Skates, S.J.; Cramer, D.W.; Berkowitz, R.S.; Mok, S.C. Identification of Epithelial Cell Adhesion Molecule Autoantibody in Patients with Ovarian Cancer. Clin. Cancer Res. 2003, 9, 4782–4791. [Google Scholar] [PubMed]
- Osta, W.A.; Chen, Y.; Mikhitarian, K.; Mitas, M.; Salem, M.; Hannun, Y.A.; Cole, D.J.; Gillanders, W.E. EpCAM Is Overexpressed in Breast Cancer and Is a Potential Target for Breast Cancer Gene Therapy. Cancer Res. 2004, 64, 5818–5824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herlyn, M.; Steplewski, Z.; Herlyn, D.; Koprowski, H. Colorectal carcinoma-specific antigen: Detection by means of monoclonal antibodies. Proc. Natl. Acad. Sci. USA 1979, 76, 1438–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Scheulen, M.E.; Dittrich, C.; Obrist, P.; Marschner, N.; Dirix, L.; Rüttinger, D.; Schuler, M.; Reinhardt, C.; Awada, A. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann. Oncol. 2010, 21, 275–282. [Google Scholar] [CrossRef]
- Lopes, N.; Bergsland, C.; Bruun, J.; Bjørnslett, M.; Vieira, A.F.; Mesquita, P.; Pinto, R.; Gomes, R.; Cavadas, B.; Bennett, E. A panel of intestinal differentiation markers (CDX2, GPA33, and LI-cadherin) identifies gastric cancer patients with favourable prognosis. Gastric Cancer 2020, 23, 811–823. [Google Scholar] [CrossRef]
- Heath Joan , K.; White Sara , J.; Johnstone, C.N.; Catimel, B.; Simpson, R.J.; Moritz, R.L.; Tu, G.-F.; Ji, H.; Whitehead, R.H.; Groenen, L.C.; et al. The human A33 antigen is a transmembrane glycoprotein and a novel member of the immunoglobulin superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Fan, Q.; Cai, H.; Yang, H.; Wan, L.; Li, L.; Lu, X. CF750-A33scFv-Fc-based optical imaging of subcutaneous and orthotopic xenografts of GPA33-positive colorectal cancer in mice. BioMed Res. Int. 2015, 2015, 505183. [Google Scholar] [CrossRef]
- Sawada, N.; Taguchi, E.; Takahashi, M. In vitro and in vivo activities of KRN330, a fully human monoclonal antibody against colon cancer. J. Clin. Oncol. 2011, 29, 432. [Google Scholar] [CrossRef]
- Berlin, J.D.; Infante, J.R.; Taguchi, E.; Goff, L.W.; Jones, S.F.; Chan, E.; Bendell, J.C.; Rothenberg, M.L.; Burris, H.A. In vivo antibody binding to tumor in xenograft rodent models and colorectal cancer patients treated with anti-A33 antibody KRN330. Cancer Res. 2010, 70, 2431. [Google Scholar]
- Iqbal, N.; Iqbal, N.J.M. Human epidermal growth factor receptor 2 (HER2) in cancers: Overexpression and therapeutic implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef] [PubMed]
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 1996, 16, 5276–5287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- SiShi, L.; Buchbinder, E.; Wu, L.; Bjorge, J.D.; Fujita, D.J.; Zhu, S. EGFR and HER2 levels are frequently elevated in colon cancer cells. Discov. Rep. 2014, 1, 1. [Google Scholar]
- Lemoine, N.R.; Jain, S.; Silvestre, F.; Lopes, C.; Hughes, C.M.; McLelland, E.; Gullick, W.J.; Filipe, M.I. Amplification and overexpression of the EGF receptor and c-erbB-2 proto-oncogenes in human stomach cancer. Brit. J. Cancer 1991, 64, 79. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.J.; Grosshans, D.R.; Wong, S.G.; Slamon, D.J. Identification of differentially expressed genes associated with HER-2/neu overexpression in human breast cancer cells. Nucleic Acids Res. 1999, 27, 4008–4017. [Google Scholar] [CrossRef] [Green Version]
- Tai, W.; Qin, B.; Cheng, K. Inhibition of breast cancer cell growth and invasiveness by dual silencing of HER-2 and VEGF. Mol. Pharm. 2010, 7, 543–556. [Google Scholar] [CrossRef]
- Roh, H.; Pippin, J.; Drebin, J.A. Down-regulation of HER2/neu expression induces apoptosis in human cancer cells that overexpress HER2/neu. Cancer Res. 2000, 60, 560–565. [Google Scholar]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Haridas, D.; Ponnusamy, M.P.; Chugh, S.; Lakshmanan, I.; Seshacharyulu, P.; Batra, S.K. MUC16: Molecular analysis and its functional implications in benign and malignant conditions. FASEB J. 2014, 28, 4183–4199. [Google Scholar] [CrossRef]
- Aithal, A.; Rauth, S.; Kshirsagar, P.; Shah, A.; Lakshmanan, I.; Junker, W.M.; Jain, M.; Ponnusamy, M.P.; Batra, S.K. MUC16 as a novel target for cancer therapy. Expert Opin. Tar. 2018, 22, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Felder, M.; Kapur, A.; Gonzalez-Bosquet, J.; Horibata, S.; Heintz, J.; Albrecht, R.; Fass, L.; Kaur, J.; Hu, K.; Shojaei, H.; et al. MUC16 (CA125): Tumor biomarker to cancer therapy, a work in progress. Mol. Cancer 2014, 13, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comamala, M.; Pinard, M.; Thériault, C.; Matte, I.; Albert, A.; Boivin, M.; Beaudin, J.; Piché, A.; Rancourt, C. Downregulation of cell surface CA125/MUC16 induces epithelial-to-mesenchymal transition and restores EGFR signalling in NIH:OVCAR3 ovarian carcinoma cells. Brit. J. Cancer 2011, 104, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, L.; Luo, N.; Liu, Q.; Liu, L.; Chen, D.; Cheng, Z.; Xi, X.J.E.; Medicine, T. Inflammatory signals induce MUC16 expression in ovarian cancer cells via NF-κB activation. Exp. Ther. Med. 2021, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Taylor, P.T.; Gordon, A.; Cunningham, M.J.; Finkler, N.; Orr, J., Jr.; Rivkin, S.; Schultes, B.C.; Whiteside, T.L.; Nicodemus, C.F. Randomized, placebo-controlled study of oregovomab for consolidation of clinical remission in patients with advanced ovarian cancer. J. Clin. Oncol. 2004, 22, 3507–3516. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Wu, A.W.; Hu, Y.Q.; Tao, C.J.; Wang, J.M.; Lu, Y.Y.; Xing, R. Mucin 17 inhibits the progression of human gastric cancer by limiting inflammatory responses through a MYH9-p53-RhoA regulatory feedback loop. J. Exp. Clin. Canc. Res. 2019, 38, 283. [Google Scholar] [CrossRef]
- Lordick, F.; Orra, E.B.; Cervantes, A.; Dayyani, F.; Rocha-Lima, C.; Greil, R.; van Laarhoven, H.; Lorenzen, S.; Kischel, R.; Shitara, K. P-76 A phase 1 study of AMG 199, a half-life extended bispecific T-cell engager (HLE BiTE®) immune therapy, targeting MUC17 in patients with gastric and gastroesophageal junction cancer. Ann. Oncol. 2020, 31, S114. [Google Scholar] [CrossRef]
- Panwar, H.; Rokana, N.; Aparna, S.V.; Kaur, J.; Singh, A.; Singh, J.; Singh, K.S.; Chaudhary, V.; Puniya, A.K. Gastrointestinal stress as innate defence against microbial attack. J. Appl. Microbiol. 2021, 130, 1035–1061. [Google Scholar] [CrossRef]
- Junker, W.M. Molecular and Biological Studies of MUC17; University of Nebraska Medical Center: Omaha, NE, USA, 2008. [Google Scholar]
- MUC17. Available online: https://www.amgenoncology.com/targets/MUC17.html (accessed on 14 April 2022).
- Chang, S.S. Overview of prostate-specific membrane antigen. Rev. Urol. 2004, 6, S13. [Google Scholar]
- Wang, F.; Li, Z.; Feng, X.; Yang, D.; Lin, M. Advances in PSMA-targeted therapy for prostate cancer. Prostate Cancer Prostatic Dis. 2021, 25, 1–16. [Google Scholar]
- Oh, S.W.; Cheon, G.J. Prostate-specific membrane antigen PET imaging in prostate cancer: Opportunities and challenges. Korean J. Radiol. 2018, 19, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Bostwick, D.G.; Pacelli, A.; Blute, M.; Roche, P.; Murphy, G.P. Prostate specific membrane antigen expression in prostatic intraepithelial neoplasia and adenocarcinoma. Cancer 1998, 82, 2256–2261. [Google Scholar] [CrossRef]
- Ross, J.S.; Sheehan, C.E.; Fisher, H.A.G.; Kaufman, R.P., Jr.; Kaur, P.; Gray, K.; Webb, I.; Gray, G.S.; Mosher, R.; Kallakury, B.V.S. Correlation of Primary Tumor Prostate-Specific Membrane Antigen Expression with Disease Recurrence in Prostate Cancer. Clin. Cancer Res. 2003, 9, 6357–6362. [Google Scholar]
- Trover, J.K.; Beckett, M.L.; Wright, G.L., Jr. Detection and characterization of the prostate-specific membrane antigen (PSMA) in tissue extracts and body fluids. Int. J. Cancer 1995, 62, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Vallabhajosula, S.; Christos, P.J.; Jhanwar, Y.S.; Batra, J.S.; Lam, L.; Osborne, J.; Beltran, H.; Molina, A.M.; Goldsmith, S.J.J.C. Phase 1/2 study of fractionated dose lutetium-177–labeled anti–prostate-specific membrane antigen monoclonal antibody J591 (177Lu-J591) for metastatic castration-resistant prostate cancer. Cancer 2019, 125, 2561–2569. [Google Scholar] [CrossRef]
- Baek, J.H.; Park, D.J.; Kim, G.Y.; Cheon, J.; Kang, B.W.; Cha, H.J.; Kim, J. Clinical implications of Claudin18. 2 expression in patients with gastric cancer. Anticancer. Res. 2019, 39, 6973–6979. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Yagi, K.; Kondoh, M. Current progress in a second-generation claudin binder, anti-claudin antibody, for clinical applications. Drug Discov. Today 2016, 21, 1711–1718. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Zhang, R. Abstract B73: Development of anti-human CLDN18. 2 monoclonal antibody as cancer therapeutics. Cancer Immunol. Res. 2020, 8, B73. [Google Scholar]
- Sanada, Y.; Oue, N.; Mitani, Y.; Yoshida, K.; Nakayama, H.; Yasui, W. Down-regulation of the claudin-18 gene, identified through serial analysis of gene expression data analysis, in gastric cancer with an intestinal phenotype. J. Pathol. 2006, 208, 633–642. [Google Scholar] [CrossRef]
- Singh, P.; Toom, S.; Huang, Y.J. Anti-claudin 18.2 antibody as new targeted therapy for advanced gastric cancer. J. Hematol. Oncol. 2017, 10, 105. [Google Scholar] [CrossRef]
- Barroca-Ferreira, J.; Cruz-Vicente, P.; Santos, M.F.; Rocha, S.M.; Santos-Silva, T.; Maia, C.J.; Passarinha, L.A. Enhanced Stability of Detergent-Free Human Native STEAP1 Protein from Neoplastic Prostate Cancer Cells upon an Innovative Isolation Procedure. Int. J. Mol. Sci. 2021, 22, 10012. [Google Scholar] [CrossRef]
- Hubert Rene, S.; Vivanco, I.; Chen, E.; Rastegar, S.; Leong, K.; Mitchell Steve, C.; Madraswala, R.; Zhou, Y.; Kuo, J.; Raitano Arthur, B.; et al. STEAP: A prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 14523–14528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.-Y.; Jiang, J.-N.; Fang, X.-D.; Ji, F.-J.J. STEAP1 regulates tumorigenesis and chemoresistance during peritoneal metastasis of gastric cancer. Front. Physiol. 2018, 9, 1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.-J.; Wu, H.-T.; Li, C.-L.; Lin, Y.-K.; Fang, Z.-X.; Lin, W.-T.; Liu, J. Regulatory Roles of Six-Transmembrane Epithelial Antigen of the Prostate Family Members in the Occurrence and Development of Malignant Tumors. Front. Cell Dev. Biol. 2021, 9, 2988. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.; Barroca-Ferreira, J.; Passarinha, L.; Socorro, S.; Maia, C.J.E.P. The Usefulness of STEAP Proteins in Prostate Cancer Clinical Practice. Exon Publ. 2021, 10, 139–153. [Google Scholar]
- Activity of Anti-STEAP1 Antibody-Drug Conjugate in Patients With Metastatic Castration-Resistant Prostate Cancer. Available online: https://ascopost.com/news/november-2019/activity-of-anti-steap1-antibody-drug-conjugate-in-patients-with-mcrpc/ (accessed on 30 March 2022).
- Lehman, J.; Hoeksema, M.; Chen, H.; Shi, C.; Eisenberg, R.; Massion, P.P. Loss of somatostatin receptor 2 expression and lung cancer growth. J. Clin. Oncol. 2015, 33, 7569. [Google Scholar] [CrossRef]
- Lehman, J.M.; Hoeksema, M.D.; Staub, J.; Qian, J.; Harris, B.; Callison, J.C.; Miao, J.; Shi, C.; Eisenberg, R.; Chen, H. Somatostatin receptor 2 signaling promotes growth and tumor survival in small-cell lung cancer. Int. J. Cancer 2019, 144, 1104–1114. [Google Scholar] [CrossRef]
- Reisine, T.; Bell, G.I. Molecular biology of somatostatin receptors. Endocr. Rev. 1995, 16, 427–442. [Google Scholar]
- Si, Y.; Kim, S.; Ou, J.; Lu, Y.; Ernst, P.; Chen, K.; Whitt, J.; Carter, A.M.; Markert, J.M.; Bibb, J.A. Anti-SSTR2 antibody-drug conjugate for neuroendocrine tumor therapy. Cancer Gene Ther. 2021, 28, 799–812. [Google Scholar] [CrossRef]
- Kammerer, R.; Zimmermann, W. Coevolution of activating and inhibitory receptors within mammalian carcinoembryonic antigen families. BMC Biol. 2010, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Benchimol, S.; Fuks, A.; Jothy, S.; Beauchemin, N.; Shirota, K.; Stanners, C.P. Carcinoembryonic antigen, a human tumor marker, functions as an intercellular adhesion molecule. Cell 1989, 57, 327–334. [Google Scholar] [CrossRef]
- Oikawa, S.; Inuzuka, C.; Kuroki, M.; Matsuoka, Y.; Kosaki, G.; Nakazato, H. Cell adhesion activity of non-specific cross-reacting antigen (NCA) and carcinoembryonic antigen (CEA) expressed on CHO cell surface: Homophilic and heterophilic adhesion. Biochem. Biophys. Res. Commun. 1989, 164, 39–45. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, S.-W. The roles of carcinoembryonic antigen in liver metastasis and therapeutic approaches. Gastroenterol. Res. Pract. 2017, 2017, 7521987. [Google Scholar] [CrossRef]
- Deng, K.; Yang, L.; Hu, B.; Wu, H.; Zhu, H.; Tang, C. The Prognostic Significance of Pretreatment Serum CEA Levels in Gastric Cancer: A Meta-Analysis Including 14651 Patients. PLoS ONE 2015, 10, e0124151. [Google Scholar] [CrossRef]
- Pakdel, A.; Malekzadeh, M.; Naghibalhossaini, F. The association between preoperative serum CEA concentrations and synchronous liver metastasis in colorectal cancer patients. Cancer Biomark. 2016, 16, 245–252. [Google Scholar] [CrossRef]
- Auclin, E.; Taieb, J.; Lepage, C.; Aparicio, T.; Faroux, R.; Mini, E.; Folprecht, G.; Salazar, R.; Benetkiewicz, M.; Banzi, M.J.C.E.; et al. Carcinoembryonic antigen levels and survival in stage III colon cancer: Post hoc analysis of the MOSAIC and PETACC-8 trials. Cancer Epidemiol. Prev. Biomark. 2019, 28, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Campos-da-Paz, M.; Dórea, J.G.; Galdino, A.S.; Lacava, Z.G.M.; de Fatima, M.A.S. Carcinoembryonic antigen (CEA) and hepatic metastasis in colorectal cancer: Update on biomarker for clinical and biotechnological approaches. Recent Pat. Biotechnol. 2018, 12, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Beauchemin, N.; Arabzadeh, A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013, 32, 643–671. [Google Scholar] [CrossRef]
- Thomas, P.; Gangopadhyay, A.; Steele, G., Jr.; Andrews, C.; Nakazato, H.; Oikawa, S.; Jessup, J.M. The effect of transfection of the CEA gene on the metastatic behavior of the human colorectal cancer cell line MIP-101. Cancer Lett. 1995, 92, 59–66. [Google Scholar] [CrossRef]
- Stein, R.; Goldenberg, D.M. A humanized monoclonal antibody to carcinoembryonic antigen, labetuzumab, inhibits tumor growth and sensitizes human medullary thyroid cancer xenografts to dacarbazine chemotherapy. Mol. Cancer Ther. 2004, 3, 1559–1564. [Google Scholar]
- Segal, N.H.; Verghis, J.; Govindan, S.; Maliakal, P.; Sharkey, R.M.; Wegener, W.A.; Goldenberg, D.M.; Saltz, L.B. Abstract LB-159: A Phase I study of IMMU-130 (labetuzumab-SN38) anti-CEACAM5 antibody-drug conjugate (ADC) in patients with metastatic colorectal cancer (mCRC). Cancer Res. 2013, 73, LB-159. [Google Scholar]
- Govindan, S.V.; Cardillo, T.M.; Rossi, E.A.; McBride, W.J.; Sharkey, R.M.; Goldenberg, D.M. IMMU-130, a unique antibody-drug conjugate (ADC) of SN-38 targeting CEACAM5 antigen: Preclinical basis for clinical activity in metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2015, 33, 625. [Google Scholar] [CrossRef]
- Li, N.; Spetz, M.R.; Ho, M. The Role of Glypicans in Cancer Progression and Therapy. J. Histochem. Cytochem. 2020, 68, 841–862. [Google Scholar] [CrossRef] [PubMed]
- Ofuji, K.; Saito, K.; Yoshikawa, T.; Nakatsura, T. Critical analysis of the potential of targeting GPC3 in hepatocellular carcinoma. J. Hepatocell. Carcinoma 2014, 1, 35. [Google Scholar] [PubMed] [Green Version]
- Capurro, M.I.; Xiang, Y.-Y.; Lobe, C.; Filmus, J. Glypican-3 Promotes the Growth of Hepatocellular Carcinoma by Stimulating Canonical Wnt Signaling. Cancer Res. 2005, 65, 6245–6254. [Google Scholar] [CrossRef] [Green Version]
- Aviel-Ronen, S.; Lau, S.K.; Pintilie, M.; Lau, D.; Liu, N.; Tsao, M.S.; Jothy, S. Glypican-3 is overexpressed in lung squamous cell carcinoma, but not in adenocarcinoma. Mod. Pathol. 2008, 21, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Maeda, D.; Ota, S.; Takazawa, Y.; Aburatani, H.; Nakagawa, S.; Yano, T.; Taketani, Y.; Kodama, T.; Fukayama, M. Glypican-3 expression in clear cell adenocarcinoma of the ovary. Mod. Pathol. 2009, 22, 824–832. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, T.; Sugimoto, M.; Kinoshita, Y.; Miyazaki, Y.; Nakano, K.; Tsunoda, H.; Sugo, I.; Ohizumi, I.; Aburatani, H.; Hamakubo, T. Anti–glypican 3 antibody as a potential antitumor agent for human liver cancer. Cancer Res. 2008, 68, 9832–9838. [Google Scholar] [CrossRef] [Green Version]
- Doronin, I.I.; Vishnyakova, P.A.; Kholodenko, I.V.; Ponomarev, E.D.; Ryazantsev, D.Y.; Molotkovskaya, I.M.; Kholodenko, R.V. Ganglioside GD2 in reception and transduction of cell death signal in tumor cells. BMC Cancer 2014, 14, 295. [Google Scholar] [CrossRef] [Green Version]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Dobrenkov, K.; Cheung, N.-K.V. GD2-targeted immunotherapy and radioimmunotherapy. Semin. Oncol. 2014, 41, 589–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GD2 Ganglioside. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/GD2-Ganglioside (accessed on 15 April 2022).
- Yoshida, S.; Fukumoto, S.; Kawaguchi, H.; Sato, S.; Ueda, R.; Furukawa, K. Ganglioside GD2 in small cell lung cancer cell lines: Enhancement of cell proliferation and mediation of apoptosis. Cancer Res. 2001, 61, 4244–4252. [Google Scholar]
- Navid, F.; Santana, V.M.; Barfield, R.C. Anti-GD2 antibody therapy for GD2-expressing tumors. Curr. Cancer Drug Targets 2010, 10, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Cheresh, D.A.; Pierschbacher, M.D.; Herzig, M.A.; Mujoo, K. Disialogangliosides GD2 and GD3 are involved in the attachment of human melanoma and neuroblastoma cells to extracellular matrix proteins. J. Cell Biol. 1986, 102, 688–696. [Google Scholar] [CrossRef]
- Cheung, N.; Kushner, B.H.; Yeh, S.; Larson, S.M. 3F8 monoclonal antibody treatment of patients with stage 4 neuroblastoma: A phase II study. Int. J. Oncol. 1998, 12, 1299–1605. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef]
- Schiemann, B.; Gommerman Jennifer, L.; Vora, K.; Cachero Teresa, G.; Shulga-Morskaya, S.; Dobles, M.; Frew, E.; Scott Martin, L. An Essential Role for BAFF in the Normal Development of B Cells Through a BCMA-Independent Pathway. Science 2001, 293, 2111–2114. [Google Scholar] [CrossRef]
- Yu, G.; Boone, T.; Delaney, J.; Hawkins, N.; Kelley, M.; Ramakrishnan, M.; McCabe, S.; Qiu, W.-R.; Kornuc, M.; Xia, X.-Z.; et al. APRIL and TALL-1 and receptors BCMA and TACI: System for regulating humoral immunity. Nat. Immunol. 2000, 1, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.-T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.R.; Cenik, C.; Jiang, D.; Mizuno, K.; Li, G.C.; Zhao, H.; Thakker, K.; Diep, A.; Xu, J.Y.; Zhang, X.E.; et al. Aberrant BCMA Signaling Promotes Tumor Growth by Altering Protein Translation Machinery, a Therapeutic Target for the Treatment of Relapse/Refractory Multiple Myeloma. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sanchez, E.; Li, M.; Kitto, A.; Li, J.; Wang, C.S.; Kirk, D.T.; Yellin, O.; Nichols, C.M.; Dreyer, M.P.; Ahles, C.P.; et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br. J. Haematol. 2012, 158, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Ketchum, E.B.; Clarke, A.; Clemmons, A.B. Belantamab Mafodotin-blmf: A Novel Antibody-Drug Conjugate for Treatment of Patients With Relapsed/Refractory Multiple Myeloma. J. Adv. Pract. Oncol. 2022, 13, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti–B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Tedder, T.F.; Zhou, L.-J.; Engel, P. The CD19/CD21 signal transduction complex of B lymphocytes. Immunol. Today 1994, 15, 437–442. [Google Scholar] [CrossRef]
- Van Zelm, M.C.; Reisli, I.; van der Burg, M.; Castaño, D.; van Noesel, C.J.M.; van Tol, M.J.D.; Woellner, C.; Grimbacher, B.; Patiño, P.J.; van Dongen, J.J.M. An antibody-deficiency syndrome due to mutations in the CD19 gene. N. Engl. J. Med. 2006, 354, 1901–1912. [Google Scholar] [CrossRef] [Green Version]
- Carter, R.H.; Fearon, D.T. CD19: Lowering the threshold for antigen receptor stimulation of B lymphocytes. Science 1992, 256, 105–107. [Google Scholar] [CrossRef]
- Anderson, K.C.; Bates, M.P.; Slaughenhoupt, B.L.; Pinkus, G.S.; Schlossman, S.F.; Nadler, L.M. Expression of human B cell-associated antigens on leukemias and lymphomas: A model of human B cell differentiation. Blood 1984, 63, 1424–1433. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Kläsener, K.; Iype, J.M.; Becker, M.; Maity, P.C.; Cavallari, M.; Nielsen, P.J.; Yang, J.; Reth, M. Continuous signaling of CD 79b and CD 19 is required for the fitness of Burkitt lymphoma B cells. EMBO J. 2018, 37, e97980. [Google Scholar] [CrossRef]
- Jurczak, W.; Zinzani, P.L.; Gaidano, G.; Goy, A.; Provencio, M.; Nagy, Z.; Robak, T.; Maddocks, K.; Buske, C.; Ambarkhane, S.; et al. Phase IIa study of the CD19 antibody MOR208 in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma. Ann. Oncol. 2018, 29, 1266–1272. [Google Scholar] [CrossRef]
- Walker, J.A.; Smith, K.G.C. CD22: An inhibitory enigma. Immunology 2008, 123, 314–325. [Google Scholar] [CrossRef]
- Nitschke, L. The role of CD22 and other inhibitory co-receptors in B-cell activation. Curr. Opin. Immunol. 2005, 17, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Stevenson, M.S.; Yuan, C.M.; Richards, K.; Delbrook, C.; Kreitman, R.J.; Pastan, I.; Wayne, A.S. Characterization of CD22 expression in acute lymphoblastic leukemia. Pediatric Blood Cancer 2015, 62, 964–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasper, G.A.; Arun, I.; Venzon, D.; Kreitman, R.J.; Wayne, A.S.; Yuan, C.M.; Marti, G.E.; Stetler-Stevenson, M. Variables affecting the quantitation of CD22 in neoplastic B cells. Cytom. Part B Clin. Cytom. 2011, 80B, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, A.; Wöhner, M.; Prescher, H.; Brossmer, R.; Nitschke, L. Targeting of CD 22-positive B-cell lymphoma cells by synthetic divalent sialic acid analogues. Eur. J. Immunol. 2012, 42, 2792–2802. [Google Scholar] [CrossRef]
- Ereño-Orbea, J.; Sicard, T.; Cui, H.; Mazhab-Jafari, M.T.; Benlekbir, S.; Guarné, A.; Rubinstein, J.L.; Julien, J.-P. Molecular basis of human CD22 function and therapeutic targeting. Nat. Commun. 2017, 8, 764. [Google Scholar] [CrossRef]
- Leonard, J.P.; Coleman, M.; Ketas, J.; Ashe, M.; Fiore, J.M.; Furman, R.R.; Niesvizky, R.; Shore, T.; Chadburn, A.; Horne, H.; et al. Combination Antibody Therapy With Epratuzumab and Rituximab in Relapsed or Refractory Non-Hodgkin’s Lymphoma. J. Clin. Oncol. 2005, 23, 5044–5051. [Google Scholar] [CrossRef]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, Present, and Future of Rituximab—The World’s First Oncology Monoclonal Antibody Therapy. Front. Oncol. 2018, 8, 168. [Google Scholar] [CrossRef]
- Nagata, S.; Ise, T.; Onda, M.; Nakamura, K.; Ho, M.; Raubitschek, A.; Pastan, I.H. Cell membrane-specific epitopes on CD30: Potentially superior targets for immunotherapy. Proc. Natl. Acad. Sci. USA 2005, 102, 7946–7951. [Google Scholar] [CrossRef] [Green Version]
- Horie, R.; Watanabe, T.; Morishita, Y.; Ito, K.; Ishida, T.; Kanegae, Y.; Saito, I.; Higashihara, M.; Mori, S.; Kadin, M.E.; et al. Ligand-independent signaling by overexpressed CD30 drives NF-κB activation in Hodgkin–Reed-Sternberg cells. Oncogene 2002, 21, 2493–2503. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Sun, A.; Wang, W.; He, J.; Hou, J.; Zhou, P.; Chen, Z. TRAF1 is involved in the classical NF-κB activation and CD30-induced alternative activity in Hodgkin’s lymphoma cells. Mol. Immunol. 2009, 46, 2441–2448. [Google Scholar] [CrossRef]
- Barta, S.K.; Gong, J.Z.; Porcu, P. Brentuximab vedotin in the treatment of CD30+ PTCL. Blood 2019, 134, 2339–2345. [Google Scholar] [CrossRef] [PubMed]
- Ranuhardy, D.; Suzanna, E.; Sari, R.M.; Hadisantoso, D.W.; Andalucia, R.; Abdillah, A. CD30, CD15, CD50, and PAX5 expressions as diagnostic markers for Hodgkin lymphoma (HL) and systemic anaplastic large cell lymphoma (sALCL). Acta Med. Indones. 2018, 50, 104–109. [Google Scholar] [PubMed]
- Lawrence, C.E.; Hammond, P.W.; Zalevsky, J.; Horton, H.; Chu, S.; Karki, S.; Desjarlais, J.R.; Carmichael, D.F. XmAb™2513, an Fc Engineered Humanized Anti-CD30 Monoclonal Antibody, Has Potent In Vitro and In Vivo Activities, and Has the Potential for Treating Hematologic Malignancies. Blood 2007, 110, 2340. [Google Scholar] [CrossRef]
- Álvarez, B.; Escalona, Z.; Uenishi, H.; Toki, D.; Revilla, C.; Yuste, M.; del Moral, M.G.; Alonso, F.; Ezquerra, A.; Domínguez, J. Molecular and functional characterization of porcine Siglec-3/CD33 and analysis of its expression in blood and tissues. Dev. Comp. Immunol. 2015, 51, 238–250. [Google Scholar] [CrossRef]
- Crocker, P.R.; McMillan, S.J.; Richards, H.E. CD33-related siglecs as potential modulators of inflammatory responses. Ann. New York Acad. Sci. 2012, 1253, 102–111. [Google Scholar] [CrossRef]
- Läubli, H.; Alisson-Silva, F.; Stanczak, M.A.; Siddiqui, S.S.; Deng, L.; Verhagen, A.; Varki, N.; Varki, A. Lectin galactoside-binding soluble 3 binding protein (LGALS3BP) is a tumor-associated immunomodulatory ligand for CD33-related Siglecs. J. Biol. Chem. 2014, 289, 33481–33491. [Google Scholar] [CrossRef] [Green Version]
- De Propris, M.S.; Raponi, S.; Diverio, D.; Milani, M.L.; Meloni, G.; Falini, B.; Foà, R.; Guarini, A. High CD33 expression levels in acute myeloid leukemia cells carrying the nucleophosmin (NPM1) mutation. Haematologica 2011, 96, 1548. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Tong, J.; Yang, H. Targeting CD33 for acute myeloid leukemia therapy. BMC Cancer 2022, 22, 24. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.; Hunyadkurti, J.; Desilets, S.; Leyton, J.V.; Keating, A. Antibody therapies for acute myeloid leukemia: Unconjugated, toxin-conjugated, radio-conjugated and multivalent formats. J. Clin. Med. 2019, 8, 1261. [Google Scholar] [CrossRef] [Green Version]
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Löwenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef]
- Gao, Y.; Mehta, K. N-linked glycosylation of CD38 is required for its structure stabilization but not for membrane localization. Mol. Cell. Biochem. 2007, 295, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Liu, H.; Fang, C.; Li, C.; Xhyliu, F.; Dysert, H.; Bodo, J.; Habermehl, G.; Russell, B.E.; Li, W. Targeting of CD38 by the tumor suppressor miR-26a serves as a novel potential therapeutic agent in multiple myeloma. Cancer Res. 2020, 80, 2031–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irimia, R.M.; Gerke, M.B.; Thakar, M.; Ren, Z.; Helmenstine, E.; Imus, P.H.; Ghiaur, G.; Leone, R.; Gocke, C.B. CD38 Is a Key Regulator of Tumor Growth By Modulating the Metabolic Signature of Malignant Plasma Cells. Blood 2021, 138, 2652. [Google Scholar] [CrossRef]
- Sanchez, L.; Wang, Y.; Siegel, D.S.; Wang, M.L. Daratumumab: A first-in-class CD38 monoclonal antibody for the treatment of multiple myeloma. J. Hematol. Oncol. 2016, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Hoseini, S.S.; Cheung, N.K. Acute myeloid leukemia targets for bispecific antibodies. Blood Cancer J. 2017, 7, e522. [Google Scholar] [CrossRef]
- Angelova, E.; Audette, C.; Kovtun, Y.; Daver, N.; Wang, S.A.; Pierce, S.; Konoplev, S.N.; Khogeer, H.; Jorgensen, J.L.; Konopleva, M.; et al. CD123 expression patterns and selective targeting with a CD123-targeted antibody-drug conjugate (IMGN632) in acute lymphoblastic leukemia. Haematologica 2019, 104, 749–755. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Mughal, T.I.; Brooks, C.; Lindsay, R.; Pemmaraju, N. Targeting CD123 in hematologic malignancies: Identifying suitable patients for targeted therapy. Leuk. Lymphoma 2021, 62, 2568–2586. [Google Scholar] [CrossRef]
- Muñoz, L.; Nomdedéu, J.F.; López, O.; Carnicer, M.J.; Bellido, M.; Aventín, A.; Brunet, S.; Sierra, J. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica 2001, 86, 1261–1269. [Google Scholar]
- Aldinucci, D.; Poletto, D.; Gloghini, A.; Nanni, P.; Degan, M.; Perin, T.; Ceolin, P.; Rossi, F.M.; Gattei, V.; Carbone, A.; et al. Expression of Functional Interleukin-3 Receptors on Hodgkin and Reed-Sternberg Cells. Am. J. Pathol. 2002, 160, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Lee, E.M.; Ramshaw, H.S.; Busfield, S.J.; Peoppl, A.G.; Wilkinson, L.; Guthridge, M.A.; Thomas, D.; Barry, E.F.; Boyd, A. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor α chain, eliminates human acute myeloid leukemic stem cells. Cell. Stem. Cell. 2009, 5, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.; Jones, G.E.; Adams, S.; Harvey, L.; Audette, C.A.; Wilhelm, A.; Bai, C.; Rui, L.; Laleau, R.; Liu, F.; et al. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv. 2018, 2, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting CLL-1 for acute myeloid leukemia therapy. J. Hematol. Oncol. 2019, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Neumann, K.; Castiñeiras-Vilariño, M.; Höckendorf, U.; Hannesschläger, N.; Lemeer, S.; Kupka, D.; Meyermann, S.; Lech, M.; Anders, H.-J.; Kuster, B.; et al. Clec12a Is an Inhibitory Receptor for Uric Acid Crystals that Regulates Inflammation in Response to Cell Death. Immunity 2014, 40, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Bakker, A.B.H.; van den Oudenrijn, S.; Bakker, A.Q.; Feller, N.; van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.C.; Visser, T.J.; Bijl, N.; Geuijen, C.A.W.; et al. C-Type Lectin-Like Molecule-1: A Novel Myeloid Cell Surface Marker Associated with Acute Myeloid Leukemia. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [CrossRef] [Green Version]
- Haubner, S.; Perna, F.; Köhnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2019, 33, 64–74. [Google Scholar] [CrossRef]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Kim, M.; Soderquist, C.; Bagg, A.; Singh, R.; Richardson, C.; Young, R. Targeting CLEC12A with chimeric antigen receptor T cells can overcome the chemotherapy refractoriness of leukemia stem cells. Biol. Blood Marrow Transplant. 2017, 23, S247–S248. [Google Scholar] [CrossRef]
- Xiaoxian, Z.; Shweta, S.; Cecile, P.; Jingsong, Z.; Eric, D.H.; Arie, A.; Wouter, K. Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica 2010, 95, 71–78. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [Green Version]
- Rasko, J.E.; Metcalf, D.; Rossner, M.T.; Begley, C.G.; Nicola, N.A. The flt3/flk-2 ligand: Receptor distribution and action on murine haemopoietic cell survival and proliferation. Leukemia 1995, 9, 2058–2066. [Google Scholar] [PubMed]
- Rusten, L.S.; Lyman, S.D.; Veiby, O.P.; Jacobsen, S.E. The FLT3 ligand is a direct and potent stimulator of the growth of primitive and committed human CD34+ bone marrow progenitor cells in vitro. Blood 1996, 87, 1317–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poubel, C.P.; Mansur, M.B.; Boroni, M.; Emerenciano, M. FLT3 overexpression in acute leukaemias: New insights into the search for molecular mechanisms. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2019, 1872, 80–88. [Google Scholar] [CrossRef]
- Radich, J.P.; Kopecky, K.J.; Willman, C.L.; Weick, J.; Head, D.; Appelbaum, F.; Collins, S.J. N-ras mutations in adult de novo acute myelogenous leukemia: Prevalence and clinical significance. Blood 1990, 76, 801–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Sanford, D.; Blum, W.G.; Ravandi, F.; Klisovic, R.B.; Borthakur, G.; Walker, A.R.; Garcia-Manero, G.; Marcucci, G.; Wierda, W.G.; Whitman, S.P.; et al. Efficacy and safety of an anti-FLT3 antibody (LY3012218) in patients with relapsed acute myeloid leukemia. J. Clin. Oncol. 2015, 33, 7059. [Google Scholar] [CrossRef]
- Piloto, O.; Griesemer, M.; Nguyen, B.; Li, L.; Li, Y.; Witte, L.; Hicklin, D.J.; Small, D. The Anti-FLT3 Monoclonal Antibody EB10 Is Cytotoxic to FLT3 Inhibitor Resistant Cells In Vivo. Blood 2005, 106, 1511. [Google Scholar] [CrossRef]
- Furusawa, Y.; Kaneko, M.K.; Kato, Y. Establishment of C20Mab-11, a novel anti-CD20 monoclonal antibody, for the detection of B cells. Oncol. Lett. 2020, 20, 1961–1967. [Google Scholar] [CrossRef]
- Stashenko, P.; Nadler, L.M.; Hardy, R.; Schlossman, S.F. Characterization of a human B lymphocyte-specific antigen. J. Immunol. 1980, 125, 1678–1685. [Google Scholar]
- Prevodnik, V.K.; Lavrenčak, J.; Horvat, M.; Novakovič, B.J. The predictive significance of CD20 expression in B-cell lymphomas. Diagn. Pathol. 2011, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Weaver, L.S.; Liewehr, D.; Venzon, D.; Stetler-Stevenson, M.; Yuan, C.M. Increased expression of CD20 and CD45 and diminished expression of CD19 are features of follicular lymphoma. Pathol. Lab. Med. Int. 2013, 5, 21. [Google Scholar]
- Leonard, J.P.; Trneny, M.; Izutsu, K.; Fowler, N.H.; Hong, X.; Zhu, J.; Zhang, H.; Offner, F.; Scheliga, A.; Nowakowski, G.S.; et al. AUGMENT: A Phase III Study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2019, 37, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Bräuner-Osborne, H.; Jensen, A.A.; Sheppard, P.O.; Brodin, B.; Krogsgaard-Larsen, P.; O’Hara, P. Cloning and characterization of a human orphan family C G-protein coupled receptor GPRC5D1GenBank accession Nos. for GPRC5C: AF207989, for Gprc5d: AF218809 and for GPRC5D: AF209923.1. Biochim. Et Biophys. Acta (BBA)-Gene Struct. Expr. 2001, 1518, 237–248. [Google Scholar] [CrossRef]
- Pillarisetti, K.; Edavettal, S.; Mendonça, M.; Li, Y.; Tornetta, M.; Babich, A.; Majewski, N.; Husovsky, M.; Reeves, D.; Walsh, E.; et al. A T-cell–redirecting bispecific G-protein–coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood 2020, 135, 1232–1243. [Google Scholar] [CrossRef]
- Smith Eric, L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long Thomas, J.; Ng Khong, Y.; Ghoddusi, M.; Purdon Terence, J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, X.; Yan, H.; Zeng, J.; Ma, S.; Niu, Y.; Zhou, G.; Jiang, Y.; Chen, Y. Comparative Transcriptome Analysis of Fetal Skin Reveals Key Genes Related to Hair Follicle Morphogenesis in Cashmere Goats. PLoS ONE 2016, 11, e0151118. [Google Scholar] [CrossRef] [Green Version]
- Suurs, F.V.; Lub-de Hooge, M.N.; de Vries, E.G.E.; de Groot, D.J.A. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 2019, 201, 103–119. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef]
- Loffler, A.; Kufer, P.; Lutterbuse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmuller, G.; Dorken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [CrossRef]
- Frankel, S.R.; Baeuerle, P.A. Targeting T cells to tumor cells using bispecific antibodies. Curr. Opin. Chem. Biol. 2013, 17, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and production of bispecific antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holliger, P.; Prospero, T.; Winter, G. “Diabodies”: Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Wen, W.; Qin, W. Bispecific Antibodies as a Development Platform for New Concepts and Treatment Strategies. Int. J. Mol. Sci. 2016, 18, 48. [Google Scholar] [CrossRef] [Green Version]
- Rader, C. DARTs take aim at BiTEs. Blood 2011, 117, 4403–4404. [Google Scholar] [CrossRef]
- Reusch, U.; Burkhardt, C.; Fucek, I.; Le Gall, F.; Le Gall, M.; Hoffmann, K.; Knackmuss, S.H.; Kiprijanov, S.; Little, M.; Zhukovsky, E.A. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. mAbs 2014, 6, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Reusch, U.; Duell, J.; Ellwanger, K.; Herbrecht, C.; Knackmuss, S.H.; Fucek, I.; Eser, M.; McAleese, F.; Molkenthin, V.; Gall, F.L.; et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. mAbs 2015, 7, 584–604. [Google Scholar] [CrossRef] [Green Version]
- Choe-Juliak, C.; Alexis, K.M.; Schwarz, S.; Garcia, L.; Sawas, A. A phase II open-label multicenter study to assess the efficacy and safety of AFM13 in patients with relapsed or refractory CD30-positive peripheral T-cell lymphoma or transformed mycosis fungoides: The REDIRECT study design and rationale. J. Clin. Oncol. 2020, 38, TPS3148. [Google Scholar] [CrossRef]
- Gong, S.; Wu, C. Generation of Fabs-in-tandem immunoglobulin molecules for dual-specific targeting. Methods 2019, 154, 87–92. [Google Scholar] [CrossRef]
- Gong, S.; Ren, F.; Wu, D.; Wu, X.; Wu, C. Fabs-in-tandem immunoglobulin is a novel and versatile bispecific design for engaging multiple therapeutic targets. mAbs 2017, 9, 1118–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Ph1/2 Study of EMB-06 in Participants with Relapsed or Refractory Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT04735575 (accessed on 19 April 2022).
- Santich, B.H.; Park, J.A.; Tran, H.; Guo, H.F.; Huse, M.; Cheung, N.V. Interdomain spacing and spatial configuration drive the potency of IgG-[L]-scFv T cell bispecific antibodies. Sci. Transl. Med. 2020, 12, eaax1315. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Guo, H.-F.; Xu, H.; Cheung, N.-K.V. Development of a Tetravalent Anti-GPA33/Anti-CD3 Bispecific Antibody for Colorectal Cancers. Mol. Cancer Ther. 2018, 17, 2164–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoseini, S.S.; Guo, H.; Wu, Z.; Hatano, M.N.; Cheung, N.-K.V. A potent tetravalent T-cell–engaging bispecific antibody against CD33 in acute myeloid leukemia. Blood Adv. 2018, 2, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, M.; Guo, H.; Chen, Y.; Huse, M.; Cheung, N.K. Retargeting T cells to GD2 pentasaccharide on human tumors using Bispecific humanized antibody. Cancer Immunol. Res. 2015, 3, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zekri, L.; Vogt, F.; Osburg, L.; Müller, S.; Kauer, J.; Manz, T.; Pflügler, M.; Maurer, A.; Heitmann, J.S.; Hagelstein, I. An IgG-based bispecific antibody for improved dual targeting in PSMA-positive cancer. EMBO Mol. Med. 2021, 13, e11902. [Google Scholar] [CrossRef]
- Heitmann, J.S.; Pfluegler, M.; Jung, G.; Salih, H.R. Bispecific Antibodies in Prostate Cancer Therapy: Current Status and Perspectives. Cancers 2021, 13, 549. [Google Scholar] [CrossRef]
- Heitmann, J.S.; Walz, J.S.; Pflügler, M.; Kauer, J.; Schlenk, R.F.; Jung, G.; Salih, H.R. Protocol of a prospective, multicentre phase I study to evaluate the safety, tolerability and preliminary efficacy of the bispecific PSMAxCD3 antibody CC-1 in patients with castration-resistant prostate carcinoma. BMJ Open 2020, 10, e039639. [Google Scholar] [CrossRef]
- Uckun, F.M.; Lin, T.L.; Mims, A.S.; Patel, P.; Lee, C.; Shahidzadeh, A.; Shami, P.J.; Cull, E.; Cogle, C.R.; Watts, J. A Clinical Phase 1B Study of the CD3xCD123 Bispecific Antibody APVO436 in Patients with Relapsed/Refractory Acute Myeloid Leukemia or Myelodysplastic Syndrome. Cancers 2021, 13, 4113. [Google Scholar] [CrossRef]
- Watts, J.M.; Lin, T.; Wang, E.S.; Mims, A.S.; Cull, E.H.; Patel, P.A.; Shami, P.J.; Walter, R.B.; Cogle, C.R.; Chenault, R.A.; et al. Preliminary Results from a Phase 1 Study of APVO436, a Novel Anti-CD123 x Anti-CD3 Bispecific Molecule, in Relapsed/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome. Blood 2020, 136, 11–12. [Google Scholar] [CrossRef]
- Han, L.; Chen, J.; Ding, K.; Zong, H.; Xie, Y.; Jiang, H.; Zhang, B.; Lu, H.; Yin, W.; Gilly, J.; et al. Efficient generation of bispecific IgG antibodies by split intein mediated protein trans-splicing system. Sci. Rep. 2017, 7, 8360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duivelshof, B.L.; Beck, A.; Guillarme, D.; D’Atri, V. Bispecific antibody characterization by a combination of intact and site-specific/chain-specific LC/MS techniques. Talanta 2022, 236, 122836. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Meesters, J.I.; de Goeij, B.E.; van den Bremer, E.T.; Neijssen, J.; van Kampen, M.D.; Strumane, K.; Verploegen, S.; Kundu, A.; Gramer, M.J.; et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 5145–5150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suresh, M.R.; Cuello, A.C.; Milstein, C. Bispecific monoclonal antibodies from hybrid hybridomas. Methods Enzymol. 1986, 121, 210–228. [Google Scholar] [CrossRef]
- Shuptrine, C.W.; Surana, R.; Weiner, L.M. Monoclonal antibodies for the treatment of cancer. Semin. Cancer Biol. 2012, 22, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Chelius, D.; Ruf, P.; Gruber, P.; Ploscher, M.; Liedtke, R.; Gansberger, E.; Hess, J.; Wasiliu, M.; Lindhofer, H. Structural and functional characterization of the trifunctional antibody catumaxomab. mAbs 2010, 2, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Lindhofer, H.; Mocikat, R.; Steipe, B.; Thierfelder, S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J. Immunol. 1995, 155, 219–225. [Google Scholar]
- Viardot, A.; Bargou, R. Bispecific antibodies in haematological malignancies. Cancer Treat. Rev. 2018, 65, 87–95. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Carter, P. Bispecific human IgG by design. J. Immunol. Methods 2001, 248, 7–15. [Google Scholar] [CrossRef]
- Xu, Y.; Lee, J.; Tran, C.; Heibeck, T.H.; Wang, W.D.; Yang, J.; Stafford, R.L.; Steiner, A.R.; Sato, A.K.; Hallam, T.J.; et al. Production of bispecific antibodies in “knobs-into-holes” using a cell-free expression system. mAbs 2015, 7, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.; Sustmann, C.; Thomas, M.; Stubenrauch, K.; Croasdale, R.; Schanzer, J.; Brinkmann, U.; Kettenberger, H.; Regula, J.T.; Schaefer, W. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. mAbs 2012, 4, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraiwa, H.; Narita, A.; Kamata-Sakurai, M.; Ishiguro, T.; Sano, Y.; Hironiwa, N.; Tsushima, T.; Segawa, H.; Tsunenari, T.; Ikeda, Y.; et al. Engineering a bispecific antibody with a common light chain: Identification and optimization of an anti-CD3 epsilon and anti-GPC3 bispecific antibody, ERY974. Methods 2019, 154, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Sano, Y.; Komatsu, S.-I.; Kamata-Sakurai, M.; Kaneko, A.; Kinoshita, Y.; Shiraiwa, H.; Azuma, Y.; Tsunenari, T.; Kayukawa, Y. An anti–glypican 3/CD3 bispecific T cell–redirecting antibody for treatment of solid tumors. Sci. Transl. Med. 2017, 9, eaal4291. [Google Scholar] [CrossRef] [Green Version]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef] [Green Version]
- Van der Neut Kolfschoten, M.; Schuurman, J.; Losen, M.; Bleeker Wim, K.; Martínez-Martínez, P.; Vermeulen, E.; den Bleker Tamara, H.; Wiegman, L.; Vink, T.; Aarden Lucien, A.; et al. Anti-Inflammatory Activity of Human IgG4 Antibodies by Dynamic Fab Arm Exchange. Science 2007, 317, 1554–1557. [Google Scholar] [CrossRef] [Green Version]
- Van den Bremer, E.T.J.; Labrijn, A.F.; van den Boogaard, R.; Priem, P.; Scheffler, K.; Melis, J.P.M.; Schuurman, J.; Parren, P.W.H.I.; de Jong, R.N. Cysteine-SILAC Mass Spectrometry Enabling the Identification and Quantitation of Scrambled Interchain Disulfide Bonds: Preservation of Native Heavy-Light Chain Pairing in Bispecific IgGs Generated by Controlled Fab-arm Exchange. Anal. Chem. 2017, 89, 10873–10882. [Google Scholar] [CrossRef]
- Engelberts, P.J.; Hiemstra, I.H.; de Jong, B.; Schuurhuis, D.H.; Meesters, J.; Beltran Hernandez, I.; Oostindie, S.C.; Neijssen, J.; van den Brink, E.N.; Horbach, G.J.; et al. DuoBody-CD3xCD20 induces potent T-cell-mediated killing of malignant B cells in preclinical models and provides opportunities for subcutaneous dosing. EbioMedicine 2020, 52, 102625. [Google Scholar] [CrossRef]
- Syed, Y.Y. Amivantamab: First Approval. Drugs 2021, 81, 1349–1353. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Kim, T.M.; Alonso, A.; Prince, M.; Taszner, M.; Cho, S.-G.; Stevens, D.A.; Poon, M.; Lim, F.; Le Gouill, S.; Carpio, C.; et al. A Phase 2 Study of Odronextamab (REGN1979), a CD20 x CD3 Bispecific Antibody, in Patients with Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma. Blood 2020, 136, 28–29. [Google Scholar] [CrossRef]
- Surowka, M.; Schaefer, W.; Klein, C. Ten years in the making: Application of CrossMab technology for the development of therapeutic bispecific antibodies and antibody fusion proteins. mAbs 2021, 13, 1967714. [Google Scholar] [CrossRef] [PubMed]
- Hertzberg, M.; Ku, M.; Catalani, O.; Althaus, B.; Simko, S.; Gregory, G.P. A phase III trial evaluating glofitamab in combination with gemcitabine plus oxaliplatin versus rituximab in combination with gemcitabine and oxaliplatin in patients with relapsed/refractory (R/R) diffuse large B-cell lymphoma (DLBCL). J. Clin. Oncol. 2021, 39, TPS7575. [Google Scholar] [CrossRef]
- Li, Y. IgG-like bispecific antibody platforms with built-in purification-facilitating elements. Protein Expr. Purif. 2021, 188, 105955. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.-H.; Kim, J.-E.; Kim, Y.-S. Immunoglobulin Fc Heterodimer Platform Technology: From Design to Applications in Therapeutic Antibodies and Proteins. Front. Immunol. 2016, 7, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yi, J.; Zhou, P. Development of bispecific antibodies in China: Overview and prospects. Antib. Ther. 2020, 3, 126–145. [Google Scholar] [CrossRef]
- Safety and Efficacy of XmAb18087 ± Pembrolizumab in Advanced Merkel Cell Carcinoma or Extensive-Stage Small Cell Lung Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04590781?term=NCT04590781&draw=2&rank=1 (accessed on 15 April 2022).
- You, G.; Won, J.; Lee, Y.; Moon, D.; Park, Y.; Lee, S.H.; Lee, S.-W. Bispecific Antibodies: A Smart Arsenal for Cancer Immunotherapies. Vaccines 2021, 9, 724. [Google Scholar] [CrossRef]
- Voynov, V.; Adam, P.J.; Nixon, A.E.; Scheer, J.M. Discovery Strategies to Maximize the Clinical Potential of T-Cell Engaging Antibodies for the Treatment of Solid Tumors. Antibodies 2020, 9, 65. [Google Scholar] [CrossRef]
- Moore, P.A.; Shah, K.; Yang, Y.; Alderson, R.; Roberts, P.; Long, V.; Liu, D.; Li, J.C.; Burke, S.; Ciccarone, V. Development of MGD007, a gpA33 x CD3-bispecific DART protein for T-cell immunotherapy of metastatic colorectal cancer. Mol. Cancer Ther. 2018, 17, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Melero, I.; Ros, W.; Argiles, G.; Marabelle, A.; Rodriguez-Ruiz, M.E.; Albanell, J.; Calvo, E.; Moreno, V.; Cleary, J.M. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, 35, 3002. [Google Scholar] [CrossRef]
- Bailis, J.M.; Lutterbuese, P.; Thomas, O.; Locher, K.; Harrold, J.; Boyle, M.; Wahl, J.; Li, S.; Sternjak, A.; Henn, A. Preclinical evaluation of BiTE® immune therapy targeting MUC17 or CLDN18. 2 for gastric cancer. Cancer Res. 2020, 80, 3364. [Google Scholar]
- Lordick, F.; Chao, J.; Buxò, E.; van Laarhoven, H.; Lima, C.; Lorenzen, S.; Dayyani, F.; Heinemann, V.; Greil, R.; Stienen, S. 1496TiP Phase I study evaluating safety and tolerability of AMG 910, a half-life extended bispecific T cell engager targeting claudin-18.2 (CLDN18. 2) in gastric and gastroesophageal junction (G/GEJ) adenocarcinoma. Ann. Oncol. 2020, 31, S928–S929. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Champiat, S.; Johnson, M.L.; Govindan, R.; Izumi, H.; Lai, W.V.V.; Borghaei, H.; Boyer, M.J.; Boosman, R.J.; Hummel, H.-D. Updated results from a phase 1 study of AMG 757, a half-life extended bispecific T-cell engager (BiTE) immuno-oncology therapy against delta-like ligand 3 (DLL3), in small cell lung cancer (SCLC). J. Clin. Oncol. 2021, 39, 8510. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727. [Google Scholar]
- Lakhani, N.; Johnson, M.; Groisberg, R.; Han, H.; Casey, K.; Li, S.; Skokos, D.; Seebach, F.; Lowy, I.; Fury, M. 535 A phase I/II study of REGN7075 (EGFRxCD28 costimulatory bispecific antibody) in combination with cemiplimab (anti–PD-1) in patients with advanced solid tumors. J. Immunother. Cancer 2021, 9, A565. [Google Scholar] [CrossRef]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef]
- Xu, J.; Zhao, C.; Jia, Y.; Wang, S.; Ma, X.; Wang, T.; Huang, S.; Pei, M.; Wang, X.; Zhou, P. 539P Interim results of a phase I study of M701, a recombinant anti-EpCAM and anti-CD3 bispecific antibody in EpCAM-positive cancer patients with malignant ascites. Annals of Oncology 2021, 32, S603. [Google Scholar] [CrossRef]
- Yankelevich, M.; Kondadasula, S.V.; Thakur, A.; Buck, S.; Cheung, N.-K.V.; Lum, L.G. Anti-CD3 × anti-GD2 bispecific antibody redirects T-cell cytolytic activity to neuroblastoma targets. Pediatric Blood Cancer 2012, 59, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Elshiaty, M.; Schindler, H.; Christopoulos, P. Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer. Int. J. Mol. Sci. 2021, 22, 5632. [Google Scholar] [CrossRef]
- Hurwitz, H.; Crocenzi, T.; Lohr, J.; Bonvini, E.; Johnson, S.; Moore, P.; Wigginton, J. A Phase I, first-in-human, open label, dose escalation study of MGD007, a humanized gpA33× CD3 dual-affinity re-targeting (DART®) protein in patients with relapsed/refractory metastatic colorectal carcinoma. J. Immunother. Cancer 2014, 2, 86. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Dees, S.; Grewal, I. Overcoming the challenges associated with CD3+ T-cell redirection in cancer. Br. J. Cancer 2021, 124, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Ogita, Y.; Weiss, D.; Sugaya, N.; Nakamura, M.; Ito, H.; Ishiguro, T.; Shimada, S.; Ueda, M.; Matsushima, J.; Gianella-Borradori, A. A phase 1 dose escalation (DE) and cohort expansion (CE) study of ERY974, an anti-Glypican 3 (GPC3)/CD3 bispecific antibody, in patients with advanced solid tumors. J. Clin. Oncol. 2018, 36, TPS2599. [Google Scholar] [CrossRef]
- A Phase I Study of ERY974 in Patients With Hepatocellular Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT05022927?term=NCT05022927&draw=2&rank=1 (accessed on 15 April 2022).
- Yu, S.; Zhang, J.; Yan, Y.; Yao, X.; Fang, L.; Xiong, H.; Liu, Y.; Chu, Q.; Zhou, P.; Wu, K.; et al. A novel asymmetrical anti-HER2/CD3 bispecific antibody exhibits potent cytotoxicity for HER2-positive tumor cells. J. Exp. Clin. Cancer Res. 2019, 38, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Study of REGN4018 Administered Alone or in Combination with Cemiplimab in Adult Patients with Recurrent Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03564340?term=NCT03564340&draw=2&rank=1 (accessed on 18 February 2022).
- Einsele, H.; Borghaei, H.; Orlowski, R.Z.; Subklewe, M.; Roboz, G.J.; Zugmaier, G.; Kufer, P.; Iskander, K.; Kantarjian, H.M.J.C. The BiTE (bispecific T-cell engager) platform: Development and future potential of a targeted immuno-oncology therapy across tumor types. Cancer 2020, 126, 3192–3201. [Google Scholar] [CrossRef]
- Subudhi, S.K.; Siddiqui, B.A.; Maly, J.J.; Nandagopal, L.; Lam, E.T.; Whang, Y.E.; Minocha, M.; Gupta, V.; Penny, X.; Cooner, F. Safety and efficacy of AMG 160, a half-life extended BiTE immune therapy targeting prostate-specific membrane antigen (PSMA), and other therapies for metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2021, 39, TPS5088. [Google Scholar] [CrossRef]
- Tran, B.; Horvath, L.; Dorff, T.B.; Greil, R.; Machiels, J.-P.H.; Roncolato, F.; Autio, K.A.; Rettig, M.; Fizazi, K.; Lolkema, M.P. Phase I study of AMG 160, a half-life extended bispecific T-cell engager (HLE BiTE) immune therapy targeting prostate-specific membrane antigen (PSMA), in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, TPS261. [Google Scholar] [CrossRef]
- Buelow, B.; Dalvi, P.; Dang, K.; Patel, A.; Johal, K.; Pham, D.; Panchal, S.; Liu, Y.; Fong, L.; Sartor, A.O. TNB585. 001: A multicenter, phase 1, open-label, dose-escalation and expansion study of tnb-585, a bispecific T-cell engager targeting PSMA in subjects with metastatic castrate resistant prostate cancer. J. Clin. Oncol. 2021, 39, TPS5092. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O. Abstract DDT02-03: AMG 509: A novel, humanized, half-Life extended, bispecific STEAP1 × CD3 T cell recruiting XmAb® 2+1 antibody. Cancer Res. 2020, 80, DDT02–DDT03. [Google Scholar] [CrossRef]
- Lee, S.-H.; Chu, S.Y.; Rashid, R.; Phung, S.; Leung, I.W.; Muchhal, U.S.; Moore, G.L.; Bernett, M.J.; Schubbert, S.; Ardila, C.J.C.R. Anti-SSTR2× anti-CD3 bispecific antibody induces potent killing of human tumor cells in vitro and in mice, and stimulates target-dependent T cell activation in monkeys: A potential immunotherapy for neuroendocrine tumors. Cancer Res. 2017, 77, 3633. [Google Scholar]
- Pillarisetti, K.; Powers, G.; Luistro, L.; Babich, A.; Baldwin, E.; Li, Y.; Zhang, X.; Mendonça, M.; Majewski, N.; Nanjunda, R.; et al. Teclistamab is an active T cell–redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020, 4, 4538–4549. [Google Scholar] [CrossRef]
- Krishnan, A.Y.; Garfall, A.L.; Mateos, M.-V.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.; Oriol, A.; Rosiñol, L.; Chari, A.; Bhutani, M. Updated phase 1 results of teclistamab, a B-cell maturation antigen (BCMA)× CD3 bispecific antibody, in relapsed/refractory multiple myeloma (MM). Blood 2021, 136, 27. [Google Scholar] [CrossRef]
- A Study of Teclistamab in Combination With Daratumumab Subcutaneously (SC) (Tec-Dara) Versus Daratumumab SC, Pomalidomide, and Dexamethasone (DPd) or Daratumumab SC, Bortezomib, and Dexamethasone (DVd) in Participants With Relapsed or Refractory Multiple Myeloma (MajesTEC-3). Available online: https://clinicaltrials.gov/ct2/show/NCT05083169?term=NCT05083169&draw=2&rank=1 (accessed on 18 February 2022).
- Caraccio, C.; Krishna, S.; Phillips, D.J.; Schürch, C.M. Bispecific Antibodies for Multiple Myeloma: A Review of Targets, Drugs, Clinical Trials, and Future Directions. Front. Immunol. 2020, 11, 501. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.; Warcel, D.; Fielding, A.K. Blinatumomab, a bispecific B-cell and T-cell engaging antibody, in the treatment of B-cell malignancies. Hum. Vaccines Immunother. 2019, 15, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Viardot, A.; Hess, G.; Bargou, R.C.; Morley, N.; Gritti, G.; Iskander, K.; Cohan, D.; Zhang, A.; Franklin, J.; Coyle, L. Durability of complete response after blinatumomab therapy for refractory/relapsed aggressive B-cell non-Hodgkin lymphoma. J. Clin. Oncol. 2019, 37, e19041. [Google Scholar] [CrossRef]
- Study to Evaluate Safety and Efficacy of Blinatumomab in Subjects with Relapsed/Refractory (R/R) Aggressive B-Cell NHL. Available online: https://clinicaltrials.gov/ct2/show/NCT02910063 (accessed on 15 April 2022).
- Katz, D.A.; Morris, J.D.; Chu, M.P.; David, K.A.; Thieblemont, C.; Morley, N.J.; Khan, S.S.; Viardot, A.; Martín García-Sancho, A.; Rodríguez-García, G.; et al. Open-label, phase 2 study of blinatumomab after frontline R-chemotherapy in adults with newly diagnosed, high-risk DLBCL. Leuk. Lymphoma 2022, 1–11. [Google Scholar] [CrossRef]
- A Study of TNB-486 in Subjects With Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma. Available online: https://clinicaltrials.gov/ct2/show/NCT04594642?term=NCT04594642&draw=2&rank=1 (accessed on 18 February 2022).
- Schuster, S.J. Bispecific antibodies for the treatment of lymphomas: Promises and challenges. Hematol. Oncol. 2021, 39, 113–116. [Google Scholar] [CrossRef]
- Budde, L.E.; Sehn, L.H.; Matasar, M.J.; Schuster, S.J.; Assouline, S.; Giri, P.; Kuruvilla, J.; Canales, M.; Dietrich, S.; Fay, K.; et al. Mosunetuzumab Monotherapy Is an Effective and Well-Tolerated Treatment Option for Patients with Relapsed/Refractory (R/R) Follicular Lymphoma (FL) Who Have Received ≥2 Prior Lines of Therapy: Pivotal Results from a Phase I/II Study. Blood 2021, 138, 127. [Google Scholar] [CrossRef]
- Salvaris, R.; Ong, J.; Gregory, G.P. Bispecific Antibodies: A Review of Development, Clinical Efficacy and Toxicity in B-Cell Lymphomas. J. Pers. Med. 2021, 11, 355. [Google Scholar] [CrossRef]
- Patel, K.; Michot, J.-M.; Chanan-Khan, A.; Ghesquieres, H.; Bouabdallah, K.; Byrd, J.C.; Cartron, G.; Portell, C.A.; Solh, M.; Tilly, H.; et al. Safety and Anti-Tumor Activity of Plamotamab (XmAb13676), an Anti-CD20 x Anti-CD3 Bispecific Antibody, in Subjects with Relapsed/Refractory Non-Hodgkin’s Lymphoma. Blood 2021, 138, 2494. [Google Scholar] [CrossRef]
- A Study of JNJ-75348780 in Participants With Non-Hodgkin Lymphoma (NHL) and Chronic Lymphocytic Leukemia (CLL). Available online: https://clinicaltrials.gov/ct2/show/NCT04540796?term=NCT04540796&draw=2&rank=1 (accessed on 19 February 2022).
- Wu, J.; Fu, J.; Zhang, M.; Liu, D. AFM13: A first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J. Hematol. Oncol. 2015, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Affimed Announces 100% Objective Response Rate at Highest Dose in Phase 1-2 Study of Cord Blood-Derived Natural Killer Cells Pre-Complexed with Innate Cell Engager AFM13 for CD30-Positive Lyphomas. Available online: https://www.affimed.com/affimed-announces-100-objective-response-rate-at-highest-dose-in-phase-1-2-study-of-cord-blood-derived-natural-killer-cells-pre-complexed-with-innate-cell-engager-afm13-for-cd30-positive-lymphomas/ (accessed on 19 February 2022).
- Ravandi, F.; Stein, A.S.; Kantarjian, H.M.; Walter, R.B.; Paschka, P.; Jongen-Lavrencic, M.; Ossenkoppele, G.J.; Yang, Z.; Mehta, B.; Subklewe, M. A phase 1 first-in-human study of AMG 330, an anti-CD33 bispecific T-cell engager (BiTE®) antibody construct, in relapsed/refractory acute myeloid leukemia (R/R AML). Blood 2018, 132, 25. [Google Scholar] [CrossRef]
- Nair-Gupta, P.; Diem, M.; Reeves, D.; Wang, W.; Schulingkamp, R.; Sproesser, K.; Mattson, B.; Heidrich, B.; Mendonça, M.; Joseph, J.; et al. A novel C2 domain binding CD33xCD3 bispecific antibody with potent T-cell redirection activity against acute myeloid leukemia. Blood Adv. 2020, 4, 906–919. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.R.; Landgren, C.O.; Kauh, J.S.; Back, J.; Salhi, Y.; Reddy, V.; Bayever, E.; Berdeja, J.G. Phase 1, multicenter, open-label study of single-agent bispecific antibody t-cell engager GBR 1342 in relapsed/refractory multiple myeloma. J. Clin. Oncol. 2018, 36, TPS3132. [Google Scholar] [CrossRef]
- Simoneaux, R. Positive Results for Bispecific DART Flotetuzumab in Patients With AML. Oncol. Times 2021, 43, 15. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2021. mAbs 2021, 12, 1860476. [Google Scholar] [CrossRef]
- Slade, M.J.; Uy, G.L. CD123 bi-specific antibodies in development in AML: What do we know so far? Best Pract. Res. Clin. Haematol. 2020, 33, 101219. [Google Scholar] [CrossRef]
- Van Loo, P.F.; Hangalapura, B.N.; Thordardottir, S.; Gibbins, J.D.; Veninga, H.; Hendriks, L.J.A.; Kramer, A.; Roovers, R.C.; Leenders, M.; de Kruif, J.; et al. MCLA-117, a CLEC12AxCD3 bispecific antibody targeting a leukaemic stem cell antigen, induces T cell-mediated AML blast lysis. Expert Opin. Biol. Ther. 2019, 19, 721–733. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Huls, G.; Venditti, A.; Breems, D.; De Botton, S. Update from the ongoing phase I multinational study of MCLA-117, a bispecific CLEC12A x CD3 T-cell engager, in patients (pts) with acute myelogenous leukemia (AML). Eur. Hematol. Assoc. 2020. [Google Scholar]
- Mehta, N.K.; Pfluegler, M.; Meetze, K.; Li, B.; Sindel, I.; Vogt, F.; Marklin, M.; Heitmann, J.S.; Kauer, J.; Osburg, L.; et al. A novel IgG-based FLT3xCD3 bispecific antibody for the treatment of AML and B-ALL. J. Immunother. Cancer 2022, 10, e003882. [Google Scholar] [CrossRef]
- Meetze, K.; Mehta, N.K.; Pfluegler, M.; Li, B.; Baeuerle, P.A.; Michaelson, J.S.; Jung, G.; Salih, H. CLN-049 is a bispecific T cell engaging IgG-like antibody targeting FLT3 on AML cells [abstract]. In Proceedings of the 113th Annual Meeting of the American Association for Cancer Research, New Orleans, LA, USA, 8–13 April 2021. [Google Scholar]
- CLN-049 in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML). Available online: https://clinicaltrials.gov/ct2/show/NCT05143996 (accessed on 14 April 2022).
- Safety, Tolerability, PK, PD, and Efficacy of AMG 427 in Subjects with Relapsed/Refractory Acute Myeloid Leukemia (20170528). Available online: https://clinicaltrials.gov/ct2/show/NCT03541369 (accessed on 19 April 2022).
- Berdeja, J.G.; Krishnan, A.Y.; Oriol, A.; van de Donk, N.W.C.J.; Rodríguez-Otero, P.; Askari, E.; Mateos, M.-V.; Minnema, M.C.; Costa, L.J.; Verona, R.; et al. Updated results of a phase 1, first-in-human study of talquetamab, a G protein-coupled receptor family C group 5 member D (GPRC5D) × CD3 bispecific antibody, in relapsed/refractory multiple myeloma (MM). J. Clin. Oncol. 2021, 39, 8008. [Google Scholar] [CrossRef]
- A Study of Talquetamab in Participants With Relapsed or Refractory Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/NCT04634552?term=NCT04634552&draw=2&rank=1 (accessed on 18 February 2022).
- Lorenczewski, G.; Friedrich, M.; Kischel, R.; Dahlhoff, C.; Anlahr, J.; Balazs, M.; Rock, D.; Boyle, M.C.; Goldstein, R.; Coxon, A. Generation of a half-life extended anti-CD19 BiTE® antibody construct compatible with once-weekly dosing for treatment of CD19-positive malignancies. Blood 2017, 130, 2815. [Google Scholar]
- Borlak, J.; Länger, F.; Spanel, R.; Schöndorfer, G.; Dittrich, C. Immune-mediated liver injury of the cancer therapeutic antibody catumaxomab targeting EpCAM, CD3 and Fcγ receptors. Oncotarget 2016, 7, 28059. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.R.; Sukumaran, S.; Hristopoulos, M.; Totpal, K.; Stainton, S.; Lu, E.; Wong, A.; Tam, L.; Newman, R.; Vuillemenot, B.R.; et al. An anti-CD3/anti–CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood 2017, 129, 609–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandikian, D.; Takahashi, N.; Lo, A.A.; Li, J.; Eastham-Anderson, J.; Slaga, D.; Ho, J.; Hristopoulos, M.; Clark, R.; Totpal, K. Relative target affinities of T-cell–dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol. Cancer Ther. 2018, 17, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Schlothauer, T.; Herter, S.; Koller, C.F.; Grau-Richards, S.; Steinhart, V.; Spick, C.; Kubbies, M.; Klein, C.; Umaña, P.; Mössner, E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 2016, 29, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Moore, G.L.; Bernett, M.J.; Rashid, R.; Pong, E.W.; Nguyen, D.-H.T.; Jacinto, J.; Eivazi, A.; Nisthal, A.; Diaz, J.E.; Chu, S.Y. A robust heterodimeric Fc platform engineered for efficient development of bispecific antibodies of multiple formats. Methods 2019, 154, 38–50. [Google Scholar] [CrossRef]
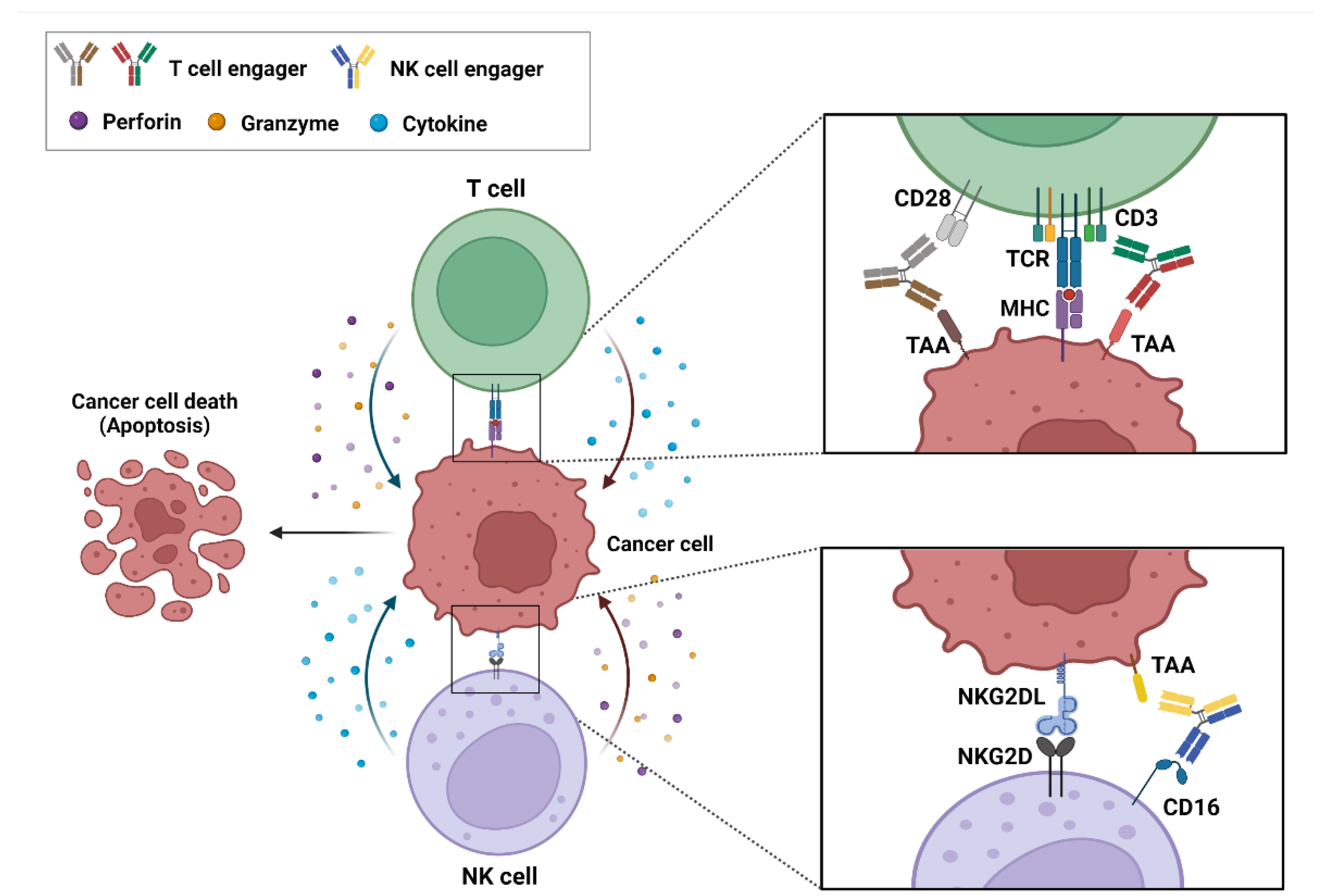



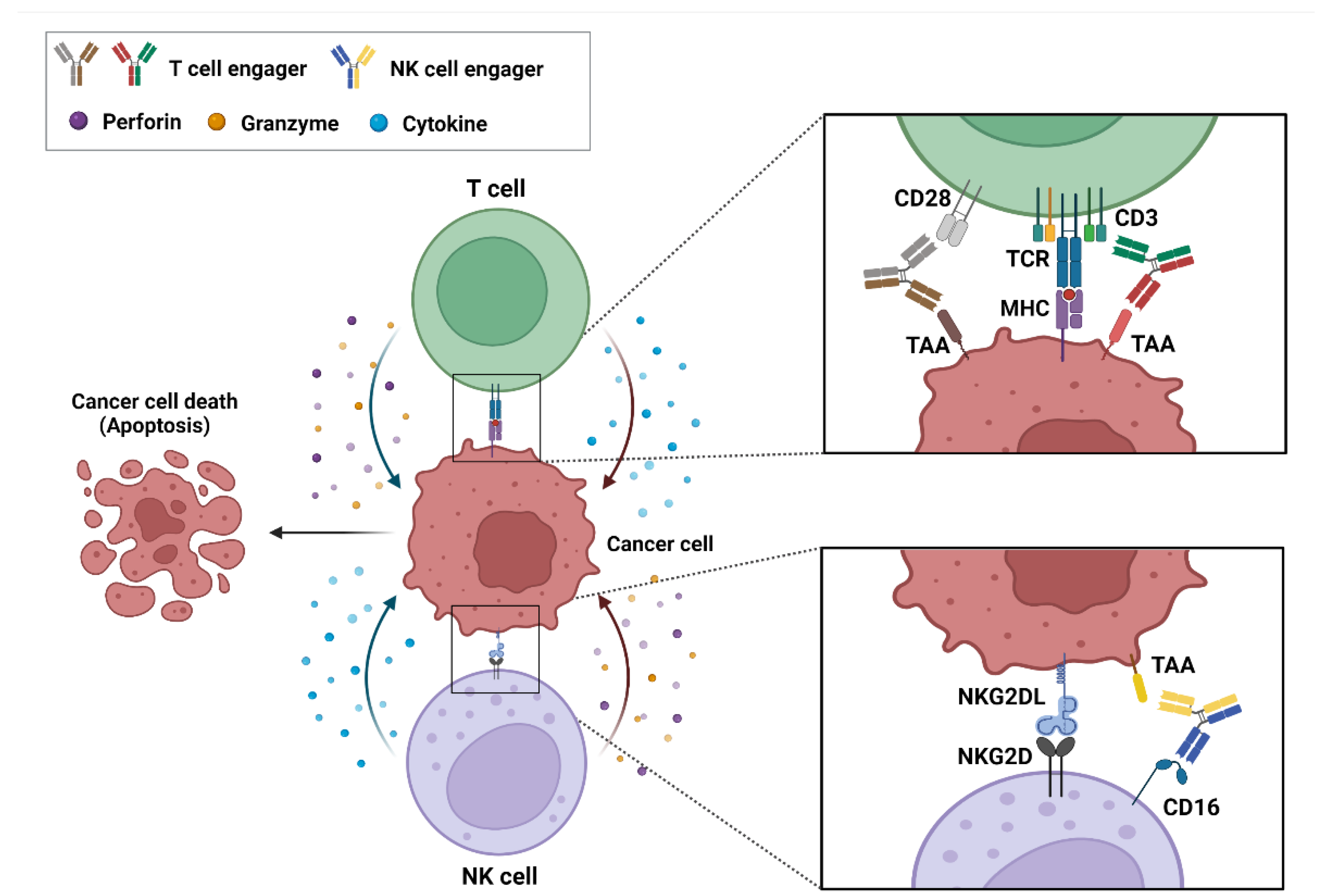{kind=link}
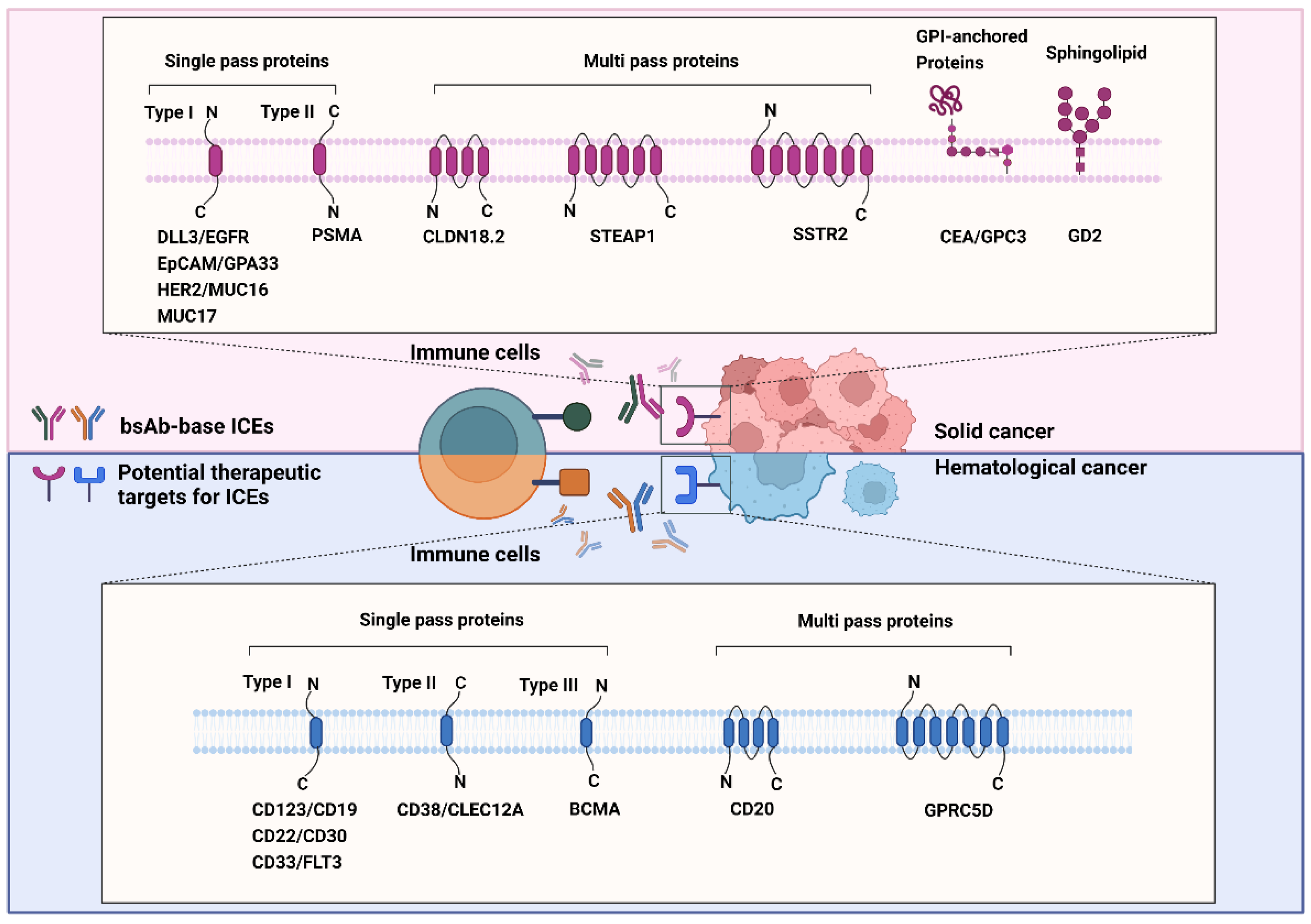{kind=link}
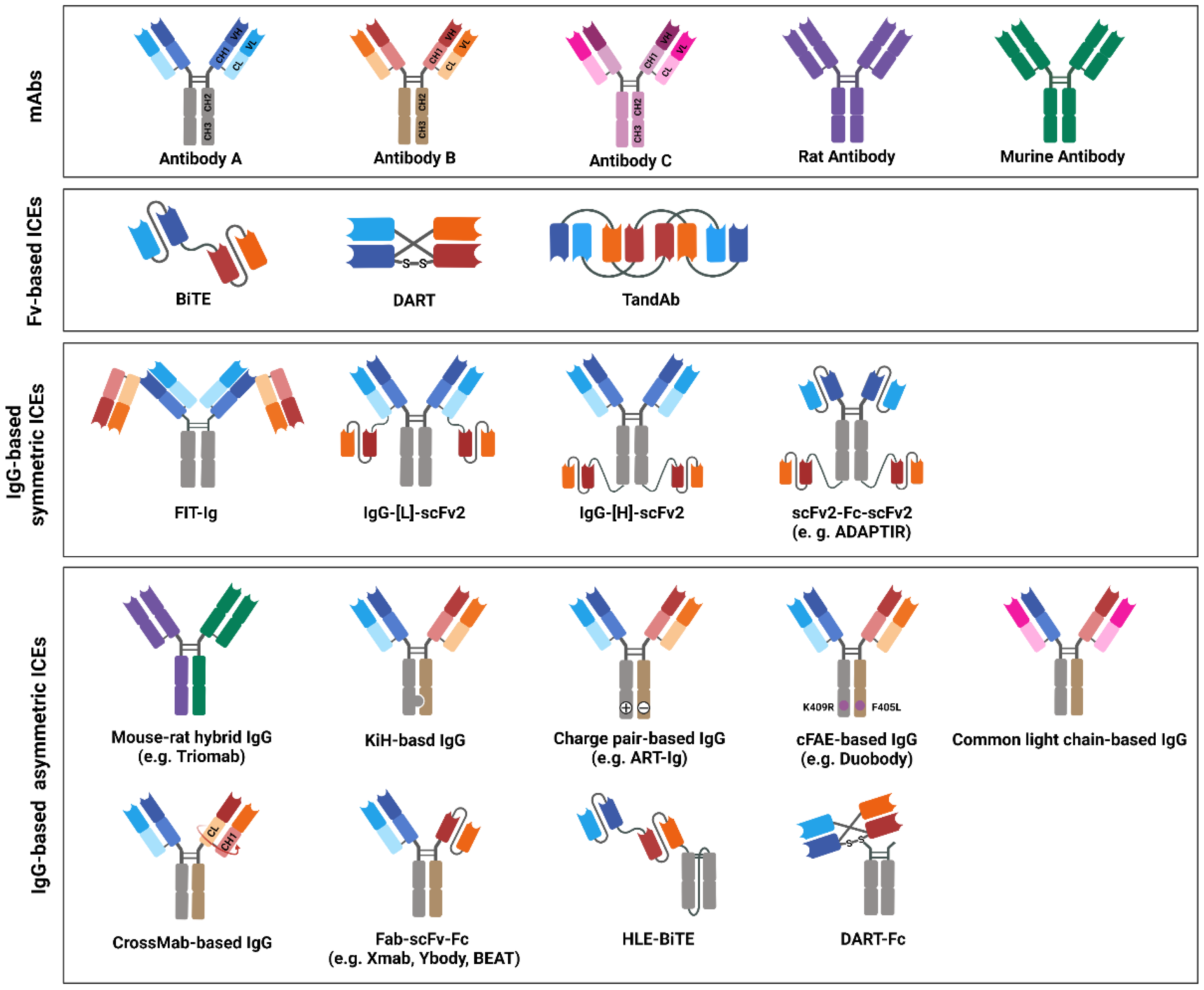{kind=link}
| BsAb Names | Company | Target | Role | Development Stage (Selected) |
|---|---|---|---|---|
| AFM13 | Affimed | CD16 × CD30 | NK cell engager | Phase II (NCT04101331) |
| AMG 160 | Amgen | CD3 × PSMA | T-cell engager | Phase I (NCT03792841) |
| AMG 199 | Amgen | CD3 × MUC17 | T-cell engager | Phase I (NCT04117958) |
| AMG 330 | Amgen | CD3 × CD33 | T-cell engager | Phase I (NCT02520427) |
| AMG 427 | Amgen | CD3 × FLT3 | T-cell engager | Phase I (NCT03541369) |
| AMG 509 | Amgen | CD3 × STEAP1 | T-cell engager | Phase I (NCT04221542) |
| AMG 701 | Amgen | CD3 × BCMA | T-cell engager | Phase I (NCT03287908) |
| AMG 910 | Amgen | CD3 × CLDN18.2 | T-cell engager | Phase I (NCT04260191) |
| APVO414/ES414/MOR209 | Aptevo Therapeutics | CD3 × PSMA | T-cell engager | Phase I (NCT02262910) |
| APVO436 | Aptevo Therapeutics | CD3 × CD123 | T-cell engager | Phase I (NCT03647800) |
| Blinatumomab/Blincyto | Amgen | CD3 × CD19 | T-cell engager | Marketed |
| Catumaxomab/Removab | Fresenius Biotechand and Trion Pharma | CD3 × EpCAM | T-cell engager | Withdrawn from the market |
| CC-1 | University of Tubingen | CD3 × PSMA | T-cell engager | Phase I (NCT04104607) |
| CC-93269/EM801 | Celgene | CD3 × BCMA | T-cell engager | Phase I (NCT03486067) |
| Cibisatamab/RG7802/RO6958688 | Roche | CD3 × CEA | T-cell engager | Phase I (NCT02324257, NCT02650713) |
| CLN-049 | Cullinan Oncology | CD3 × FLT3 | T-cell engager | Phase I (NCT05143996) |
| Elranatamab/PF-06863135 | Pfizer | CD3 × BCMA | T-cell engager | Phase II (NCT04649359) |
| EMB-06 | EpimAb Biotherapeutics | CD3 × BCMA | T-cell engager | Phase I/II (NCT04735575) |
| Epcoritamab/GEN3013 | Genmab A/S | CD3 × CD20 | T-cell engager | Phase III (NCT04628494) |
| ERY974 | Chugai | CD3 × GPC3 | T-cell engager | Phase I (NCT05022927) |
| Flotetuzumab/MGD006 | MacroGenics | CD3 × CD123 | T-cell engager | Phase II (NCT04582864) |
| Glofitamab/RG6026/RO7082859 | Roche | CD3 × CD20 | T-cell engager | Phase III (NCT04408638) |
| ISB 1342/GBR 1342 | Ichnos Sciences | CD3 × CD38 | T-cell engager | Phase I (NCT03309111) |
| JNJ-63709178 | Johnson & Johnson | CD3 × CD123 | T-cell engager | Phase I (NCT02715011) |
| JNJ-63898081 | Johnson & Johnson | CD3 × PSMA | T-cell engager | Phase I (NCT03926013) |
| JNJ-67571244 | Johnson & Johnson | CD3 × CD33 | T-cell engager | Phase I (NCT03915379) |
| JNJ-75348780 | Johnson & Johnson | CD3 × CD22 | T-cell engager | Phase I (NCT04540796) |
| Linvoseltamab/REGN 5458 | Regeneron Pharmaceuticals | CD3 × BCMA | T-cell engager | Phase I/II (NCT03761108) |
| M701 | YZYBio | CD3 × EpCAM | T-cell engager | Phase I (NCT04501744) |
| M802 | YZYBio | CD3 × HER2 | T-cell engager | Phase I (NCT04501770) |
| MGD007 | Macrogenics | CD3 × GPA33 | T-cell engager | Phase I/II (NCT03531632) |
| Mosunetuzumab/RG7828 | Genentech | CD3 × CD20 | T-cell engager | Phase III (NCT04712097) |
| Nivatrotamab/Hu3F8-BsAb | Y-mAbs | CD3 × GD2 | T-cell engager | Phase I/II (NCT04750239) |
| Odronextamab/REGN1979 | Regeneron Pharmaceuticals | CD3 × CD20 | T-cell engager | Phase II (NCT03888105) |
| REGN4018 | Regeneron Pharmaceuticals | CD3 × MUC16 | T-cell engager | Phase I/II (NCT03564340) |
| REGN5459 | Regeneron Pharmaceuticals | CD3 × BCMA | T-cell engager | Phase I/II (NCT04083534) |
| REGN5678 | Regeneron Pharmaceuticals | CD28 × PSMA | T-cell engager | Phase I/II (NCT03972657) |
| REGN7075 | Regeneron Pharmaceuticals | CD28 × EGFR | T-cell engager | Phase I/II (NCT04626635) |
| Talquetamab/JNJ-64407564 | Johnson & Johnson | CD3 × GPRC5D | T-cell engager | Phase II (NCT04634552) |
| Tarlatamab/AMG 757 | Amgen | CD3 × DLL-3 | T-cell engager | Phase II (NCT05060016) |
| Teclistamab/JNJ-64007957 | Johnson & Johnson | CD3 × BCMA | T-cell engager | Phase III (NCT05083169) |
| Tepoditamab/MCLA-117 | Merus | CD3 × CLEC12A | T-cell engager | Phase I (NCT03038230) |
| TNB-383B | TeneoOne | CD3 × BCMA | T-cell engager | Phase I (NCT03933735) |
| TNB-486 | TeneoTwo | CD3 × CD19 | T-cell engager | Phase I (NCT04594642) |
| TNB-585 | Amgen | CD3 × PSMA | T-cell engager | Phase I (NCT04740034) |
| XmAb13676/Plamotamab | Xencor | CD3 × CD20 | T-cell engager | Phase I (NCT02924402) |
| XmAb14045/Vibecotamab | Xencor | CD3 × CD123 | T-cell engager | Phase I (NCT02730312) |
| XmAb18087/Tidutamab | Xencor | CD3 × SSTR2 | T-cell engager | Phase I/II (NCT04590781) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, H.G.; Yang, H.R.; Yoon, A.; Lee, S. Bispecific Antibody-Based Immune-Cell Engagers and Their Emerging Therapeutic Targets in Cancer Immunotherapy. Int. J. Mol. Sci. 2022, 23, 5686. https://doi.org/10.3390/ijms23105686
Shin HG, Yang HR, Yoon A, Lee S. Bispecific Antibody-Based Immune-Cell Engagers and Their Emerging Therapeutic Targets in Cancer Immunotherapy. International Journal of Molecular Sciences. 2022; 23(10):5686. https://doi.org/10.3390/ijms23105686
Chicago/Turabian StyleShin, Ha Gyeong, Ha Rim Yang, Aerin Yoon, and Sukmook Lee. 2022. "Bispecific Antibody-Based Immune-Cell Engagers and Their Emerging Therapeutic Targets in Cancer Immunotherapy" International Journal of Molecular Sciences 23, no. 10: 5686. https://doi.org/10.3390/ijms23105686
APA StyleShin, H. G., Yang, H. R., Yoon, A., & Lee, S. (2022). Bispecific Antibody-Based Immune-Cell Engagers and Their Emerging Therapeutic Targets in Cancer Immunotherapy. International Journal of Molecular Sciences, 23(10), 5686. https://doi.org/10.3390/ijms23105686