Comparative Transcriptome Analysis of a Toxin-Producing Dinoflagellate Alexandrium catenella and Its Non-Toxic Mutant


Abstract
:1. Introduction

2. Results
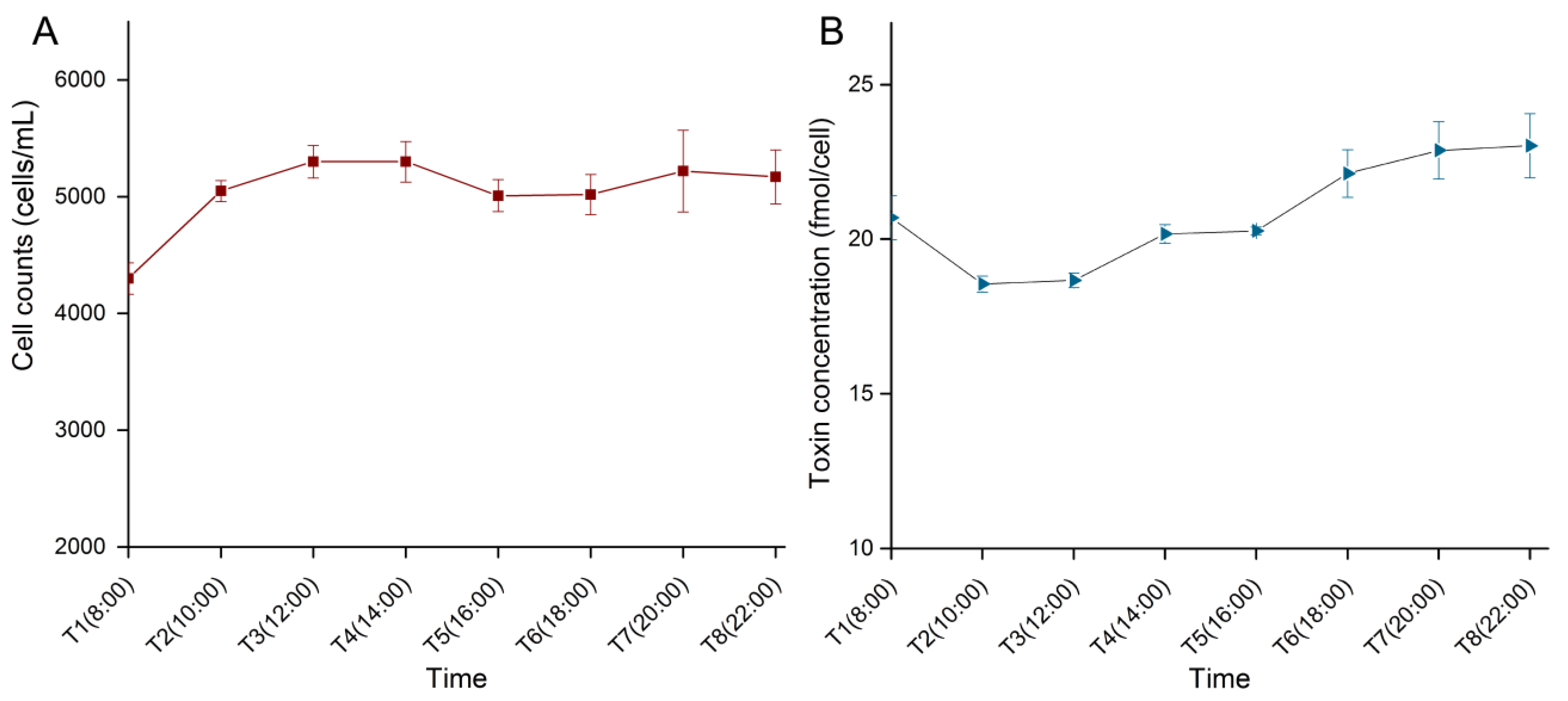
2.1. Cell Density and Toxin Content during the Light Period
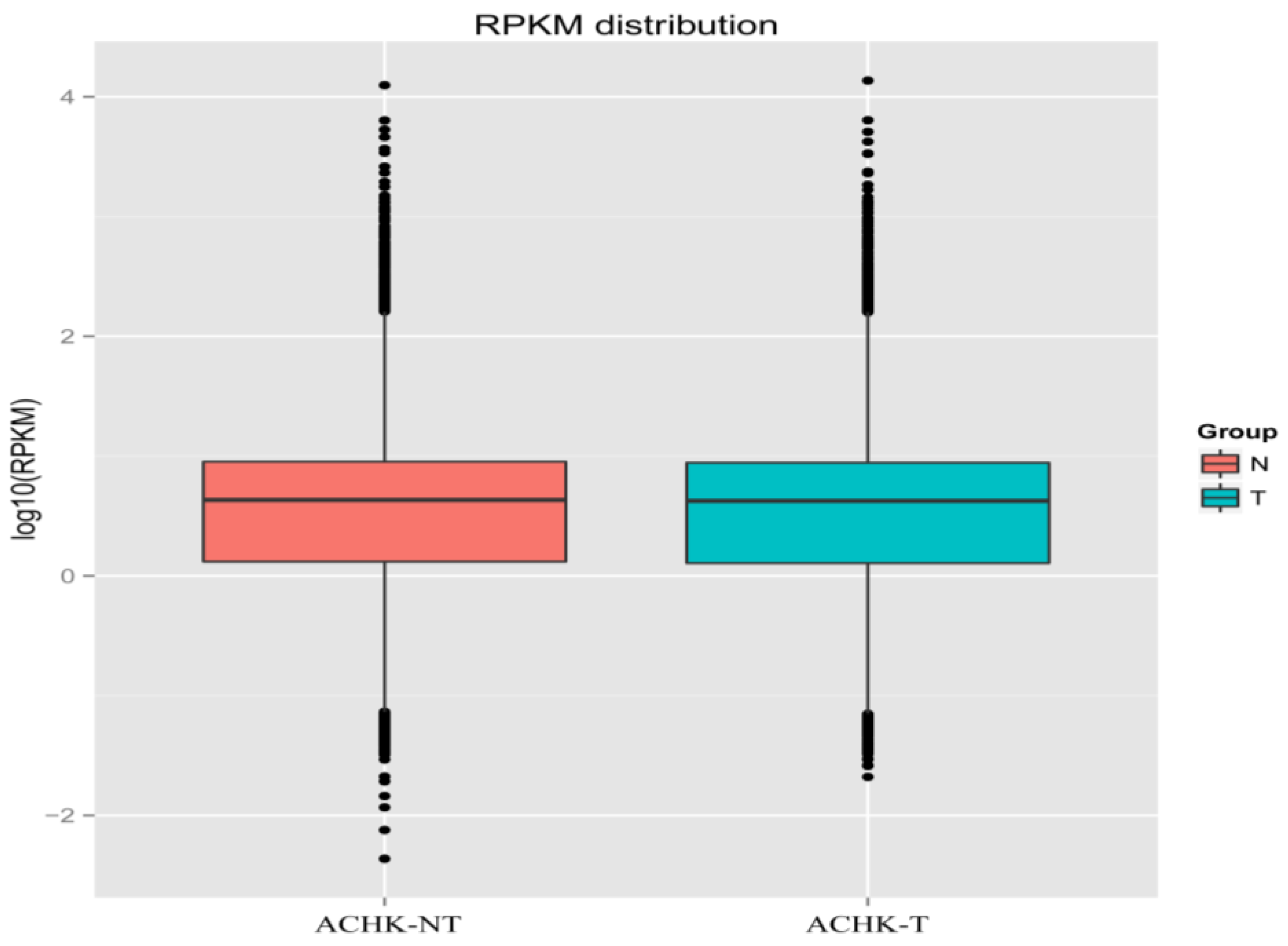
2.2. RNA-seq and de novo Assembly

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Value |
|---|---|
| Total number of raw reads | 259,503,870 |
| Total number of clean reads | 242,852,652 |
| Total clean nucleotides (Gb) | 24.28 |
| Q20 percentage (%) | 93.75~96.88 |
| GC percentage (%) | 61.07~61.40 |
| Total number of transcripts | 155,353 |
| Mean length of transcripts | 1003 |
| N50 length of transcripts | 1549 |
| Total number of unigenes | 113,674 |
| Mean length of unigenes | 901 |
| N50 length of unigenes | 1462 |
2.3. Gene Functional Annotation
| Database | Number of Unigenes |
|---|---|
| NR | 34,372 |
| NT | 2071 |
| KEGG | 10,362 |
| SwissProt | 23,511 |
| Pfam | 52,026 |
| GO | 28,146 |
| KOG | 40,535 |
| At least in one database | 66,812 |
2.4. Differential Expression Gene Analysis

| Gene_ID | N_Readcount | T_Readcount | log2.Fold_Change | NR Description |
|---|---|---|---|---|
| Down-regulation in ACHK-NT: | ||||
| comp66063_c0 | 2 | 37391.29 | −14.226 | exoglucanase 3 1,4-beta-cellobiohydrolase3 family GH6, Exoglucanase 3 (Pyrenophora tritici-repentis (strain Pt-1C-BFP)), Predicted CDS Pa_4_2420 (Podospora anserina) [Chondrus crispus] |
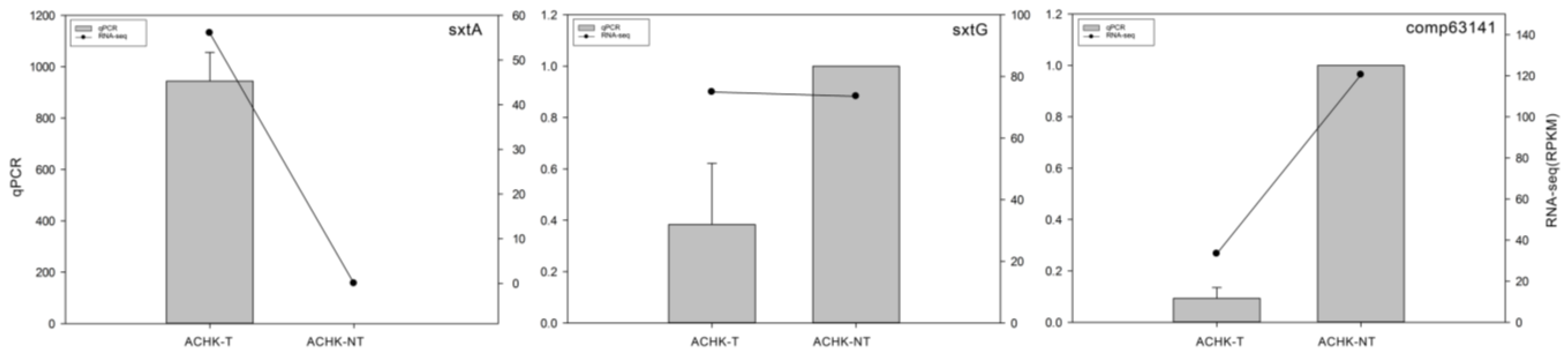
| comp66169_c0 | 1 | 12503.81 | −13.646 | SxtA long isoform precursor [Alexandrium fundyense] |
| comp57121_c1 | 3 | 15706 | −12.39 | putative uncharacterized protein [Sutterella wadsworthensis CAG:135] |
| comp66059_c0 | 3 | 3644.78 | −10.282 | hypothetical protein Pmar_PMAR010035 [Perkinsus marinus ATCC 50983] |
| comp47304_c0 | 15 | 4102.99 | −8.131 | cell wall-associated hydrolase [Burkholderia multivorans ATCC 17616] |
| comp57121_c0 | 0 | 4113 | −7.3658 | hypothetical protein Csp_D29540 [Curvibacter putative symbiont of Hydra magnipapillata] |
| comp62287_c0 | 28.75 | 3542.4 | −6.9805 | hypothetical protein, partial [Escherichia coli] |
| comp64637_c0 | 0 | 2734.05 | −6.7766 | enzymatic polyprotein; Endonuclease; Reverse transcriptase, putative [Pediculus humanus corporis] |
| comp46928_c0 | 15 | 1300.95 | −6.4739 | Protein CBR-LARP-1 [Caenorhabditis briggsae] |
| comp114480_c0 | 180 | 1354 | −2.9466 | putative S12 family peptidase [Gemmatimonas aurantiaca T-27] |
| comp66521_c0 | 11289.22 | 28698.06 | −1.3815 | Ankyrin repeats containing protein [delta proteobacterium BABL1] |
| comp60385_c0 | 2671.63 | 5630.9 | −1.1111 | -- |
| comp47283_c1 | 15773.39 | 25201.15 | −0.71144 | RecName: Full = Photosystem I P700 chlorophyll a apoprotein A2; AltName: Full = PSI-B; AltName: Full = PsaB |
| Up-regulation in ACHK-NT: | ||||
| comp64975_c0 | 119074.04 | 74056.39 | 0.64972 | cytochrome c oxidase subunit 1 [Alexandrium catenella] |
| comp65226_c1 | 19727.54 | 12087.19 | 0.67129 | light-harvesting protein, partial [Symbiodinium sp. clade C3] |
| comp54599_c0 | 40289.85 | 24201.67 | 0.69987 | -- |
| comp65353_c0 | 11048.61 | 6220.45 | 0.79333 | hypothetical protein NCLIV_047860 [Neospora caninum Liverpool] |
| comp63840_c0 | 8189.47 | 4453.25 | 0.84347 | probable high CO2 inducible periplasmic protein [Heterocapsa triquetra] |
| comp65627_c0 | 31244.01 | 16909.13 | 0.85033 | proliferating cell nuclear antigen [Alexandrium affine] |
| comp66053_c0 | 60305.52 | 30993.93 | 0.92486 | plastid C1 class II fructose bisphosphate aldolase [Heterocapsa triquetra] |
| comp66077_c1 | 8811.01 | 4410 | 0.96308 | hypothetical protein RFI_22066 [Reticulomyxa filosa] |
| comp65597_c0 | 8402.67 | 4024.29 | 1.0267 | hypothetical protein [Verrucomicrobiae bacterium DG1235] |
| comp65950_c0 | 5831.47 | 2682.38 | 1.0849 | PREDICTED: phosphoserine aminotransferase-like [Anolis carolinensis] |
| comp33299_c0 | 5150 | 2360 | 1.0903 | Eukaryotic translation initiation factor 3 subunit 9, putative [Eimeria acervulina] |
| comp70851_c0 | 2940.5 | 975 | 1.5571 | Calcium-dependent protein kinase, putative [Perkinsus marinus ATCC 50983] |
| comp71917_c0 | 2656.96 | 861 | 1.5902 | -- |
| comp62882_c0 | 14038.72 | 4456 | 1.6201 | SCO-spondin [Crassostrea gigas] |
| comp63141_c0 | 11545.27 | 3106 | 1.8587 | hypothetical protein Rcas_3705 [Roseiflexus castenholzii DSM 13941] |
| comp65143_c0 | 58658.95 | 11917.14 | 2.2639 | light-harvesting protein, partial [Symbiodinium sp. clade C3] |
| comp65099_c0 | 3250.62 | 428.7 | 2.8872 | Phosphoserine aminotransferase, putative [Perkinsus marinus ATCC 50983] |
| comp47128_c0 | 956.92 | 52 | 4.1664 | -- |
| comp64715_c0 | 1765.01 | 55 | 4.9687 | -- |
| comp57722_c1 | 872 | 0 | 5.0926 | -- |
| comp57938_c1 | 1421.88 | 19 | 6.1902 | S-adenosyl-homocysteine hydrolase like protein, partial [Alexandrium fundyense] |
| comp64901_c0 | 1290 | 10 | 6.9758 | -- |
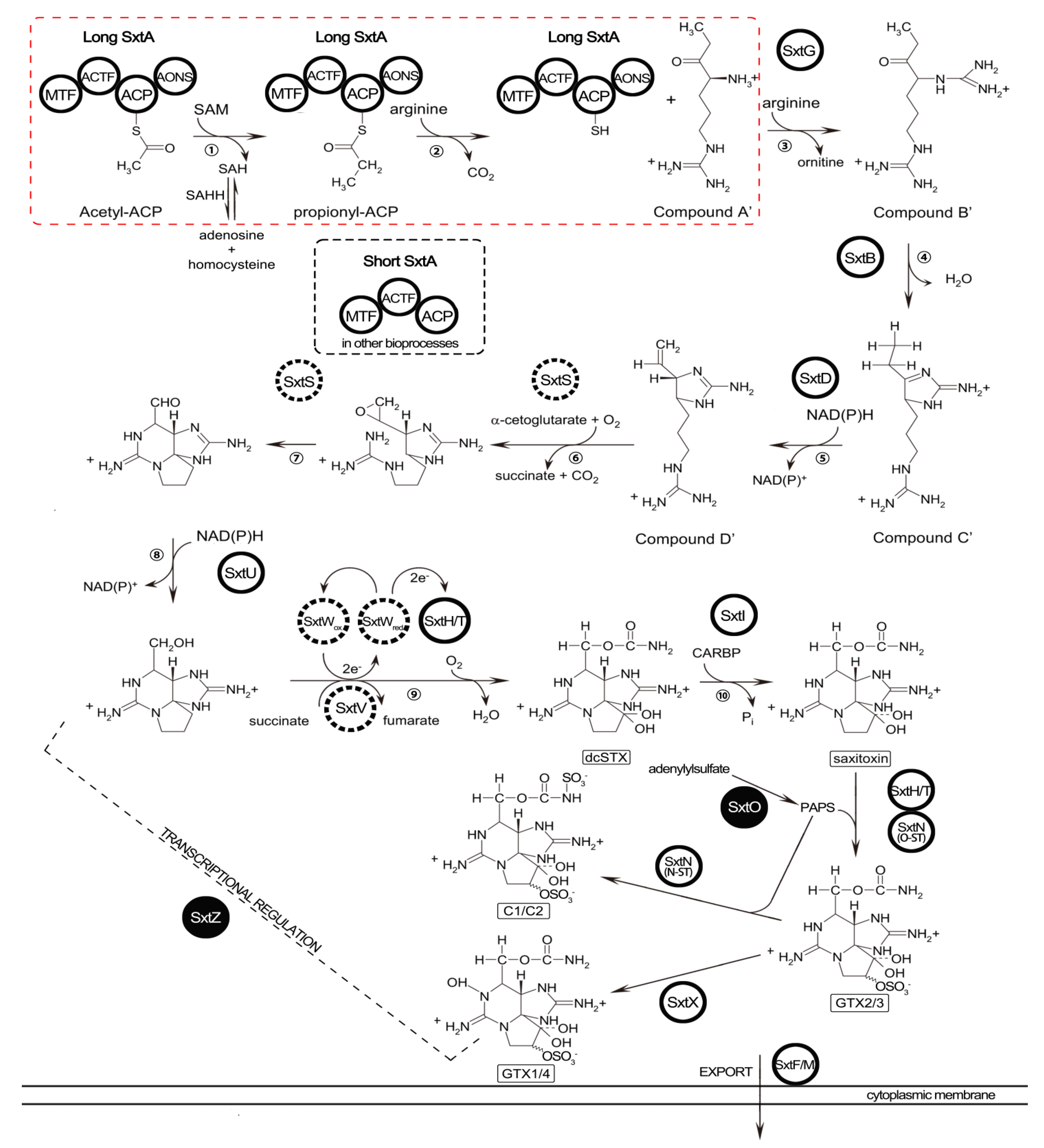
2.5. Identification of STX Genes

| STX Genes | Putative Function | A. catenella Unigenes |
|---|---|---|
| sxtA | Aspartate aminotransferase | 9 |
| sxtB | Cytidine deaminase | 2 |
| sxtD | Sterole desaturase | 1 |
| sxtF/M | Toxic compound efflux protein | 2 |
| sxtG | Amidinotransferase | 2 |
| sxtH/T | Phenylpropionate dioxygenase | 15 |
| sxtI | O-carbamoyltransferase | 2 |
| sxtN | Sulfotransferase | 1 |
| sxtO | Adenylylsulfate kinase | 1 |
| sxtU | Short-chain alcohol dehydrogenase | 59 |
| sxtX | Cephalosporin hydroxylase | 1 |
| sxtZ | Two-component sensor histidine kinase | 6 |

3. Discussion
3.1. Up-Regulated Genes in ACHK-NT
3.2. Down-Regulated Genes in ACHK-NT
3.3. STX Biosynthesis Related Genes
4. Experimental Section
4.1. Organisms and Culture Conditions
4.2. Sample Collection for Cell Density, Toxin Measurement and Cell Cycle Analysis
4.3. RNA Isolation, cDNA Library Preparation and Sequencing
4.4. Quality Control and de novo Assembly
4.5. Gene Functional Annotation and Differential Expression Analysis
4.6. Identification of STX Genes
4.7. qRT-PCR Validation of DEGs
5. Conclusions
Supplementary Files
Supplementary File 1Supplementary File 2Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wiese, M.; DʼAgostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic alkaloids: Saxitoxin and its analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar]
- Hallegraeff, G.M. Harmful algal blooms: A global overview. In Manual on Harmful Marine Microalgae; Hallegraeff, G.M., Anderson, D.M., Cembella, A.D., Eds.; International Oceanographic Commission (IOC) Manual and Guides UNESCO: Paris, France, 1995; pp. 1–22. [Google Scholar]
- Cembella, A.D. Ecophysiology and metabolism of paralytic shellfish toxins in marine microalgae. In Physiological Ecology of Harmful Algal Blooms; Hallegraeff, G.M., Anderson, D.M., Cembella, A.D., Eds.; Springer: Berlin, Germany, 1998; Volume 41, pp. 381–404. [Google Scholar]
- Orr, R.J.S.; Stüken, A.; Rundberget, T.; Eikrem, W.; Jakobsen, K.S. Improved phylogenetic resolution of toxic and non-toxic Alexandrium strains using a concatenated rDNA approach. Harmful Algae 2011, 10, 676–688. [Google Scholar] [CrossRef]
- Sivonen, K.; Jones, G. Chapter 3; Cyanobacterial Toxins. In Toxic Cyanobacteria in Water: A Guide to Their Public Health Consequences, Monitoring and Management; Chorus, I., Bartram, J., Eds.; Spon Press: London, UK, 1999. [Google Scholar]
- Chorus, I.; Falconer, I.R.; Salas, H.J.; Bartram, J. Health risks caused by freshwater cyanobacteria in recreational waters. J. Oxicol. Environ. Health. Part. B Crit. Rev. 2000, 3, 323–347. [Google Scholar] [CrossRef]
- Carmichael, W.W. Health Effects of Toxin-Producing Cyanobacteria: “The CyanoHABs”. Hum. Ecol. Risk Assess. 2001, 7, 1393–1407. [Google Scholar] [CrossRef]
- Haider, S.; Naithani, V.; Viswanathan, P.N.; Kakkar, P. Cyanobacterial toxins: A growing environmental concern. Chemosphere 2003, 52, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Kellmann, R.; Mihali, T.K.; Jeon, Y.J.; Pickford, R.; Pomati, F.; Neilan, B.A. Biosynthetic intermediate analysis and functional homology reveal a saxitoxin gene cluster in cyanobacteria. Appl. Environ. Microbiol. 2008, 74, 4044–4053. [Google Scholar] [CrossRef] [PubMed]
- Mihali, T.K.; Kellmann, R.; Neilan, B.A. Characterisation of the paralytic shellfish toxin biosynthesis gene clusters in Anabaena circinalis AWQC131C and Aphanizomenon sp. NH-5. BMC Biochem. 2009, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Mihali, T.K.; Carmichael, W.W.; Neilan, B.A. A putative gene cluster from a Lyngbya wollei bloom that encodes paralytic shellfish toxin biosynthesis. PLoS One 2011, 6, e14657. [Google Scholar] [CrossRef] [PubMed]
- Stucken, K.; John, U.; Cembella, A.; Murillo, A.A.; Soto-Liebe, K.; Fuentes-Valdés, J.J.; Friedel, M.; Plominsky, A.M.; Vásquez, M.; Glöckner, G. The Smallest Known Genomes of Multicellular and Toxic Cyanobacteria: Comparison, Minimal Gene Sets for Linked Traits and the Evolutionary Implications. PLoS One 2010, 5, e9235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y. Microalgal metabolites: A new perspective. Annu. Rev. Microbiol. 1996, 50, 431–465. [Google Scholar] [CrossRef] [PubMed]
- Kellmann, R.; Stuken, A.; Orr, R.J.; Svendsen, H.M.; Jakobsen, K.S. Biosynthesis and molecular genetics of polyketides in marine dinoflagellates. Mar. Drugs 2010, 8, 1011–1048. [Google Scholar] [PubMed]
- Kellmann, R.; Neilan, B.A. Biochemical Characterization of Paralytic Shellfish Toxin Biosynthesis in Vitro. J. Phycol. 2007, 43, 497–508. [Google Scholar] [CrossRef]
- Orr, R.J.; Stuken, A.; Murray, S.A.; Jakobsen, K.S. Evolution and distribution of saxitoxin biosynthesis in dinoflagellates. Mar. Drugs 2013, 11, 2814–2828. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hong, R.; Hastings, J.W. Three functional luciferase domains in a single polypeptide chain. Proc. Natl. Acad. Sci. USA 1997, 94, 8954–8958. [Google Scholar] [CrossRef] [PubMed]
- LaJeunesse, T.C.; Lambert, G.; Andersen, R.A.; Coffroth, M.A.; Galbraith, D.W. Symbiodinium (pyrrhophyta) genome sizes (DNA content) are smallest among dinoflagellates. J. Phycol. 2005, 41, 880–886. [Google Scholar] [CrossRef]
- Harlow, L.D.; Koutoulis, A.; Hallegraeff, G.M. S-adenosylmethionine synthetase genes from eleven marine dinoflagellates. Phycologia 2007, 46, 46–53. [Google Scholar] [CrossRef]
- Yang, I.; John, U.; Beszteri, S.; Glockner, G.; Krock, B.; Goesmann, A.; Cembella, A.D. Comparative gene expression in toxic versus non-toxic strains of the marine dinoflagellate Alexandrium minutum. BMC Genomics 2010, 11, 248. [Google Scholar] [CrossRef] [PubMed]
- Stuken, A.; Orr, R.J.; Kellmann, R.; Murray, S.A.; Neilan, B.A.; Jakobsen, K.S. Discovery of nuclear-encoded genes for the neurotoxin saxitoxin in dinoflagellates. PLoS One 2011, 6, e20096. [Google Scholar] [CrossRef] [PubMed]
- Orr, R.J.; Stuken, A.; Murray, S.A.; Jakobsen, K.S. Evolutionary Acquisition and Loss of Saxitoxin Biosynthesis in Dinoflagellates: the Second “Core” Gene, sxtG. Appl. Environ. Microbiol. 2013, 79, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.D.; Wisecaver, J.H.; Brosnahan, M.L.; Kulis, D.M.; Anderson, D.M.; Bhattacharya, D.; Plumley, F.G.; Erdner, D.L. Evolution of saxitoxin synthesis in cyanobacteria and dinoflagellates. Mol. Biol. Evol. 2013, 30, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Hiramatsu, K.; Ogawa, M.; Omura, T.; Ishimaru, T.; Oshima, Y. Non-toxic and toxic subclones obtained from a toxic clonal culture of Alexandrium tamarense (Dinophyceae): Toxicity and molecular biological feature. Harmful Algae 2008, 7, 740–751. [Google Scholar] [CrossRef]
- Wang, D.Z.; Gao, Y.; Lin, L.; Hong, H.S. Comparative proteomic analysis reveals proteins putatively involved in toxin biosynthesis in the marine dinoflagellate Alexandrium catenella. Mar. Drugs 2013, 11, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z.; Li, C.; Zhang, Y.; Wang, Y.Y.; He, Z.P.; Lin, L.; Hong, H.S. Quantitative proteomic analysis of differentially expressed proteins in the toxicity-lost mutant of Alexandrium catenella (Dinophyceae) in the exponential phase. J. Proteomics 2012, 75, 5564–5577. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Ogawa, M.; Yotsu-Yamashita, M.; Oshima, Y. Effect of 5-fluoro-2′-deoxyuridine on toxin production and cell cycle regulation in marine dinoflagellate, Alexandrium tamarense. Harmful Algae 2014, 32, 64–72. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sui, Z.; Chang, L.; Kang, K.; Ma, J.; Kong, F.; Zhou, W.; Wang, J.; Guo, L.; Geng, H.; et al. Transcriptome de novo assembly sequencing and analysis of the toxic dinoflagellate Alexandrium catenella using the Illumina Platform. Gene 2014, 537, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinfor. 2011, 12, 323. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Haiser, H.J.; Yousef, M.R.; Elliot, M.A. Cell wall hydrolases affect germination, vegetative growth, and sporulation in Streptomyces coelicolor. J. Bacteriol. 2009, 191, 6501–6512. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.A.; Mihali, T.K.; Neilan, B.A. Extraordinary conservation, gene loss, and positive selection in the evolution of an ancient neurotoxin. Mol. Biol. Evol. 2011, 28, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [PubMed]
- Hou, Y.; Lin, S. Distinct gene number-genome size relationships for eukaryotes and non-eukaryotes: Gene content estimation for dinoflagellate genomes. PLoS One 2009, 4, e6978. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.A.; Wiese, M.; Stuken, A.; Brett, S.; Kellmann, R.; Hallegraeff, G.; Neilan, B.A. sxtA-based quantitative molecular assay to identify saxitoxin-producing harmful algal blooms in marine waters. Appl. Environ. Microbiol. 2011, 77, 7050–7057. [Google Scholar] [CrossRef] [PubMed]
- Romeis, T.; Ludwig, A.A.; Martin, R.; Jones, J.D. Calcium-dependent protein kinases play an essential role in a plant defence response. EMBO J. 2001, 20, 5556–5567. [Google Scholar] [CrossRef]
- Cramer, G.R.; Van Sluyter, S.C.; Hopper, D.W.; Pascovici, D.; Keighley, T.; Haynes, P.A. Proteomic analysis indicates massive changes in metabolism prior to the inhibition of growth and photosynthesis of grapevine (Vitis vinifera L.) in response to water deficit. BMC Plant. Biol. 2013, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Taroncher-Oldenburg, G.; Anderson, D.M. Identification and characterization of three differentially expressed genes, encoding S-adenosylhomocysteine hydrolase, methionine aminopeptidase, and a histone-like protein, in the toxic dinoflagellate Alexandrium fundyense. Appl. Environ. Microbiol. 2000, 66, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Takahashi, H.; Nakano, Y.; Konishi, T.; Terauchi, R.; Takeda, T. Characterization of a cellobiohydrolase (MoCel6A) produced by Magnaporthe oryzae. Appl. Environ. Microbiol. 2010, 76, 6583–6590. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.D.; Anderson, D.M.; Erdner, D.L.; Bhattacharya, D. Dinoflagellates: A remarkable evolutionary experiment. Am. J. Bot. 2004, 91, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- Kwok, A.C.; Wong, J.T. Cellulose synthesis is coupled to cell cycle progression at G1 in the dinoflagellate Crypthecodinium cohnii. Plant. Physiol. 2003, 131, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Kwok, A.C.; Wong, J.T. The activity of a wall-bound cellulase is required for and is coupled to cell cycle progression in the dinoflagellate Crypthecodinium cohnii. Plant. Cell Online 2010, 22, 1281–1298. [Google Scholar] [CrossRef]
- Toulza, E.; Shin, M.S.; Blanc, G.; Audic, S.; Laabir, M.; Collos, Y.; Claverie, J.M.; Grzebyk, D. Gene expression in proliferating cells of the dinoflagellate Alexandrium catenella (Dinophyceae). Appl. Environ. Microbiol. 2010, 76, 4521–4529. [Google Scholar] [CrossRef] [PubMed]
- Radmer, R.; Cox, J.; Lieberman, D.; Behrens, P.; Arnett, K. Biomass recycle as a means to improve the energy efficiency of CELSS algal culture systems. Adv. Space Res. 1987, 7, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Blifernez-Klassen, O.; Klassen, V.; Doebbe, A.; Kersting, K.; Grimm, P.; Wobbe, L.; Kruse, O. Cellulose degradation and assimilation by the unicellular phototrophic eukaryote Chlamydomonas reinhardtii. Nat. Commun. 2012, 3, 1214. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.D.; Selvin, R.C.; Claus, W.; Guillard, R.R. Media for the culture of oceanic ultraphytoplankton. J. Phycol. 1987, 23, 633–638. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2009, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9945. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lin, S. Complex gene structure of the form ii rubisco in the dinoflagellate Prorocentrum minimum (dinophyceae). J. Phycol. 2003, 39, 1160–1171. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhang, S.-F.; Lin, L.; Wang, D.-Z. Comparative Transcriptome Analysis of a Toxin-Producing Dinoflagellate Alexandrium catenella and Its Non-Toxic Mutant. Mar. Drugs 2014, 12, 5698-5718. https://doi.org/10.3390/md12115698
Zhang Y, Zhang S-F, Lin L, Wang D-Z. Comparative Transcriptome Analysis of a Toxin-Producing Dinoflagellate Alexandrium catenella and Its Non-Toxic Mutant. Marine Drugs. 2014; 12(11):5698-5718. https://doi.org/10.3390/md12115698
Chicago/Turabian StyleZhang, Yong, Shu-Fei Zhang, Lin Lin, and Da-Zhi Wang. 2014. "Comparative Transcriptome Analysis of a Toxin-Producing Dinoflagellate Alexandrium catenella and Its Non-Toxic Mutant" Marine Drugs 12, no. 11: 5698-5718. https://doi.org/10.3390/md12115698
APA StyleZhang, Y., Zhang, S.-F., Lin, L., & Wang, D.-Z. (2014). Comparative Transcriptome Analysis of a Toxin-Producing Dinoflagellate Alexandrium catenella and Its Non-Toxic Mutant. Marine Drugs, 12(11), 5698-5718. https://doi.org/10.3390/md12115698