Abstract
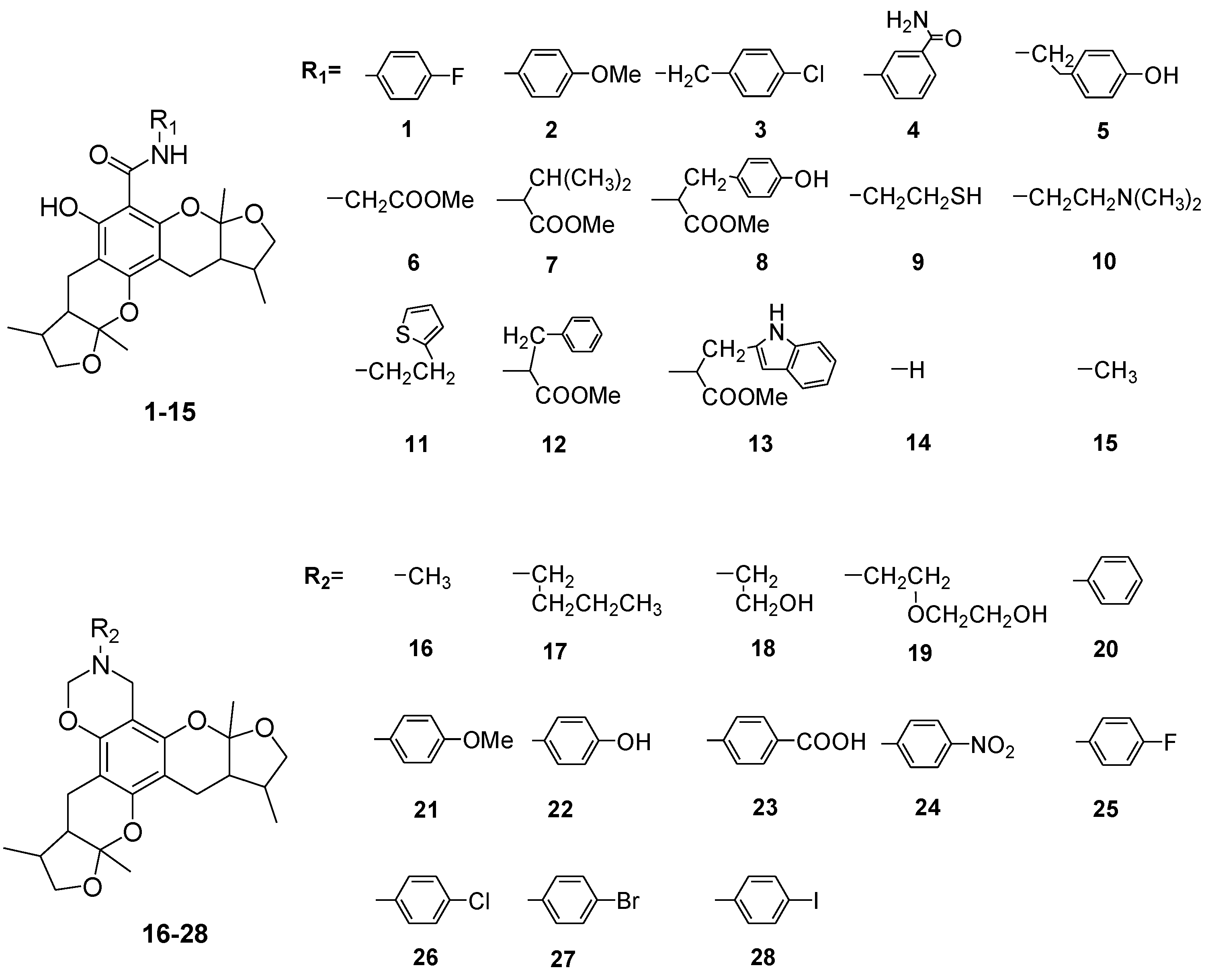
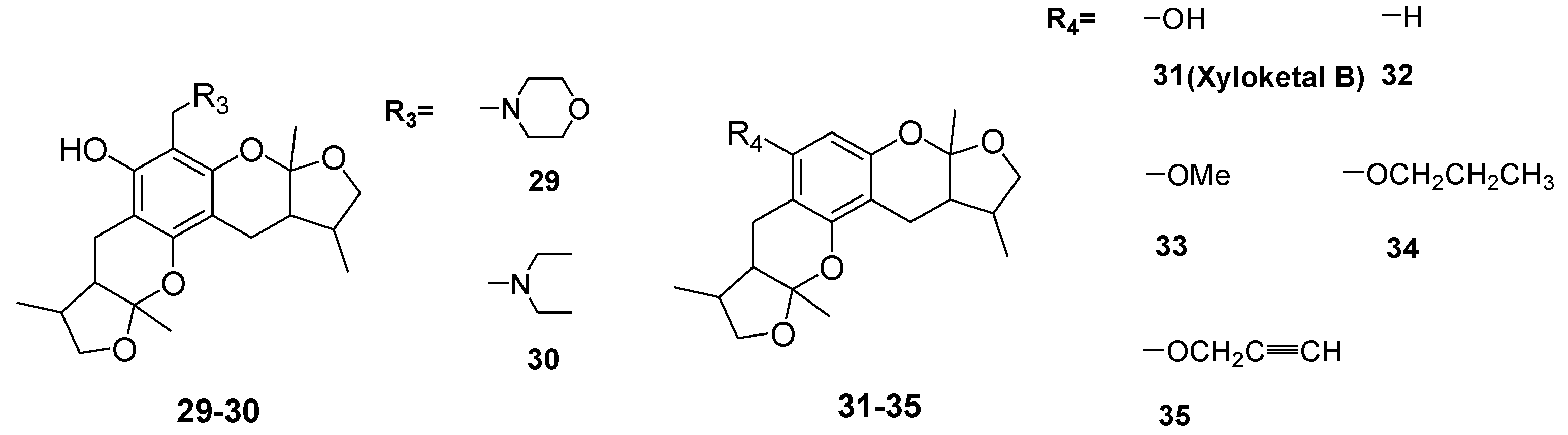
In this work, we designed and synthesized a series of amide derivatives (1–13), benzoxazine derivatives (16–28) and amino derivatives (29–30) from xyloketal B. All 28 new derivatives and seven known compounds (14, 15, 31–35) were evaluated for their protection against H2O2-induced HUVEC injury. 23 and 24 exhibited more potential protective activities than other derivatives; and the EC50 values of them and the leading compound 31 (xyloketal B) were 5.10, 3.59 and 15.97 μM, respectively. Meanwhile, a comparative molecular similarity indices analysis (CoMSIA) was constructed to explain the structural activity relationship of these xyloketal derivatives. This 3D QSAR model from CoMSIA suggested that the derived model exhibited good predictive ability in the external test-set validation. Derivative 24 fit well with the COMSIA map, therefore it possessed the highest activity of all compounds. Compounds 23, 24 and 31 (xyloketal B) were further to examine in the JC-1 mitochondrial membrane potential (MMP) assay of HUVECs using flow cytometry (FCM). The result indicated that 23 and 24 significantly inhibited H2O2-induced decrease of the cell mitochondrial membrane potential (ΔΨm) at 25 μM. Collectively, the protective effects of xyloketals on H2O2-induced endothelial cells may be generated from oxidation action by restraining ROS and reducing the MMP.
1. Introduction
Cardiovascular disease (CVD) has drawn significant attention in recent years because it has become the leading cause of mortality worldwide, affecting people from every income level. Reactive oxygen species (ROS), including H2O2, OH−, NO, and ONOO−, play key roles in the pathogenesis of many CVDs, such as hypertension and atherosclerosis (As). The ROS-induced oxidative stress in cardiac and vascular is closely connected with the endothelial dysfunction in disease initiation and progression. Reactive oxygen species (ROS) are generated under pathological conditions, such as ischemia-reperfusion and inflammation, and activate pro-apoptotic and anti-apoptotic signaling programs in endothelial cells [1]. As one of the most common ROS, hydrogen peroxide (H2O2) can easily cross the plasma membrane, produce a highly reactive radical OH·, and lead to cell and tissue damage [2,3]. The generation of H2O2 plays a key role in the atherosclerotic progression. H2O2 mediates various cellular responses. Direct or indirect stimulation by H2O2 due to its intracellular production could activate various cellular pathways, including calcium release, protein tyrosine kinase, mitogen-activated protein kinases (MAPKs), transcription factor NF-κB, and the induction of cell apoptosis [4,5,6,7]. Thus, H2O2 has been extensively used as an oxidative stimulus to induce oxidative stress in in vitro models. As the major type of endothelial cells, human umbilical vein endothelial cells (HUVECs) are commonly accepted as a model cell to explore the mechanisms involved in the pathogenesis of CVDs [8].
Mitochondrion serve as a pivotal decision center in many types of apoptotic response: they release a variety of death-promoting factors from their inter-membrane spaces into the cytosol, triggering an increase in mitochondria permeability and leading to consequences of mitochondrial dysfunction (e.g., disruption of the mitochondrial membrane potential ΔΨm) [9,10]. Mitochondria are considered the main source of ROS in the cell. Unless adequately detoxified, superoxide causes mitochondrial oxidative stress and may contribute to a decline in mitochondrial function.
Xyloketals are a type of novel compounds that possess unique molecular structures. They are isolated from the marine mangrove fungus Xylaria sp. (#2508) (Chart 1) [11,12]. We previously demonstrated that xyloketal B has protective action against a variety of pathophysiological stimuli, such as oxLDL, oxygen-glucose deprivation (OGD) and 1-methyl-4-phenylpyridinium (MPP+), in different disease models [13,14,15,16,17,18]. Thus, xyloketal B might be a good candidate for further development as an antioxidant medicine in cardiovascular diseases. However, its clinical development may be difficult due to water insolubility. Structure-activity relationship analyses in previous reports have demonstrated that the characteristic substituted groups at the C-12 or C-13 position of xyloketal B are key functional groups for its antioxidative effect. To improve the solubility and biological activity of xyloketal B, some amino groups can be introduced at the C-12 or C-13 position of this type of structure, and the corresponding acid salts could be prepared in the future. Because of the complexity of the stereoselective synthesis of xyloketals, it is difficult to provide a significant amount of optically pure samples for biological activity evaluation. We decided to begin the studies using racemic xyloketal B. In this paper, we designed and synthesized a new series of derivatives (Chart 2) from xyloketal B, including a series of C-13 xyloketal amide derivatives (1–13); xyloketal benzoxazine derivatives (16–28) using a one-pot reaction of xyloketal B, formaldehyde and different primary amines; and xyloketal amino derivatives (29–30) that C-13 substituted using different secondary amines. All 28 new derivatives and 7 known compounds (14, 15, 31–35) were evaluated for their protection against H2O2-induced HUVEC injury. Then, a comparative molecular similarity indices analysis (CoMSIA) was constructed using the SYBYL programming package (version 7.3.5) to explain the structural activity relationship of these xyloketal derivatives [19,20]. The training set and test set were randomly divided out of a total of 35 molecules. A training set of 30 molecules was used to construct the QSAR model, and a training set of five molecules was used to validate it. Mitochondria are considered the main source of reactive oxygen species (ROS) in cells [21,22]. Therefore, we investigated whether xyloketals could protect mitochondria through inhibition of ROS. Any compound with high antioxidative action was further investigated in the JC-1 mitochondrial membrane potential (MMP) assay of HUVECs using flow cytometry (FCM).
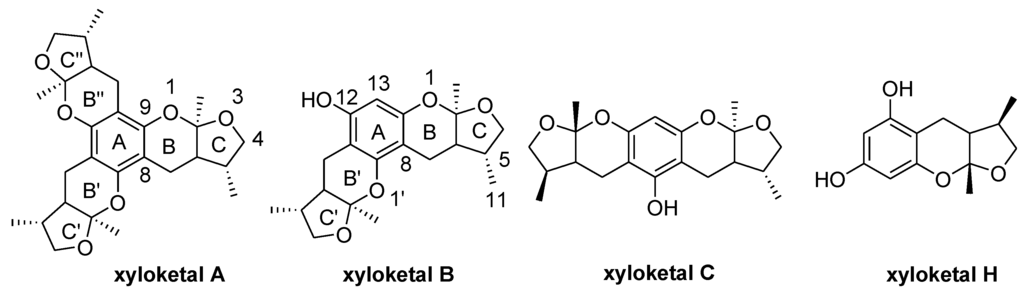
Chart 1.
Structures of xyloketal A, B, C, H.
Chart 1.
Structures of xyloketal A, B, C, H.
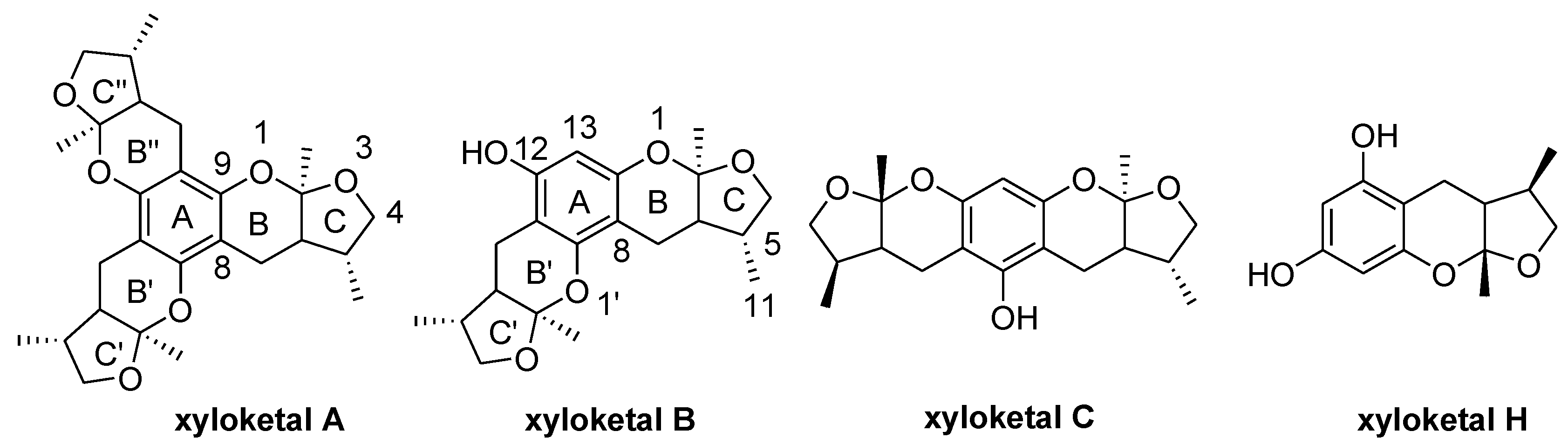
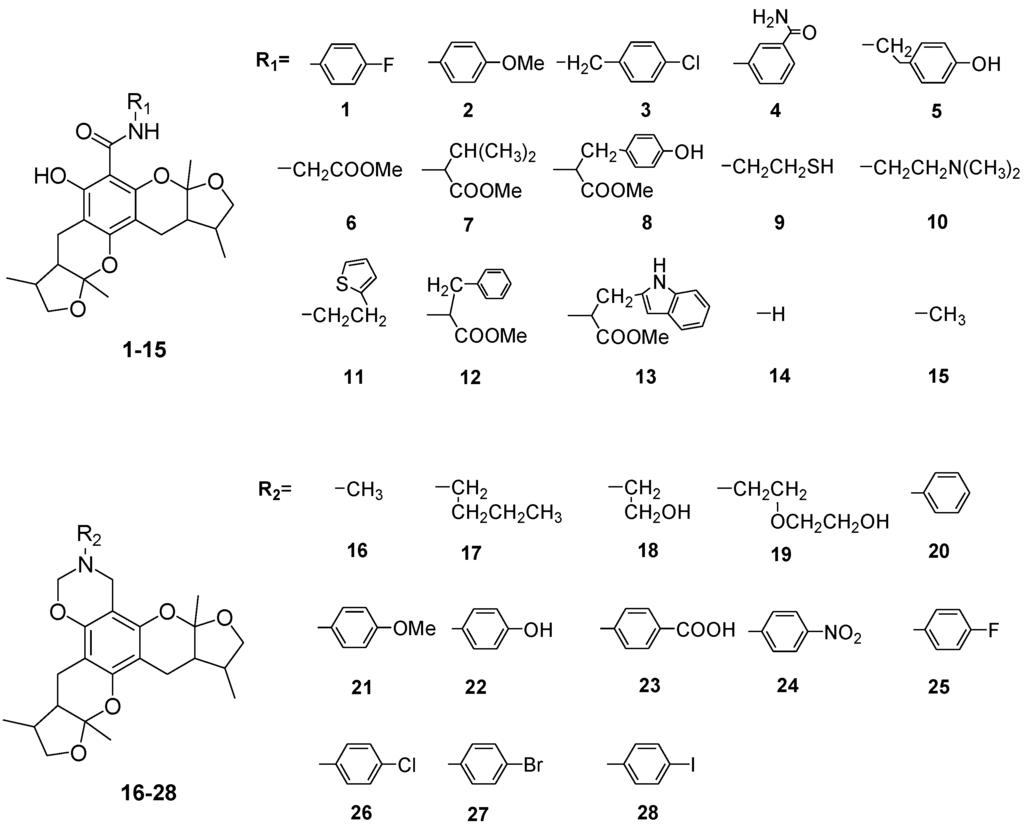
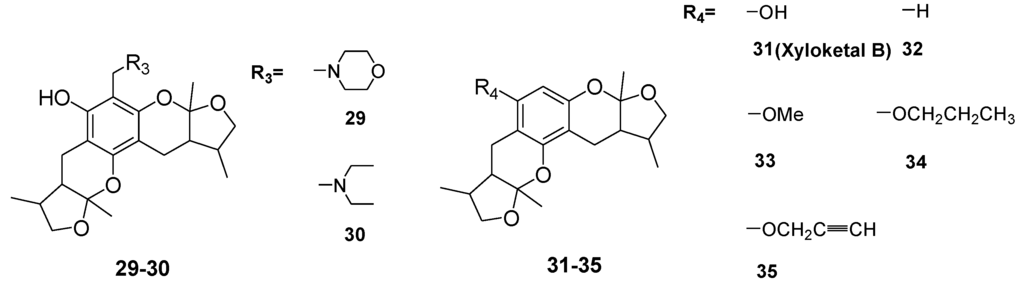
Chart 2.
Structures of xyloketal derivatives 1–35.
Chart 2.
Structures of xyloketal derivatives 1–35.
2. Results and Discussion
2.1. Chemistry
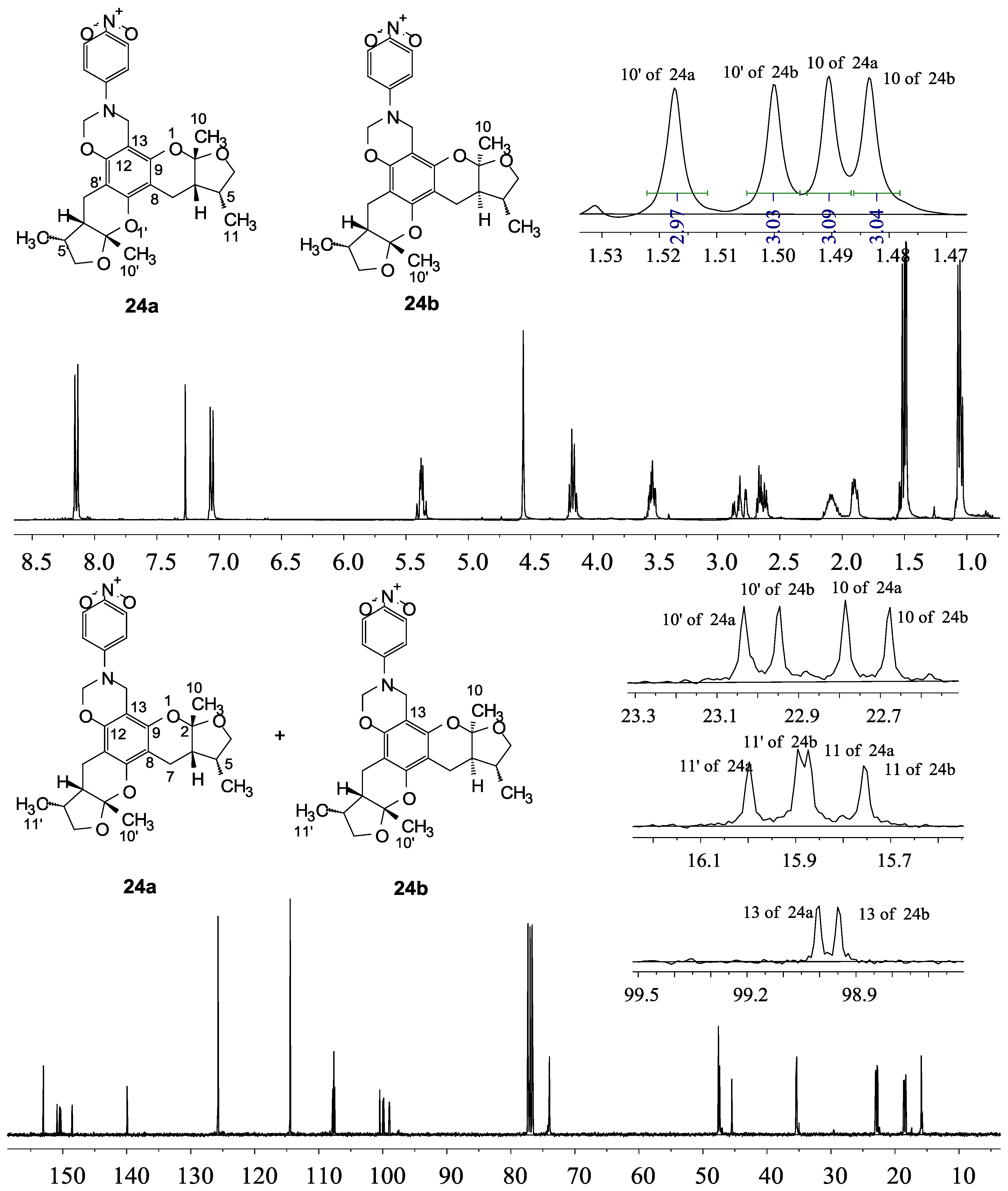
The general synthetic routes of compounds 1–35 are outlined in Scheme 1, Scheme 2 and Scheme 3. All the new compounds were prepared from xyloketal B and xyloketal B acid that were gained from synthetic way in the ordinary state without any asymmetric factors [16]. New xyloketal amides 1–13 were obtained via a condensation reaction between xyloketal B acid and the corresponding amines in the presence of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) and N,N-diisopropylethylamine (DIEA) (Scheme 1). Interestingly, the one-pot Mannich reaction of xyloketal B, formaldehyde and different primary amines afforded a series of novel xyloketal derivatives 16–28 bearing an 1,3-oxazine moiety. Instead of primary amines, the Mannich reaction of secondary amines, xyloketal B and formaldehyde generated C-13 substituted amine derivatives 29–30 (Scheme 3). All the new compounds were fully characterized using MS and NMR. Moreover, all of the examined compounds were synthesized as racemic mixtures from synthetic xyloketal B and xyloketal B acid and no asymmetric synthesis was applied. Their stereo features are the same as xyloketal B and xyloketal B acid. The stereochemistry of these xyloketal derivatives was complicated. In principle, the two oxygen-containing pyran and furan rings B and C can be connected in a cis or trans fashion. The methyl group at C-5 or C-5′ could be cis or trans with respect to the stereogenic centers at the junction at C-2 or C-2′ and C-6 or C-6′. However, previous studies indicated that rings B and C or B′ and C′ were cis for all condensations leading to xyloketal derivatives in the natural and synthetic compounds [18,23,24,25,26,27,28,29], thus only two sets of stereoisomers of xyloketals can be formed: syn, anti and syn, syn types. Moreover, C-2/C-5 methyl in cis orientation occupied dominant position both in experimental and theoretical results [29]. We previously also reported that synthetic xyloketal B and xyloketal B acid were characterized as mixtures of stereoisomers, including the enantiomers and diastereoisomers [16,18,30], and the ratio of two sets of diastereoisomers syn, anti and syn, syn was about 1:1 via NMR analysis. Similar to our previous studies, at this time, racemic mixtures of all new derivatives were consisted of two sets of diastereoisomers (Chart 3, syn, anti a and syn, syn b). Every diastereoisomer had four enantiomer pairs depending on C-5/C-5′ methyl in cis or trans, and the isomer with C-2/C-5 and C-2′/C-5′ methyl all in cis may take greatest proportion in these four enantiomer pairs in according to previous studies. The very close relationship of the diastereoisomers syn, anti and syn, syn was evident in NMR spectra. Though with overlapping of nearly identical sets of signals, two sets of signal peaks could still be detected in 1H and 13C NMR spectra assigned to isomers syn, anti and syn, syn with approximate ratio of ~1:1. Taking 24 as an example, both the 1H and 13C NMR spectra of 24 showed evidence of the diastereoisomers 24a and 24b (Figure 1). Obviously, in 1H NMR, methyl (10 and 10′) at C-2 and C-2′ showed as two peaks respectively (δ = 1.49, 1.48, and 1.52, 1.50 ppm) relative to two single peak (δ = 1.50 and 1.52 ppm) of 10 and 10′ in the natural xyloketal B [11] (Figure 1A), in addition, the integrals of the hydrogen atoms of two peaks indicated an approximately 1: 1 ratio of diastereoisomers 24a and 24b. The 13C NMR spectrum was more instructive (Figure 1B). The methyl (C-10, C-10′) and (C-11, C-11′) both presented as four closely packed peaks (δ = 23.0, 22.9, 22.8, 22.6 and 16.0, 15.9, 15.9, 15.8 ppm). Moreover, the aromatic carbon atom (C-13) also appeared as two peaks (δ = 98.9 and 99.0 ppm). These peaks all proved that compound 24 consisted of two sets of diastereoisomers. However, the enantiomers could not be found by NMR analysis because of their identical NMR spectra. Separating these stereoisomers via chromatography was very difficult. Therefore, all xyloketal derivatives were used directly in the biological screening without separating the stereoisomers this time. These compounds possessed the same structural framework; the only differences were different substituents at the C-12 or C-13 position of the aromatic ring. Although the test compounds are enantiomeric and diastereomeric mixtures, their activities and SAR analysis could be obtained.
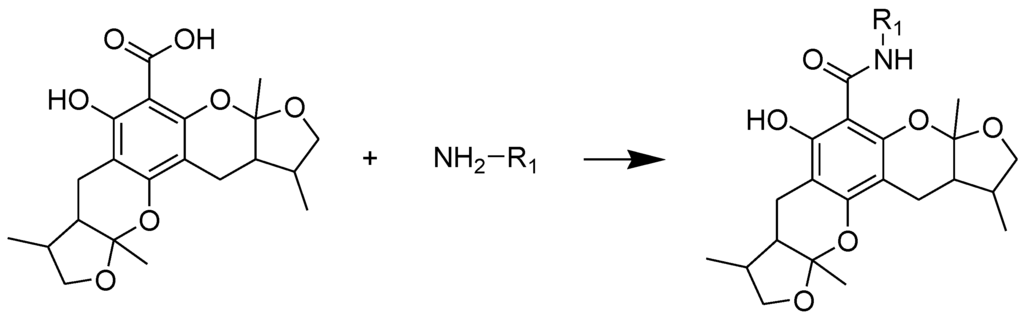
Scheme 1.
Synthesis of compounds 1–13. Reagents and conditions: BOP, DIEA, DMF, room temp.
Scheme 1.
Synthesis of compounds 1–13. Reagents and conditions: BOP, DIEA, DMF, room temp.
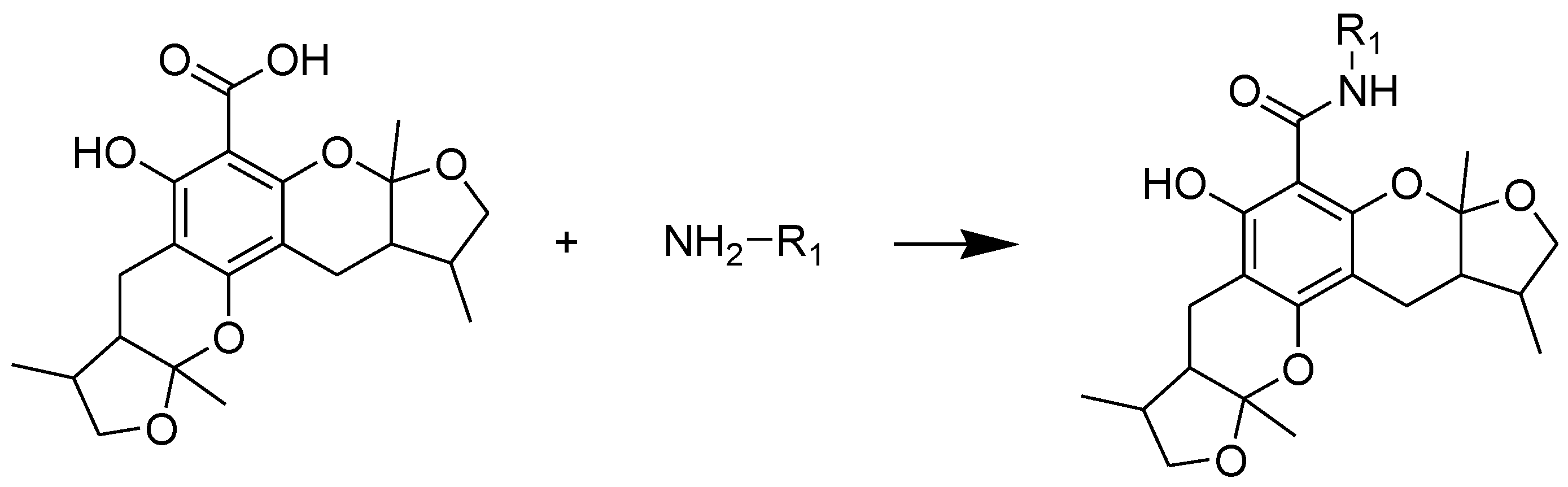
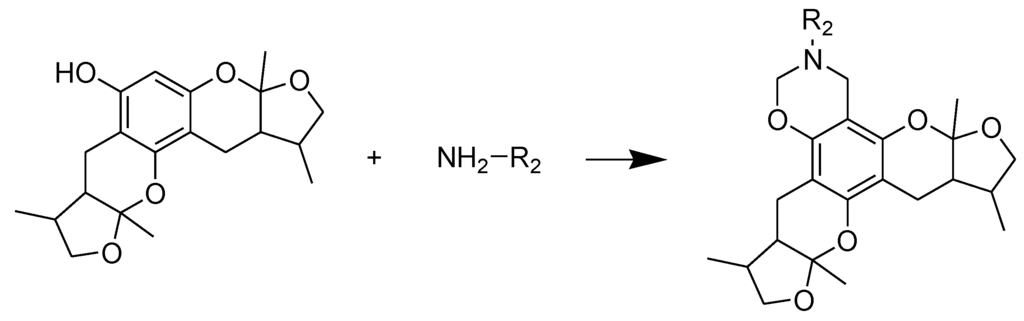
Scheme 2.
Synthesis of compounds 16–28. Reagents and conditions: THF, HCHO, room temp.
Scheme 2.
Synthesis of compounds 16–28. Reagents and conditions: THF, HCHO, room temp.
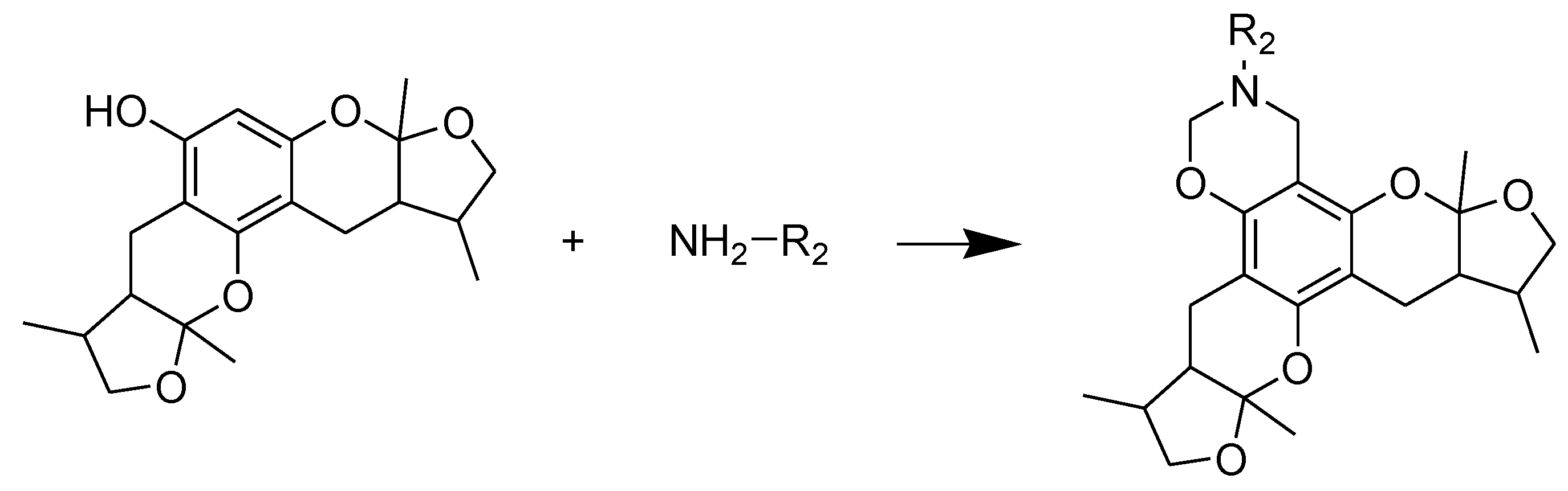
Scheme 3.
Synthesis of compounds 29–30. Reagents and conditions: THF, HCHO, room temp.
Scheme 3.
Synthesis of compounds 29–30. Reagents and conditions: THF, HCHO, room temp.
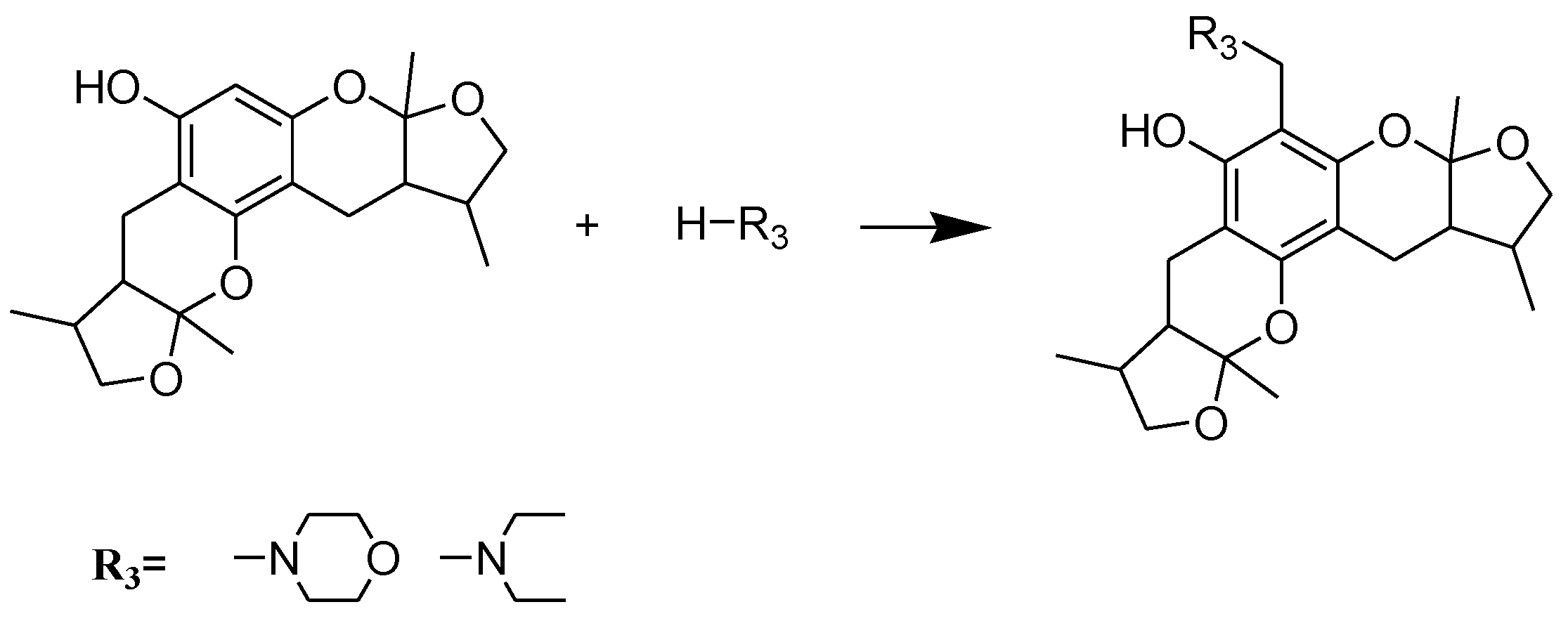
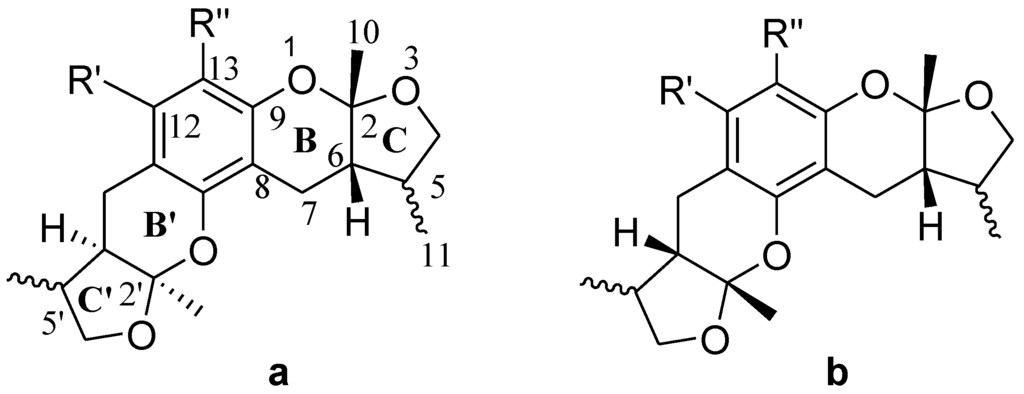
Chart 3.
All stereoisomers of synthesized xyloketal structures.
Chart 3.
All stereoisomers of synthesized xyloketal structures.
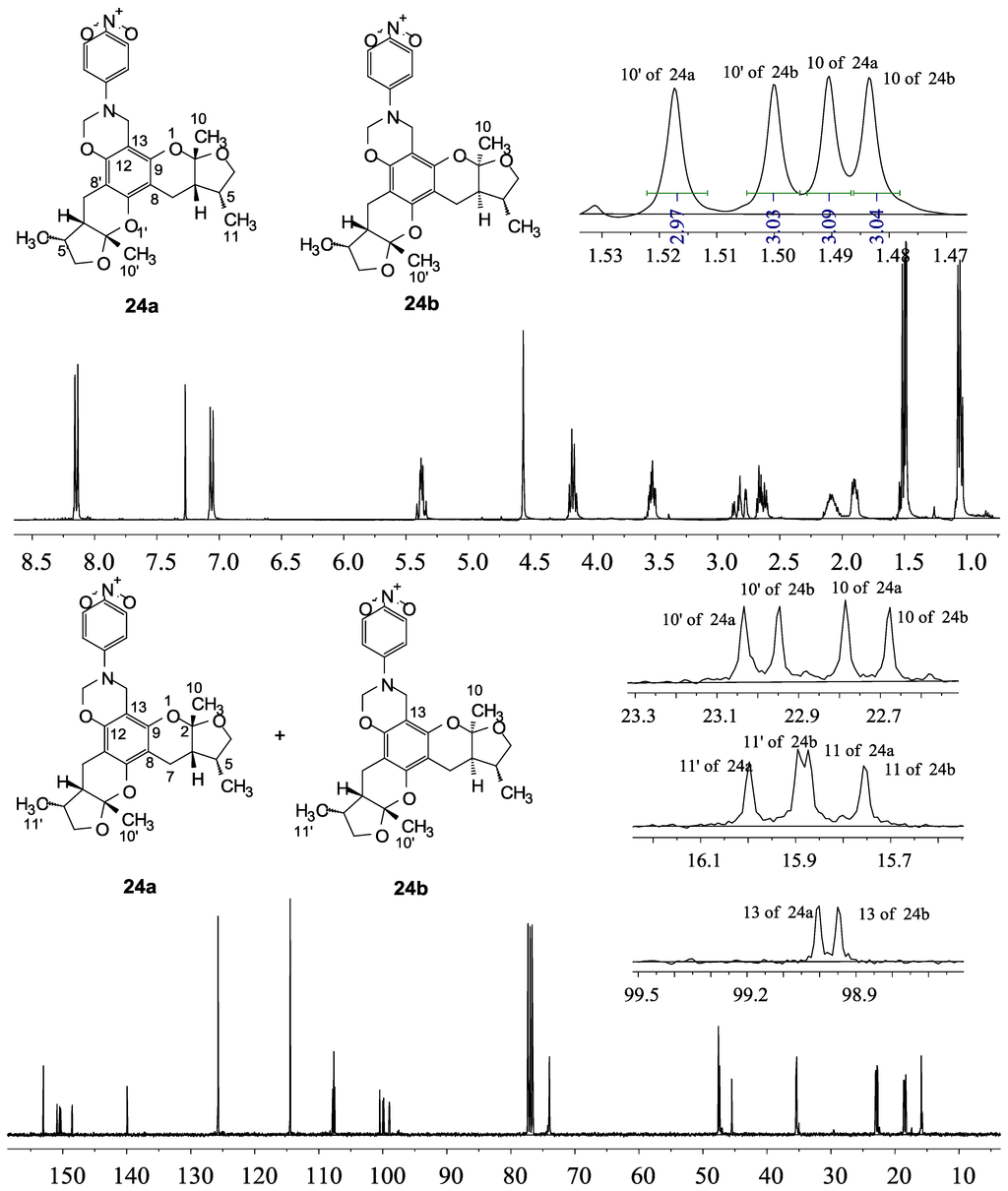
Figure 1.
1H and 13C NMR spectra of 24.
Figure 1.
1H and 13C NMR spectra of 24.
2.2. Xyloketal Derivatives Protected Endothelial Cells against H2O2-Induced Injury Assay
The apoptosis of HUVECs caused by ROS has been implicated in numerous pathophysiological processes of CVDs. An important source of endogenous ROS is generated from H2O2, and it has been proven that ROS are involved in the apoptosis of ECs [31,32]. Using a similar culture system, we have shown that xyloketals had no significant effects on cell viability up to 100 μM in the MTT assay. Accordingly, the protection of xyloketals 1–35 at concentrations of 1 and 10 μM was applied for the following H2O2 (600 μM)-induced injury of HUVECs, and apocynin (1 and 10 μM) was used as a positive control. The results (Table 1, Figure 2) showed that some compounds exhibited strong antioxidative activities, in both morphological changes and inhibition of cell apoptosis. Among them, benzo-1,3-oxazine xyloketal derivatives 23 and 24 displayed greater potential protective activities than other derivatives with cell viabilities 83.07% and 86.08%. Furthermore, to evaluate the activities of these two most significant compounds clearly, the EC50 values in HUVECs ranged from 1–50 μM of 23, 24 and the leading compound xyloketal B (31) were determined with 5.10, 3.59 and 15.97 μM, respectively (Table 2). Thus, new candidates with amino groups, which could be prepared the corresponding acid salts in the future to improve their water-insolubility, will be the promising compounds for further evaluation in the treatment of cardiovascular diseases.

Table 1.
Protective effects of xyloketal derivatives against H2O2-induced cell injury.
| No. | Cell Viability/% of Control | No. | Cell Viability/% of Control | ||
|---|---|---|---|---|---|
| 10 μM | 1 μM | 10 μM | 1 μM | ||
| 1 | 48.41 ± 4.47 | 45.38 ± 3.89 | 19 | 52.94 ± 6.80 | 57.32 ± 4.59 |
| 2 | 54.19 ± 4.11 | 45.57 ± 6.78 | 20 | 46.63 ± 1.55 | 43.77 ± 4.70 |
| 3 | 55.36 ± 7.21 | 49.33 ± 5.34 | 21 | 43.49 ± 5.28 | 54.78 ± 5.16 |
| 4 | 35.49 ± 3.90 | 48.19 ± 4.96 | 22 | 53.23 ± 6.86 | 12.85 ± 2.53 |
| 5 | 6.15 ± 1.29 | 5.96 ± 1.36 | 23 | 83.07 ± 5.01 | 59.07 ± 6.76 |
| 6 | 45.41 ± 5.29 | 50.84 ± 7.46 | 24 | 86.08 ± 4.87 | 49.95 ± 5.92 |
| 7 | 42.28 ± 6.27 | 46.64 ± 4.76 | 25 | 44.20 ± 5.95 | 50.67 ± 7.66 |
| 8 | 23.32 ± 2.22 | 8.29 ± 2.08 | 26 | 30.47 ± 2.19 | 35.97 ± 2.28 |
| 9 | 46.98 ± 4.63 | 48.61 ± 5.84 | 27 | 34.02 ± 4.76 | 39.33 ± 4.00 |
| 10 | 46.84 ± 7.17 | 46.34 ± 6.13 | 28 | 34.79 ± 4.82 | 33.18 ± 3.92 |
| 11 | 51.56 ± 8.03 | 49.66 ± 5.51 | 29 | 67.53 ± 6.68 | 48.30 ± 4.91 |
| 12 | 25.51 ± 3.94 | 23.76 ± 2.24 | 30 | 62.41 ± 7.52 | 46.04 ± 5.92 |
| 13 | 16.31 ± 2.19 | 15.91 ± 2.30 | 31 | 60.43 ± 2.89 | 44.46 ± 2.24 |
| 14 | 59.42 ± 4.76 | 53.14 ± 4.03 | 32 | 62.85 ± 7.96 | 53.00 ± 6.14 |
| 15 | 47.72 ± 5.41 | 54.35 ± 6.44 | 33 | 55.24 ± 5.09 | 48.48 ± 4.13 |
| 16 | 48.40 ± 4.02 | 48.01 ± 5.62 | 34 | 68.50 ± 2.06 | 24.06 ± 2.87 |
| 17 | 49.11 ± 6.50 | 47.62 ± 4.30 | 35 | 71.67 ± 5.28 | 63.16 ± 6.32 |
| 18 | 29.64 ± 3.88 | 30.88 ± 3.59 | Apo-cynin | 69.03 ± 0.68 | 65.48 ± 0.70 |
Datas are representative of means ± S.E.M. n = 6 wells for each group.
Table 2.
The EC50 of 23, 24 and 31 (xyloketal B).
| The EC50 of 23, 24 and 31 (xyloketal B) | |||
| No. | 23 | 24 | 31 (xyloketal B) |
| EC50 a (μM) | 5.10 | 3.59 | 15.97 |
a Each experiment was independently performed six times and expressed as means.
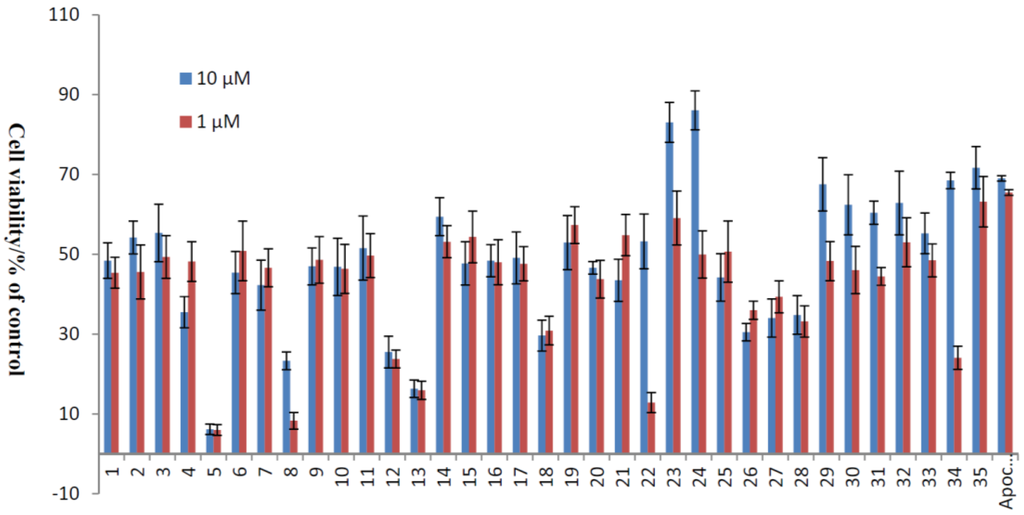
Figure 2.
Protective effects of xyloketal derivatives against H2O2-induced cell injury. HUVECs were pre-incubated with 10 μM xyloketal derivatives (blue bars) or 1 μM xyloketal derivatives (red bars) for 30 min, and 600 μM H2O2 was added to the medium. After incubation for 20 h, cell viability was determined using MTT reduction assay. Apocynin was used as positive control. Values are the mean ± SD (n = 6).
Figure 2.
Protective effects of xyloketal derivatives against H2O2-induced cell injury. HUVECs were pre-incubated with 10 μM xyloketal derivatives (blue bars) or 1 μM xyloketal derivatives (red bars) for 30 min, and 600 μM H2O2 was added to the medium. After incubation for 20 h, cell viability was determined using MTT reduction assay. Apocynin was used as positive control. Values are the mean ± SD (n = 6).
2.3. The Structural Activity Relationship of Xyloketals on a COMSIA Model
To explore the SAR of these xyloketals, a COMSIA model was constructed to explain the structural activity relationship of xyloketal B and its analogs. These compounds had the same structural framework, to unify the evaluation standard; therefore, a dominating stereoisomer of a previously reported xyloketal structure was selected for use in this SAR analysis (Chart 4) [16,18,30]. The statistical parameters of the 3D-QSAR models are shown in Table 3. For an acceptable standard of a 3D-QSAR model, the q2 training (cross-validated regression coefficient) of the training set should be greater than 0.5, and the r2 training (conventional regression coefficient) should be greater than 0.9. The LOO PLS analysis of the model gives a q2 value of 0.577 at six components, together with the conventional regression coefficient r2 of 0.988 and a standard error of estimate of 0.041.
Table 3.
Statistical parameters of the CoMSIA models.
| Training Set | |
|---|---|
| q2 | 0.577 |
| r2 | 0.988 |
| SEE a | 0.041 |
| F b | 316.828 |
| Optimal components | 6 |
| Test set | |
| qtest2 | 0.648 |
| rtest2 | 0.858 |
| k | 0.987 |
a Standard error of estimate; b F-test value.
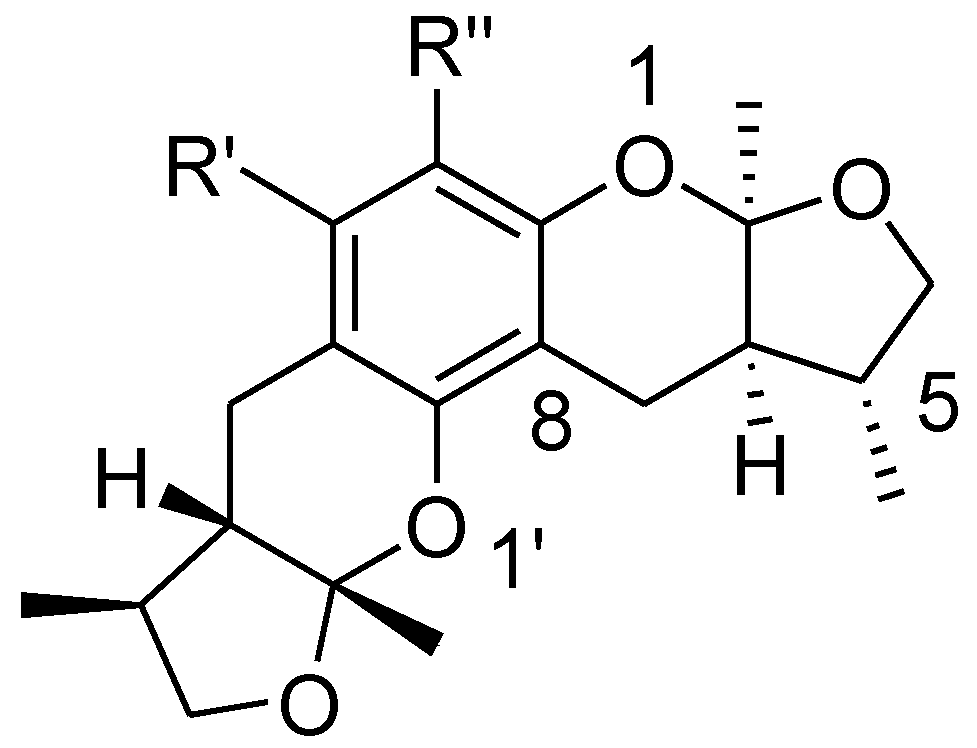
Chart 4.
A stereoisomer of synthesized xyloketal structure used to 3D SAR analysis.
Chart 4.
A stereoisomer of synthesized xyloketal structure used to 3D SAR analysis.
Furthermore, the significance and predictability of QSAR models should be further proven by an external test set using the following criteria: q2 > 0.5, r2 > 0.6 and 0.85 < k < 1.15 (k refers to the slope of the regression line between the experimental and the predicted biological activities). A graphical representation of the predicted and actual values is displayed in Figure 3. An excellent correlation between the experimental and predicted biological activities is shown in Figure 3 for the test set (q2 = 0.648, r2 = 0.858, and k = 0.987). In summary, all statistical data satisfied the recommended criteria, suggesting that the derived model exhibits good predictive ability in the external test-set validation. The predicted and actual values are shown for comparison in Table 4.
Table 4.
Structures, experimental and predicted values of the xyloketal derivatives.
| No. | Cell Viability/% of Control | Actual Value | Predicted Value | Residual Value |
|---|---|---|---|---|
| 1 | 48.41 ± 4.47 | 4.972 | 4.984 | −0.012 |
| 2 | 54.19 ± 4.11 | 5.026 | 5.029 | −0.003 |
| 3 | 55.36 ± 7.21 | 4.901 | 4.900 | 0.001 |
| 4 * | 35.49 ± 3.90 | 4.740 | 4.772 | −0.032 |
| 5 | 6.15 ± 1.29 | 4.264 | 4.276 | −0.012 |
| 6 * | 45.41 ± 5.29 | 4.935 | 5.101 | −0.166 |
| 7 | 42.28 ± 6.27 | 4.865 | 4.811 | 0.054 |
| 8 * | 23.32 ± 2.22 | 4.515 | 4.476 | 0.039 |
| 9 | 46.98 ± 4.63 | 4.947 | 4.965 | −0.018 |
| 10 | 46.84 ± 7.17 | 5.103 | 5.109 | 0.006 |
| 11 | 51.56 ± 8.03 | 4.857 | 4.886 | −0.029 |
| 12 | 25.51 ± 3.94 | 4.534 | 4.553 | −0.019 |
| 13 | 16.31 ± 2.19 | 4.290 | 4.288 | 0.002 |
| 14 * | 59.42 ± 4.76 | 5.222 | 5.038 | 0.184 |
| 15 | 47.72 ± 5.41 | 5.060 | 5.026 | 0.034 |
| 16 | 48.40 ± 4.02 | 4.972 | 5.034 | −0.062 |
| 17 | 49.11 ± 6.50 | 4.985 | 4.956 | 0.029 |
| 18 | 29.64 ± 3.88 | 4.475 | 4.454 | 0.021 |
| 19 | 52.94 ± 6.80 | 5.051 | 5.061 | −0.010 |
| 20 | 46.63 ± 1.55 | 4.941 | 4.957 | −0.016 |
| 21 | 43.49 ± 5.28 | 5.025 | 4.972 | 0.053 |
| 22 | 53.23 ± 6.86 | 4.951 | 4.976 | −0.025 |
| 23 | 83.07 ± 5.01 | 5.658 | 5.674 | −0.016 |
| 24 | 86.08 ± 4.87 | 5.801 | 5.811 | −0.010 |
| 25 | 44.20 ± 5.95 | 5.016 | 4.948 | 0.068 |
| 26 | 30.47 ± 2.19 | 4.784 | 4.889 | −0.105 |
| 27 | 34.02 ± 4.76 | 4.867 | 4.845 | 0.022 |
| 28 | 34.79 ± 4.82 | 4.781 | 4.770 | 0.011 |
| 29 | 67.53 ± 6.68 | 5.430 | 5.363 | 0.067 |
| 30 | 62.41 ± 7.52 | 5.042 | 5.054 | −0.012 |
| 31 | 60.43 ± 2.89 | 5.181 | 5.204 | −0.023 |
| 32 | 62.85 ± 7.96 | 5.311 | 5.280 | 0.031 |
| 33 | 55.24 ± 5.09 | 5.246 | 5.281 | −0.035 |
| 34 * | 68.50 ± 2.06 | 5.337 | 5.286 | 0.051 |
| 35 | 71.67 ± 5.28 | 5.314 | 5.293 | 0.021 |
* Molecules in the test set.
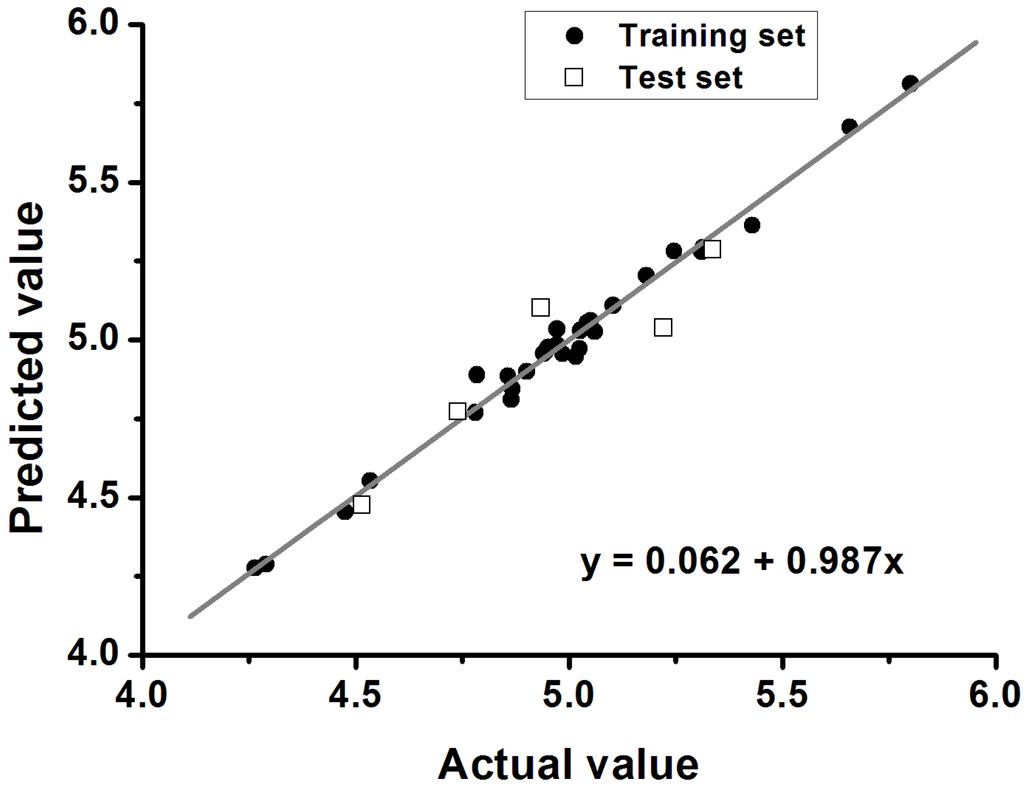
Figure 3.
Plot of predicted versus experimental values of the 3D-QSAR CoMSIA model.
Figure 3.
Plot of predicted versus experimental values of the 3D-QSAR CoMSIA model.
To determine how to modify the structure of xyloketal B, we built a model using a series of derivatives to explain the structure-activity relationship. Figure 4 shows a contour map of each field in the presence of xyloketal B. These maps indicated the favorable and unfavorable modification of the compounds in the colored regions. They are (a) an electrostatic map highlighting the regions where electropositive components were favorable (shown in blue) and unfavorable (shown in red) for the activity; (b) a hydrophobic map highlighting the regions where hydrophobic components were favorable (shown in yellow) and unfavorable (shown in white) for the activity; (c) a hydrogen donor map highlighting the regions where hydrogen donor components were favorable (shown in cyan) and unfavorable (shown in purple) for the activity; and (d) a hydrogen acceptor map highlighting the regions where hydrogen donor components were favorable (shown in magenta) and unfavorable (shown in green) for the activity.
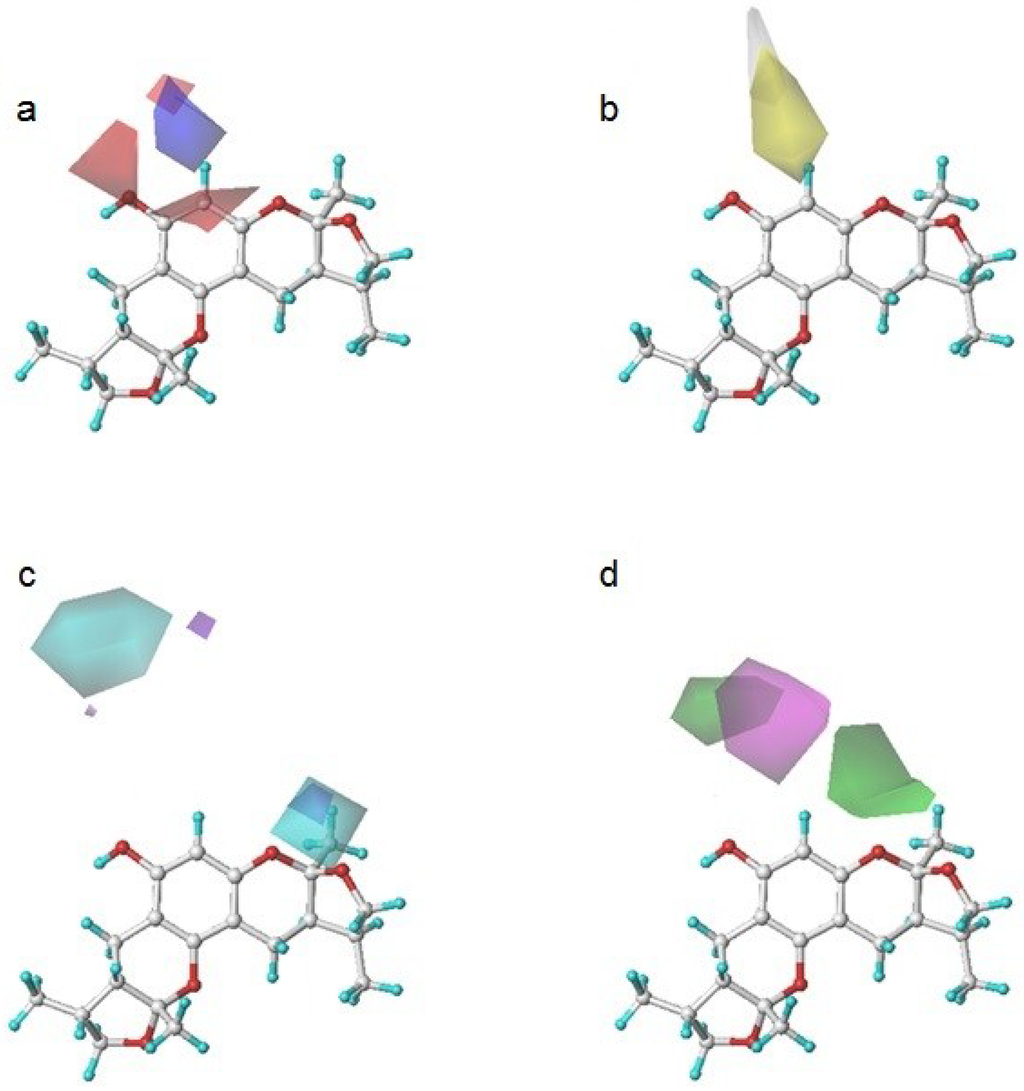
Figure 4.
Xyloketal B in CoMSIA contour maps. (a) an electrostatic map: blue and red contour referred to regions where electropositive substituents were favorable and unfavorable for the compound activity; (b) a hydrophobic map: yellow and white contour referred to regions where hydrophobic substituents were favorable and unfavorable for the compound activity; (c) a hydrogen donor map: cyan and purple contour referred to regions where hydrogen donor substituents were favorable and unfavorable for the compound activity; (d) a hydrogen acceptor map: magenta and green contour referred to regions where hydrogen acceptor substituents were favorable and unfavorable for the compound activity.
Figure 4.
Xyloketal B in CoMSIA contour maps. (a) an electrostatic map: blue and red contour referred to regions where electropositive substituents were favorable and unfavorable for the compound activity; (b) a hydrophobic map: yellow and white contour referred to regions where hydrophobic substituents were favorable and unfavorable for the compound activity; (c) a hydrogen donor map: cyan and purple contour referred to regions where hydrogen donor substituents were favorable and unfavorable for the compound activity; (d) a hydrogen acceptor map: magenta and green contour referred to regions where hydrogen acceptor substituents were favorable and unfavorable for the compound activity.
According to their structural discrepancy, all compounds can be cataloged into three groups. Their common structures are shown in Chart 5. Group A consisted of compounds 1–15 with activities ranging from 15.52 to 55.89. Group B consisted of compounds 16–28, which had activities ranging from 22.97 to 86.34. It contained a unique 6-member ring with different substitution groups. Group C consisted of compounds 29–35. No drastic modifications were made to these compounds. Therefore, their activities ranged from 52.41 to 72.89, similar to that of 31-xyloketal B (60.29).
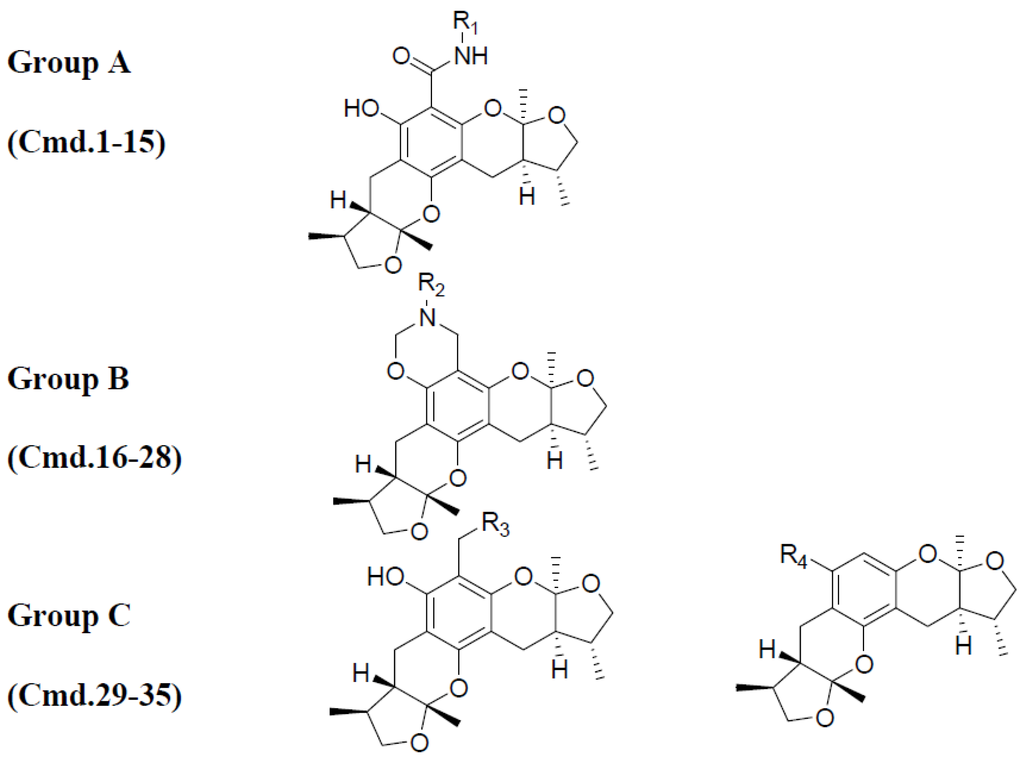
Chart 5.
All 35 molecules were divided into three groups based on their structural similarities.
Chart 5.
All 35 molecules were divided into three groups based on their structural similarities.
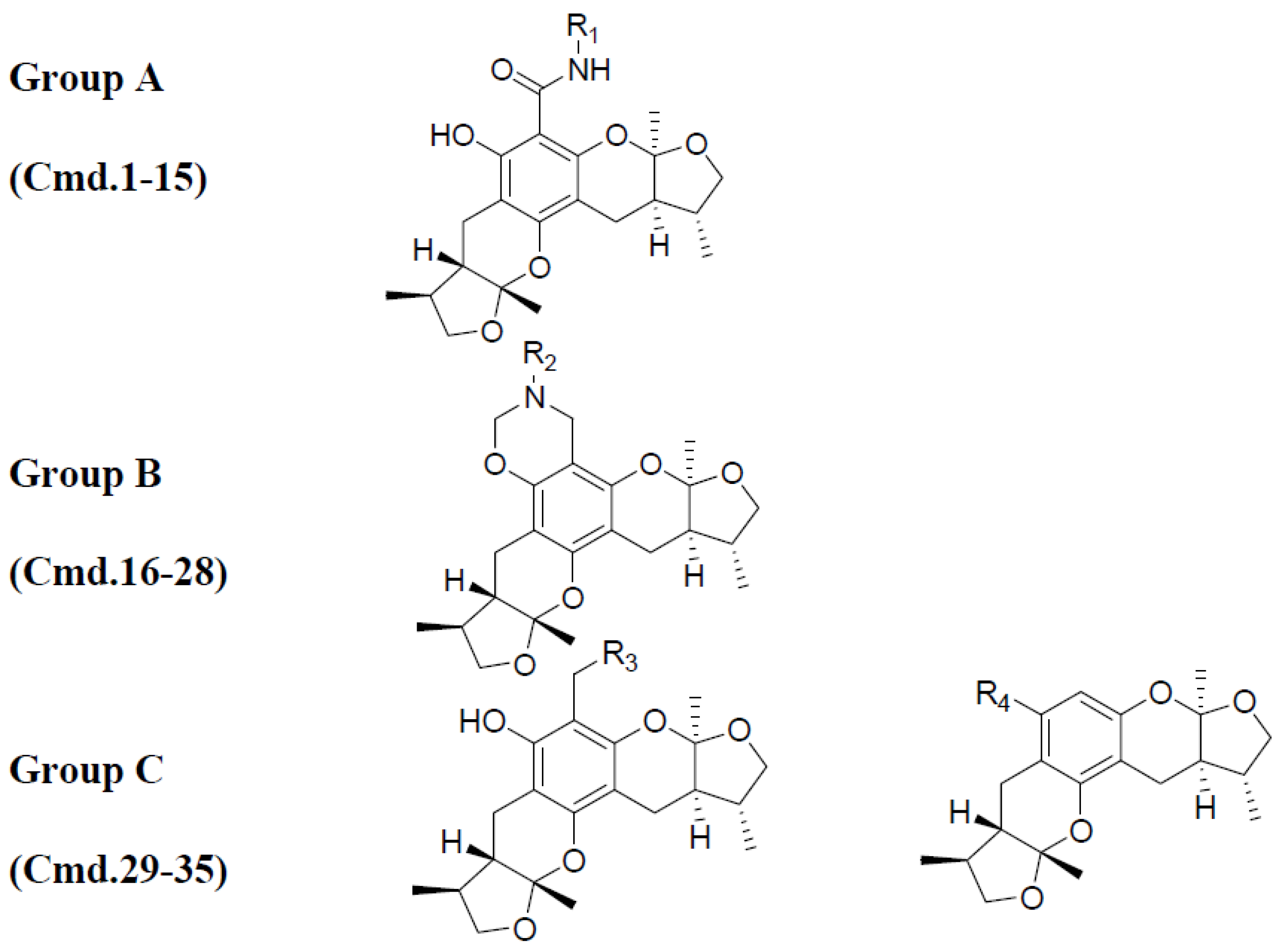
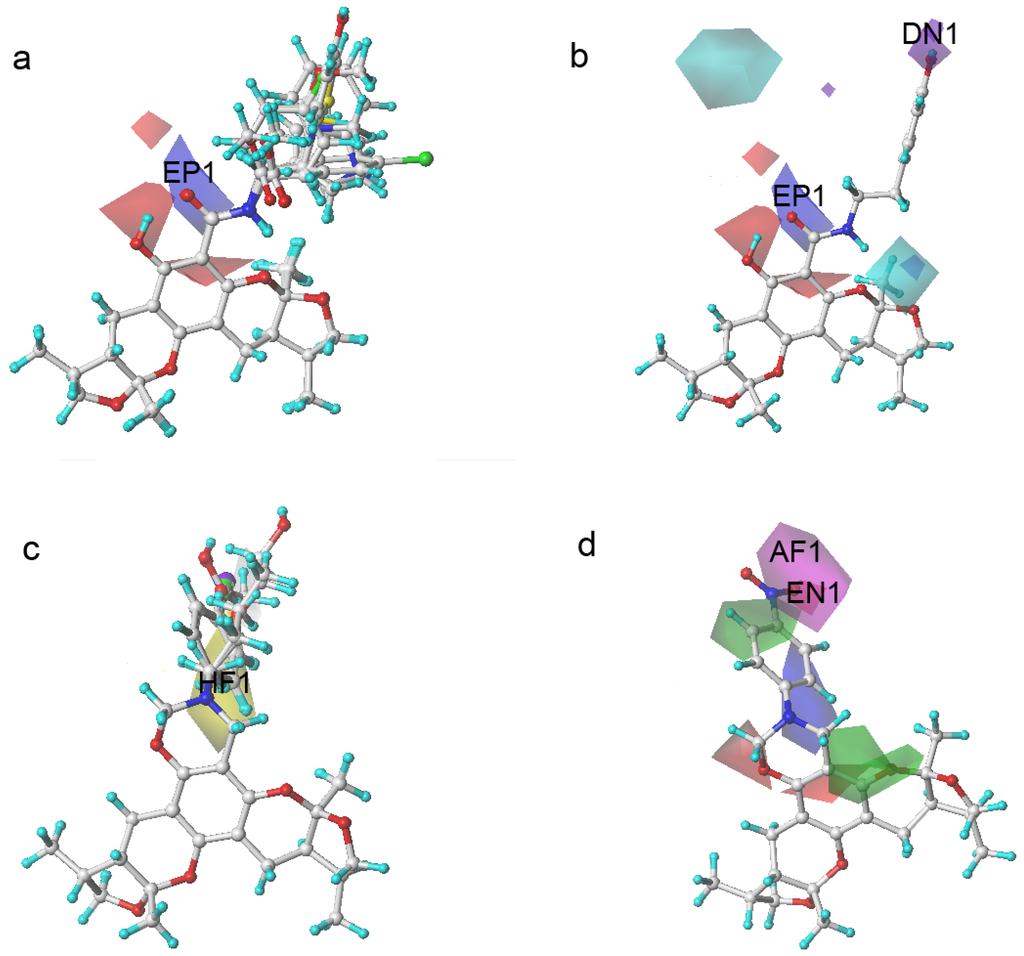
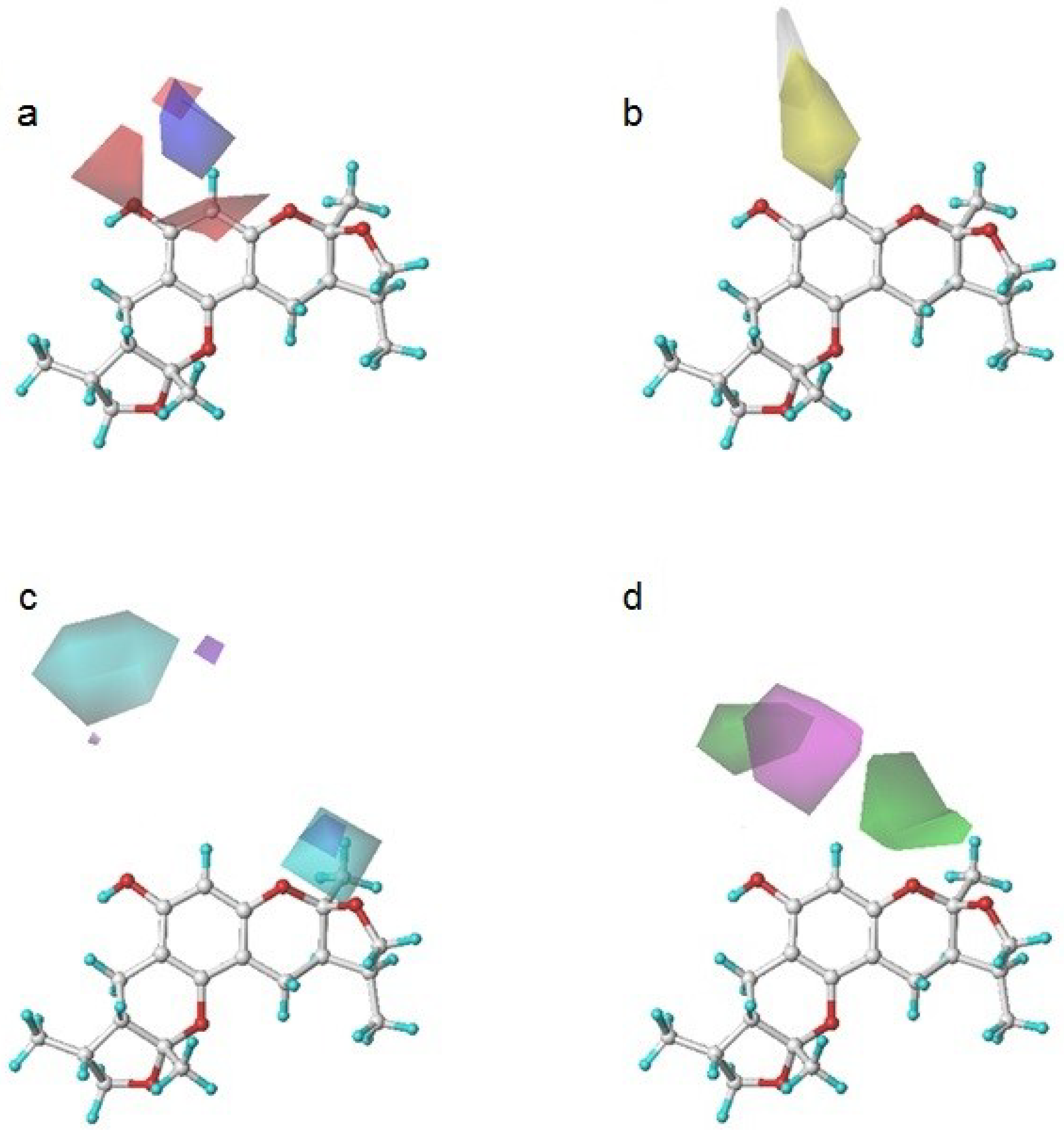
Figure 5.
Xyloketal derivatives in CoMSIA contour maps. (a) compounds of Group A overlay with electrostatic maps; (b) compound 5 (cell viability % = 15.52) with electrostatic and hydrogen donor maps; (c) compounds of Group B overlay with hydrophobic maps; (d) compound 24 (cell viability % = 86.34) with electrostatic and hydrogen bond acceptor maps.
Figure 5.
Xyloketal derivatives in CoMSIA contour maps. (a) compounds of Group A overlay with electrostatic maps; (b) compound 5 (cell viability % = 15.52) with electrostatic and hydrogen donor maps; (c) compounds of Group B overlay with hydrophobic maps; (d) compound 24 (cell viability % = 86.34) with electrostatic and hydrogen bond acceptor maps.
The compounds in Group A yielded relatively lower activities. This result could be explained by the mismatch of the CoMSIA force field with the substitution groups. As shown in Figure 5a, the overlay of the compounds in group A had a common amide group, and the oxygen fell into the blue area, EP1. As mentioned previously, the blue contour map indicates areas where positively charged components would increase the activity, but negatively charged components would decrease the activity. Take compound 5 as a more specific example; it not only had the aforementioned common amide group in EP1, but also an -OH group in the purple area DN1. Purple areas indicate that a hydrogen bond donor in the area would have a negative effect on the compound activity (Figure 5b). The above analysis justified why compound 5 had the lowest activity. Some of the compounds in Group B had stronger activities. An overlay of the compounds in Group B in Figure 5c revealed that the substitution groups of compounds in group B took a different orientation to fit in the yellow region HF1. Compound 24 extended its nitro group into the magenta area AF1 and the red area EN1, where hydrogen bond acceptor and electro-negative groups would elevate the activity of the compound, respectively. Compound 24 matched the CoMSIA map well (Figure 5d); therefore, it possessed the highest activity of all compounds. In contrast, compounds 25–28 had reduced activities due to replacing the nitro group with halogens, which were not hydrogen bond acceptors. Compounds in Group C produced similar activities to that of xyloketal B because no drastic structural changes were made to these compounds. The substitution groups did not specially fall into regions that increased or decreased the activities.
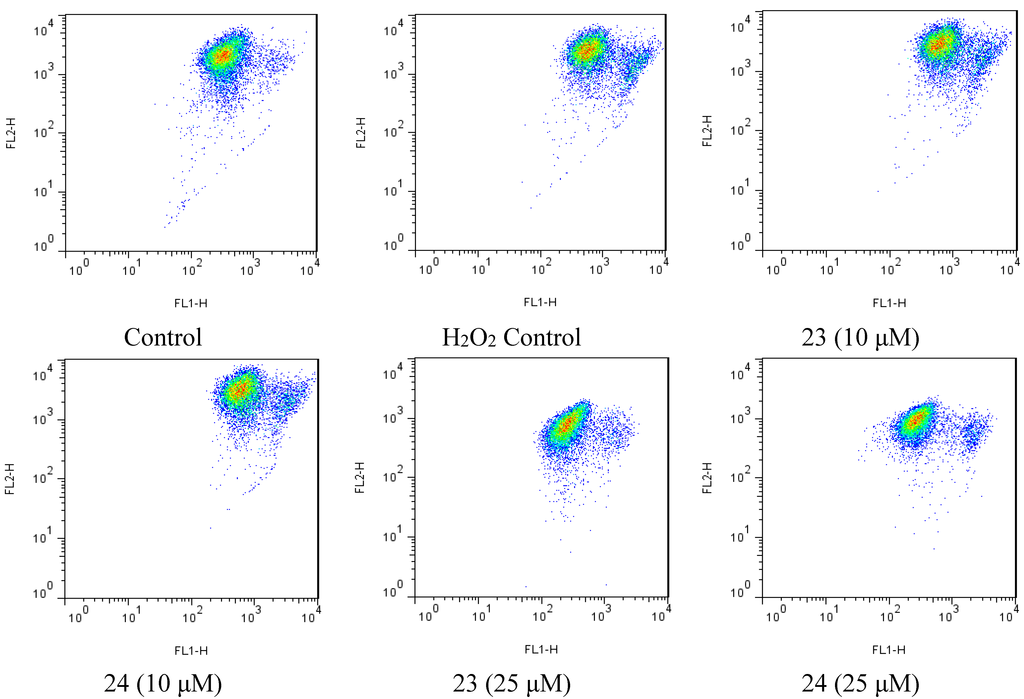
Figure 6.
Flow cytometry scatter plot was analyzed using the Flowjo (v7.6.5), showing JC-1 monomers and aggregates in different group.
Figure 6.
Flow cytometry scatter plot was analyzed using the Flowjo (v7.6.5), showing JC-1 monomers and aggregates in different group.
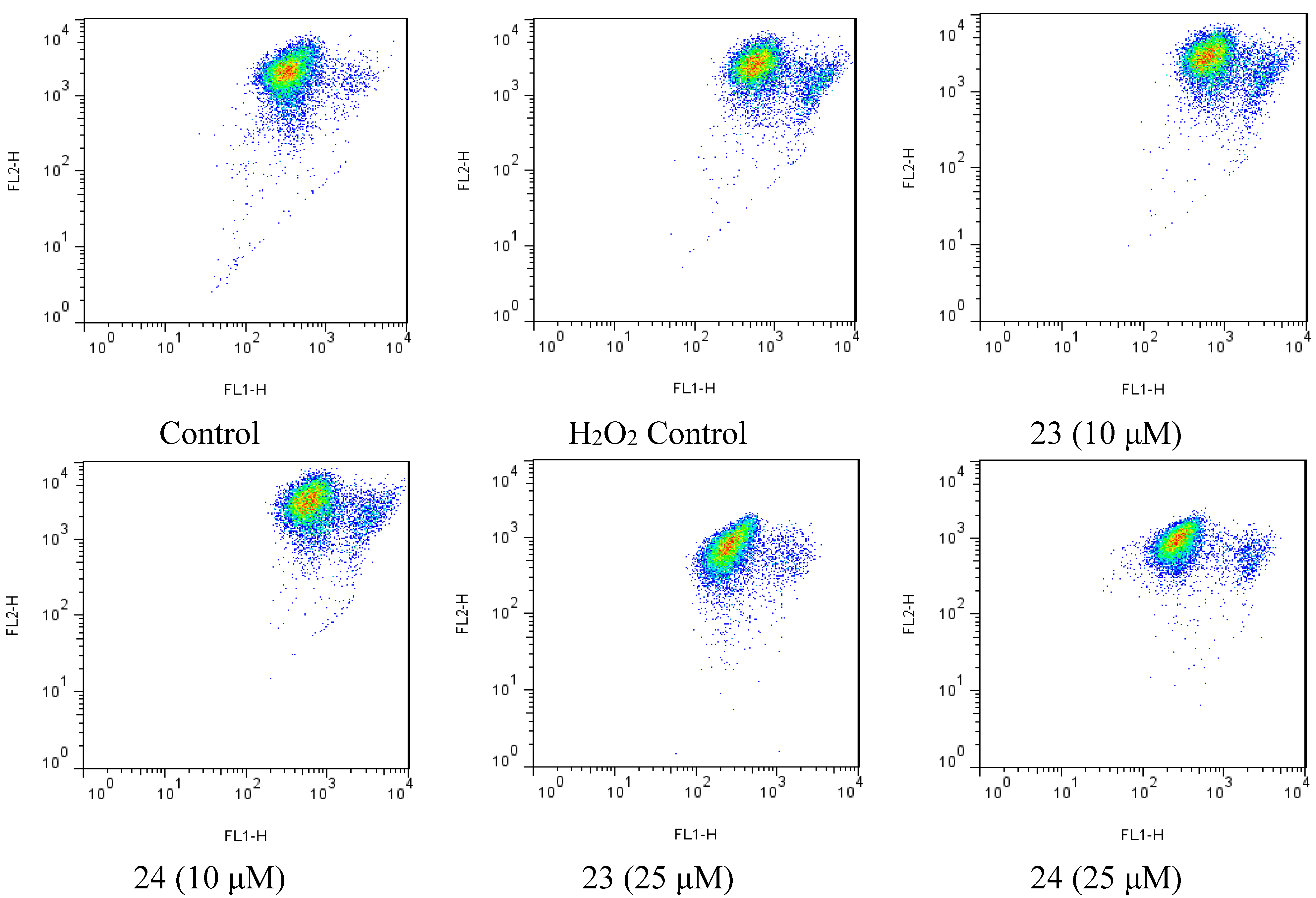
2.4. Xyloketal Derivatives Restored the H2O2-Induced Reduction of the Mitochondrial Membrane Potential (ΔΨm)
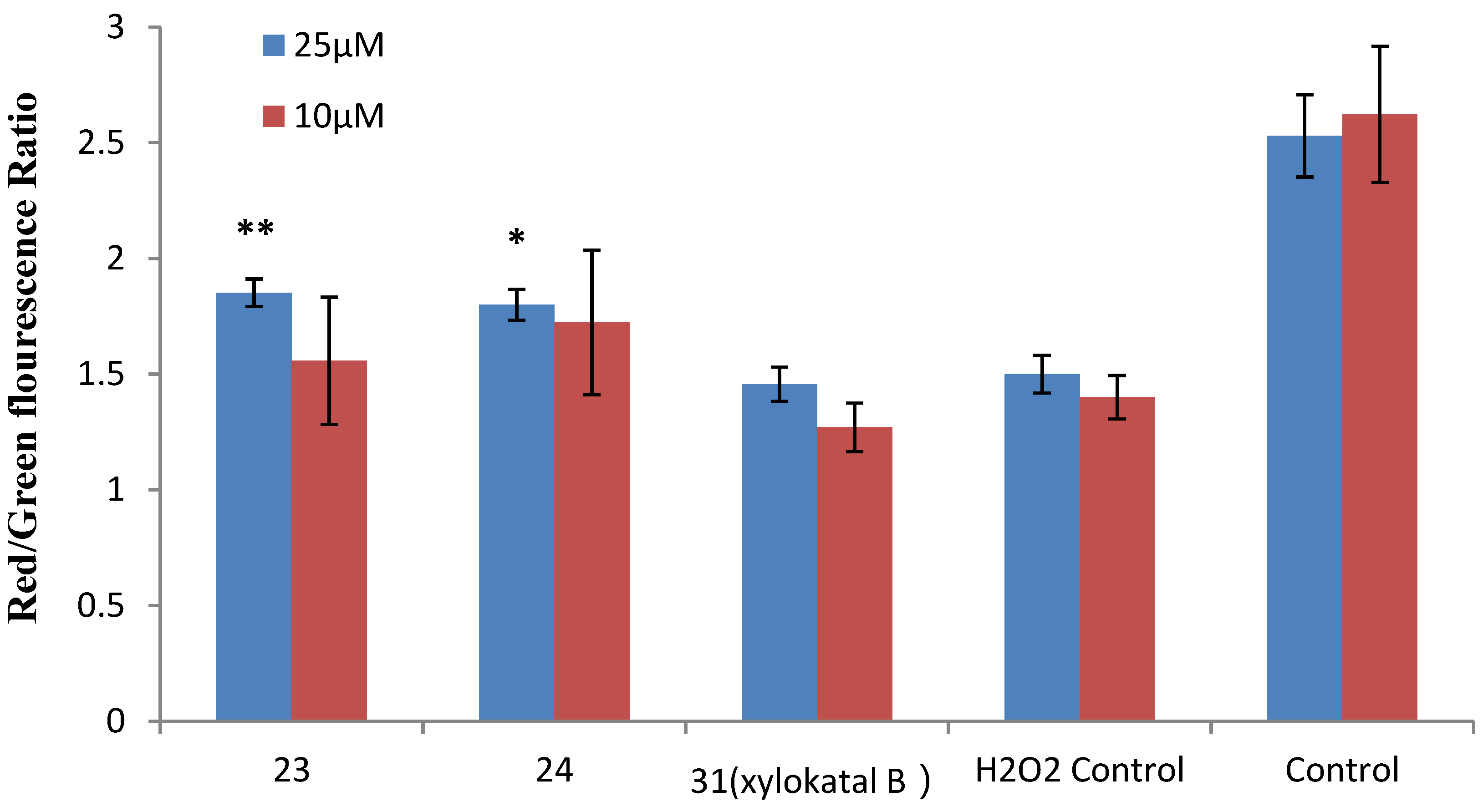
Mitochondria are considered the main source of reactive oxygen species (ROS) in cells. Therefore, we investigated whether xyloketals could protect mitochondria via inhibition of ROS. Compounds 23 and 24 were examined in the JC-1 mitochondrial membrane potential (MMP) assay of HUVECs using flow cytometry (FCM). As shown in Figure 6 and Figure 7, 23 and 24 significantly inhibited the H2O2-induced the decrease in the cell mitochondrial membrane potential (ΔΨm) at 25 μM. Collectively, 23 and 24 effectively protected HUVECs against oxidative damage and further mitochondrial membrane integrity impairment and prevented H2O2-induced apoptosis of HUVECs by regulating the ROS-mediated mitochondrial dysfunction pathway.
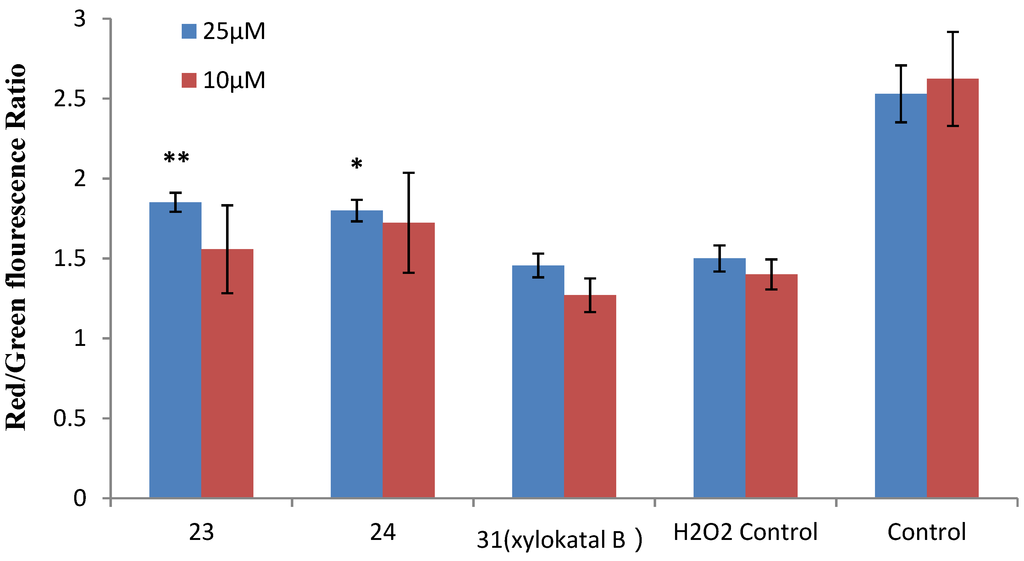
Figure 7.
Effects of xyloketal B and the derivatives 23 and 24 on H2O2-induced decrease of ΔΨm in HUVECs. HUVECs were pre-incubated with 25 μM xyloketal derivatives (blue bars) or 10 μM xyloketal derivatives (red bars) for 30 min, and 400 μM H2O2 was added to the medium. After incubation for 20 h, ΔΨm was determined by FACS analyses. Values are the mean ± SD (n = 6): * p < 0.05, ** p < 0.01 versus H2O2.
Figure 7.
Effects of xyloketal B and the derivatives 23 and 24 on H2O2-induced decrease of ΔΨm in HUVECs. HUVECs were pre-incubated with 25 μM xyloketal derivatives (blue bars) or 10 μM xyloketal derivatives (red bars) for 30 min, and 400 μM H2O2 was added to the medium. After incubation for 20 h, ΔΨm was determined by FACS analyses. Values are the mean ± SD (n = 6): * p < 0.05, ** p < 0.01 versus H2O2.
3. Experimental Section
3.1. Chemistry
All reagents and solvents were of commercial quality and used without further purification. 1H and 13C NMR data were recorded on a Bruker AVANCE 400 MB NMR spectrometer (Bruker, Fallanden, Switzerland) operating at 400 and 101 MHz for 1H and 13C respectively. All chemical shifts are in ppm (δ) with respect to tetramethylsilane (TMS) as internal standard, and coupling constants (J) are in Hz. Mass spectra were obtained on DSQ (low resolution mass spectrometer) (Thermo, San Jose, CA, USA) and MAT95XP (high resolution mass spectrometer) instruments (Thermo, San Jose, CA, USA).
3.2. General Procedure of Synthesizing Compounds
Compound 1. First, 50 mg (0.13 mmol) of xyloketal B acid [16] and 22 mg (0.20 mmol) of p-fluoroaniline were dissolved in 10 mL of DMF in a 50 mL-round-bottom flask. Then, 80 mg (0.20 mmol) of BOP and 0.40 mL (2.0 mmol) of DIEA were added and stirred at room temperature overnight. The reaction was quenched with 10 mL saturated solution of ammonium chloride in ice water. The aqueous layer was extracted with ethyl acetate, 3 × 25 mL. The combined organic extracts were washed with saturated ammonium chloride and brine, dried over anhydrous MgSO4 and concentrated in vacuo. Purification by flash chromatography (petroleum ether: ethyl acetate = 5:1~2:1) gave the title compound. Yield 75%; white solid; m. p. 68–69 °C. 1H NMR (400 MHz,CDCl3) δ 13.97 (s, 1H), 10.59 (s,1H), 7.57 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.4 Hz, 2H), 4.22–4.15 (m, 2H), 3.53–3.58 (m, 2H), 2.96–2.86 (m, 2H), 2.73–2.58(m, 2H), 2.14–2.08 (m, 2H), 2.02–1.93 (m, 2H), 1.61 (s, 3H), 1.53 (s, 3H), 1.09 (d, J = 6.0 Hz, 3H), 1.07 (d, J = 6.0 Hz, 3H). 13CNMR (101 MHz, CDCl3) δ 168.78. 161.96, 160.64, 158.22, 155.31, 150.93, 134.00, 122.44, 115.52, 110.57, 110.31, 108.43, 99.96, 98.40, 96.85, 74.63, 74.25, 47.84, 47.29, 35.68, 35.27, 23.51, 23.02, 18.61, 18.31, 15.89, 15.87. HR-EI-MS m/z 483.2057; calculated for C27H30FNO6: 483.2057.
Compound 2. The title compound was obtained from xyloketal B acid and p-anisidine with a procedure similar to that for compound 1. Yield 73%; white solid; m. p. 70–71 °C. 1H NMR (400 MHz, CDCl3) δ 14.15 (s, 1H), 10.49 (s, 1H), 7.51 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 4.18 (dd, J = 8.4 Hz, 2H), 3.81 (s, 3H), 3.56 (dd, J = 8.4 Hz, 2H), 2.96–2.85 (m, 2H), 2.72–2.58 (m, 2H), 2.16–2.08 (m, 2H), 2.00–1.93 (m, 2H), 1.62 (s, 3H), 1.52 (s, 3H), 1.08 (d, J = 6.4 Hz, 3H), 1.07 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.62, 161.91, 156.51, 155.08, 151.00, 131.05, 122.56, 114.24, 110.42, 110.17, 108.50, 108.36, 99.89, 98.53, 97.03, 74.55, 74.24, 55.54, 47.77, 47.32, 35.77, 35.28, 23.14, 22.79, 18.75, 18.34, 16.05, 15.91. HR-EI-MS m/z 495.2251; calculated for C28H33NO7: 495.2257.
Compound 3. The title compound was obtained from xyloketal B acid and 4-chlorobenzylamine with a procedure similar to that for compound 1. Yield 71%; white solid; m. p. 72–74 °C. 1H NMR (400 MHz, CDCl3) δ 14.18 (s, 1H), 8.93 (s, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 4.59 (d, J = 2.0 Hz, 2H), 4.20–4.04 (m, 2H), 3.56–3.45 (m, 2H), 2.93–2.82 (m, 2H), 2.70–2.55 (m, 2H), 2.10–2.02 (m, 2H), 1.96–1.90 (m, 2H), 1.51 (s, 3H), 1.50 (s, 3H), 1.07 (d, J = 6.4 Hz, 3H), 1.05 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.58, 161.60, 155.01, 154.83, 151.40, 137.06, 132.93, 128.71, 128.54, 110.05, 109.83, 108.47, 99.70, 98.34, 96.68, 74.41, 74.20, 47.78, 47.37, 42.32, 35.72, 35.29, 23.07, 22.78, 18.69, 18.33, 16.04, 15.89. HR-EI-MS m/z 513.1907; calculated for C28H32ClNO6: 513.1918.
Compound 4. The title compound was obtained from xyloketal B acid and m-amino-benzamide with a procedure similar to that for compound 1. Yield 78%; white solid; m. p. 108–109 °C. 1H NMR (400 MHz, CDCl3) δ 13.84 (s, 1H), 10.75 (s, 1H), 8.19 (s, 1H), 7.45 (m, 3H), 5.86 (br, 2H), 4.22–4.15 (m, 2H), 3.59–3.53 (m, 2H), 2.96–2.86 (m, 2H), 2.73–2.60 (m, 2H), 2.15–2.08 (m, 2H), 2.03–1.96 (m, 2H), 1.62 (s, 3H), 1.53 (s, 3H), 1.09 (d, J = 6.8 Hz, 3H), 1.07 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 169.10, 169.05, 162.01, 155.52, 150.98, 138.32, 134.37, 129.29, 124.14, 123.39, 119.47, 110.47, 108.49, 100.00, 98.57, 96.83, 74.67, 74.26, 47.86, 47.29, 35.76, 35.26, 23.23, 23.03, 18.71, 18.37, 16.04, 15.97. HR-EI-MS m/z 508.2201; calculated for C28H32N2O7: 508.2210.
Compound 5. The title compound was obtained from xyloketal B acid and tyramine with a procedure similar to that for compound 1. Yield 72%; white solid; m. p. 97–98 °C. 1H NMR (400 MHz, CDCl3) δ 14.43 (s, 1H), 8.56 (s, 1H), 7.08 (d, J = 8.4 Hz, 2H), 6.76 (d, J = 8.4 Hz, 2H), 6.19 (s, 1H), 4.20–4.04 (m, 2H), 3.66–3.58 (m, 2H), 3.56–3.45 (m, 2H), 2.93–2.77 (m, 4H), 2.69–2.52 (m, 2H), 2.10–2.01 (m, 2H), 1.96–1.84 (m, 2H), 1.50 (s, 3H), 1.44 (s, 3H), 1.06 (d, J = 6.4 Hz,3H), 1.03 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.61, 161.50, 154.68, 154.62, 151.34, 130.72, 129.83, 115.48, 109.76, 109.55, 108.44, 108.32, 99.55, 97.90, 96.72, 74.31, 74.23, 47.62, 47.37, 40.87, 35.46, 35.29, 34.55, 23.02, 22.83, 18.53, 18.35, 15.98, 15.86. HR-EI-MS m/z 509.2404; calculated for C29H35NO7: 509.2414.
Compound 6. The title compound was obtained from xyloketal B acid and glycine methyl ester hydrochloride with a procedure similar to that for compound 1. Yield 78%; white solid; m. p. 75–76 °C. 1H NMR (400 MHz, CDCl3) δ 13.97 (s, 1H), 9.03 (s, 1H), 4.28–4.21 (m, 2H), 4.19–4.11 (m, 2H), 3.78 (s, 3H), 3.56–3.48 (m, 2H), 2.91–2.81 (m, 2H), 2.67–2.56 (m, 2H), 2.15–2.04 (m, 2H), 1.96–1.88 (m, 2H), 1.60 (s, 3H), 1.50 (s, 3H), 1.06 (d, J = 6.4 Hz, 3H), 1.04 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.57, 170.12, 161.48, 154.93, 151.52, 109.75, 108.40, 99.66, 98.28, 96.51, 74.41, 74.17, 52.28, 47.76, 47.27, 41.31, 35.70, 35.22, 22.96, 22.75, 18.60, 18.30, 16.04, 15.85. HR-EI-MS m/z 461.2043; calculated for C24H31NO8: 461.2050.
Compound 7. The title compound was obtained from xyloketal B acid and valine methyl ester hydrochloride with a procedure similar to that for compound 1. Yield 76%; white solid; m. p. 80–81 °C. 1H NMR (400 MHz, CDCl3) δ 14.08 (s, 1H), 9.17 (s, 1H), 4.68–4.62 (m, 1H), 4.25–4.08 (m, 2H), 3.77 (s, 3H), 3.59–3.51 (m, 2H), 2.94–2.86 (m, 2H), 2.70–2.57 (m, 2H), 2.34–2.28 (m, 1H), 2.22–2.07 (m, 2H), 2.01–1.93 (m, 2H), 1.62 (s, 3H), 1.54 (s, 3H), 1.09 (d, J = 6.4 Hz, 3H), 1.07 (d, J = 6.4 Hz, 3H), 1.04 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 172.19, 170.47, 161.57, 154.90, 151.53, 109.73, 108.42, 99.49, 98.22, 96.62, 77.02, 74.37, 74.26, 57.23, 52.04, 47.53, 47.33, 35.27, 30.92, 23.20, 23.01, 22.76, 19.22, 18.66, 18.36, 17.54, 16.23, 15.88. HR-EI-MS m/z 503.2515; calculated for C27H37NO8: 503.2519.
Compound 8. The title compound was obtained from xyloketal B acid and tyrosine methyl ester hydrochloride with a procedure similar to that for compound 1. Yield 75%; white solid; m. p. 95–95 °C. 1H NMR (400 MHz, CDCl3) δ 14.06 (s, 1H), 9.01 (s, 1H), 7.04 (d, J = 8.0 Hz, 2H), 6.71 (d, J = 8.0 Hz, 2H), 5.5 (br, 1H), 4.92–4.87 (m, 1H), 4.22–3.97 (m, 2H), 3.72 (s, 3H), 3.57–3.48 (m, 2H), 3.21–3.15 (m, 1H), 3.04 (d, J = 6.8 Hz, 1H), 2.90–2.77 (m, 2H), 2.66–2.53 (m, 2H), 2.11–2.02 (m, 2H), 1.96–1.85 (m, 2H), 1.51 (s, 3H), 1.39 (s, 3H), 1.06 (d, J = 6.4 Hz, 3H), 0.98 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.99, 170.10, 161.58, 154.98, 154.82, 151.63, 130.48, 128.23, 115.46, 109.82, 109.59, 108.44, 108.33, 99.34, 97.83, 96.40, 74.25, 74.09, 54.13, 52.29, 47.56, 47.29, 37.24, 35.23, 23.04, 22.79, 19.16, 18.72, 18.30, 16.16, 15.99. HR-EI-MS m/z 567.2465; calculated for C31H37NO9: 567.2468.
Compound 9. The title compound was obtained from xyloketal B acid and cysteamine with a procedure similar to that for compound 1. Yield 73%; white solid; m. p. 72–73 °C. 1H NMR (400 MHz, CDCl3) δ 14.26 (s, 1H), 8.84 (s, 1H), 4.21–4.13 (m, 2H), 3.80–3.70 (m, 2H), 3.57–3.48 (m, 2H), 2.90 (t, J = 6.4 Hz, 2H), 2.86–2.80 (m, 2H), 2.70–2.56 (m, 2H), 2.11–2.03 (m, 2H), 1.97–1.90 (m, 2H), 1.67 (br, 1H), 1.55 (s, 3H), 1.51 (s, 3H), 1.07 (d, J = 6.4 Hz, 3H), 1.04 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.72, 161.59, 154.98, 151.45, 109.66, 108.27, 99.47, 97.80, 96.64, 74.42, 74.22, 47.65, 47.33, 38.06, 37.76, 35.62, 35.25, 23.22, 22.93, 18.60, 18.31, 16.04, 15.90. HR-EI-MS m/z 449.1869; calculated for C23H31NO6S: 449.1872.
Compound 10. The title compound was obtained from xyloketal B acid and N,N-dimethyl ethylenediamine with a procedure similar to that for compound 1. Yield 70%; white solid; m. p. 71–72 °C. 1H NMR (400 MHz, CDCl3) δ 13.49 (s, 1H), 8.94 (s, 1H), 4.17 (dd, J = 16.4, 6.8 Hz, 2H), 3.75–3.65 (m, 2H), 3.52 (q, J = 6.8 Hz, 2H), 3.27 (t, J = 5.6 Hz, 2H), 2.91 (s, 6H), 2.82 (d, J = 6.8 Hz, 2H), 2.60–2.49 (m, 2H), 2.07–2.01 (m, 2H), 1.96–1.88 (m, 2H), 1.53 (s, 3H), 1.50 (s, 3H), 1.06 (d, J = 6.8 Hz, 3H), 1.02 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.26, 161.34, 155.71, 151.49, 110.03, 108.58, 99.59, 98.46, 95.89, 74.37, 74.18, 59.20, 47.46, 47.18, 44.18, 36.69, 36.01, 35.40, 23.04, 22.75, 18.36, 18.25, 15.95, 15.81, 15.68. HR-EI-MS m/z 460.2563; calculated for C25H36N2O6: 460.2573.
Compound 11. The title compound was obtained from xyloketal B acid and thiophene ethylamine with a procedure similar to that for compound 1. Yield 76%; white solid; m. p. 75–76 °C. 1H NMR (400 MHz, CDCl3) δ 14.41 (s, 1H), 8.66 (s, 1H), 7.14 (d, J = 5.2 Hz, 1H), 6.92 (d, J = 5.2 Hz, 1H), 6.90–6.89 (m, 1H), 4.25 (dd, J = 16.4, 6.8 Hz, 2H), 3.76–3.63 (m, 2H), 3.56–3.45 (m, 2H), 3.13 (t, J = 6.8 Hz, 2H), 2.93–2.79 (m, 2H), 2.69–2.53 (m, 2H), 2.11–2.00 (m, 2H), 1.97–1.86 (m, 2H), 1.51 (s, 3H), 1.42 (s, 3H), 1.07 (d, J = 6.4 Hz, 3H), 1.03 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.67, 161.65, 154.67, 151.35, 141.59, 126.94, 125.34, 123.73, 109.46, 108.37, 99.45, 97.56, 96.64, 74.34, 74.21, 47.56, 47.34, 40.70, 35.53, 35.26, 29.80, 22.97, 22.68, 18.67, 18.38, 16.06, 15.91. HR-EI-MS m/z 499.2025; calculated for C27H33NO6S: 499.2029.
Compound 12. The title compound was obtained from xyloketal B acid and phenylalanine methyl ester hydrochloride with a procedure similar to that for compound 1. Yield 73%; white solid; m. p. 72–73 °C. 1H NMR (400 MHz, CDCl3) δ 14.05 (s, 1H), 9.03(s, 1H), 7.27–7.20 (m, 5H), 5.00–4.95 (m, 1H), 4.21–4.15 (m, 2H), 3.74 (s, 3H), 3.59–3.50 (m, 2H), 3.32–3.26 (m, 1H), 3.14 (d, J = 7.2 Hz, 1H), 2.93–2.80 (m, 2H), 2.73–2.54 (m, 2H), 2.11–2.05 (m, 2H), 1.97–1.90 (m, 2H), 1.52 (s, 3H), 1.38 (s, 3H), 1.08 (d, J = 6.4 Hz, 3H), 1.05 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.71, 170.14, 161.63, 154.96, 151.66, 136.47, 129.31, 128.49, 128.39, 126.79, 109.78, 109.57, 108.41, 99.34, 97.89, 96.42, 74.30, 74.18, 53.77, 52.16, 47.62, 47.37, 38.04, 35.66, 22.93, 22.66, 18.71, 18.64, 18.28, 16.04, 15.87. HR-EI-MS m/z 551.2512; calculated for C31H37NO8: 551.2519.
Compound 13. The title compound was obtained from xyloketal B acid and tryptophan methyl hydrochloride with a procedure similar to that for compound 1. Yield 72%; white solid; m. p. 110–111 °C. 1H NMR (400 MHz, CDCl3) δ 14.14 (s, 1H), 9.03 (s, 1H), 8.19 (s, 1H), 7.55 (t, J = 8.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.17(s, 1H), 7.13–7.11 (m, 1H), 7.06–7.00 (m, 1H), 5.10–4.99 (m, 1H), 4.15 (dd, J = 16.4, 6.8 Hz, 2H), 3.67 (s, 3H), 3.57–3.45 (m, 2H), 3.40–3.29 (m, 2H), 2.92–2.77 (m, 2H), 2.70–2.55 (m, 2H), 2.11–2.01 (m, 2H), 1.89–1.82 (m, 2H), 1.52 (s, 3H), 1.48 (s, 3H), 1.06 (d, J = 6.4 Hz, 3H), 1.02 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.18, 170.12, 161.56, 154.99, 151.52, 136.11, 127.63, 123.18, 121.81, 119.42, 118.75, 110.95, 110.20, 109.54, 108.34, 99.26, 97.71, 96.46, 74.18, 74.00, 53.34, 52.19, 47.43, 47.24, 35.44, 35.15, 27.72, 22.91, 22.71, 22.40, 18.64, 18.26, 15.82. HR-EI-MS m/z 590.2623; calculated for C33H38N2O8: 590.2628.
Compound 16. First, 50 mg (0.14 mmol) of xyloketal B [16] and 8.7 mg (0.28 mmol) of methylamine were dissolved in 10 mL of DMF and stirred, followed by adding 0.05 mL (0.28 mmol) of 40% formaldehyde solution, stirred at room temperature for 1 h. The reaction was quenched with water. The aqueous layer was extracted with ethyl acetate, 3 × 25 mL. The combined organic extracts were washed with saturated ammonium chloride and brine, dried over anhydrous MgSO4 and concentrated in vacuo. Purification by flash chromatography (petroleum ether: ethyl acetate = 5:1~2:1) gave the title compound. Yield 89%; white solid; m. p. 62–63 °C. 1H NMR (400 MHz, CDCl3) δ 4.66 (s, 2H), 4.15–4.08 (m, 2H), 3.75 (s, 2H), 3.49 (t, J = 8.4 Hz, 2H), 2.79 (dt, J = 17.2 5.2 Hz, 2H), 2.60 (d, J = 17.2 Hz, 2H), 2.53 (s, 3H), 2.11–2.04 (m, 2H), 1.87–1.81 (m, 2H), 1.47 (s, 3H), 1.44 (s, 3H), 1.04 (d, J = 6.4 Hz, 3H), 1.01 (d, J = 6.4 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 150.13, 149.50, 148.87, 107.25, 107.14, 100.01, 98.73, 98.12, 83.58, 73.85, 47.33, 47.47, 40.03, 35.42, 35.32, 22.98, 22.72, 18.73, 18.39, 16.07, 15.99. HR-EI-MS m/z 401.2196; calculated for C23H31O5N1: 401.2197.
Compound 17. The title compound was obtained from xyloketal B and butylamine with a procedure similar to that for compound 16. Yield 93%; white solid; m. p. 65–66 °C. 1H NMR (400 MHz, CDCl3) δ 4.76 (s, 2H), 4.20–4.12 (m, 2H), 3.82 (s, 2H), 3.52 (t, J = 8.4 Hz, 2H), 2.86–2.80 (m, 2H), 2.69 (t, J = 17.2 Hz, 2H), 2.67–2.59 (m, 2H), 2.17–2.11 (m, 2H), 1.91–1.83 (m, 2H), 1.58–1.51 (m, 2H), 1.49 (s, 3H), 1.48 (s, 3H), 1.00–1.30 (m, 2H), 1.06 (d, J = 6.4 Hz, 3H), 1.03 (d, J = 6.4 Hz, 3H), 0.92 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 150.75, 149.40, 148.95, 107.33, 107.24, 100.49, 98.72, 98.25, 82.68, 73.96, 51.64, 47.59, 47.59, 45.91, 35.55, 35.44, 30.29, 23.06, 22.79, 20.48, 18.85, 18.506, 16.081, 16.00, 14.05. HR-EI-MS m/z 443.2665; calculated for C26H37O5N1: 443.2666.
Compound 18. The title compound was obtained from xyloketal B and ethanolamine with a procedure similar to that for compound 16. Yield 79%; white solid; m. p. 112–113 °C. 1H NMR (400 MHz, CDCl3) δ 4.80 (s, 2H), 4.2 (s, 1H), 4.16 (dd, J = 16.4, 6.4 Hz, 2H), 3.86 (s, 2H), 3.68 (t, J = 5.2 Hz, 2H), 3.54–3.47 (m, 2H), 2.92–2.89 (m, 2H), 2.86–2.75 (m, 2H), 2.66–2.59 (m, 2H), 2.15–2.07 (m, 2H), 1.91–1.82 (m, 2H), 1.49 (s, 3H), 1.47 (s, 3H), 1.06 (d, J = 6.4 Hz, 3H), 1.03 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 101 MHz) δ 150.55, 149.64, 148.83, 107.27, 99.91, 98.82, 98.21, 82.79, 73.90, 59.02, 53.77, 47.36, 47.26, 45.39, 35.31, 22.93, 22.67, 18.63, 18.38, 15.95. HR-EI-MS m/z 431.2301; calculated for C24H33O6N1: 431.2302.
Compound 19. The title compound was obtained from xyloketal B and diglycolamine with a procedure similar to that for compound 16. Yield 72%; white solid; m. p. 126–127 °C. 1H NMR (400 MHz, CDCl3) δ 4.82 (s, 2H), 4.14 (dd, J = 16.4, 6.4 Hz, 2H), 3.89 (s, 2H), 3.73 (t, J = 5.2 Hz, 2H), 3.68 (t, J = 5.2 Hz, 2H), 3.61 (t, J = 5.2 Hz, 2H), 3.55–3.48 (m, 2H), 2.97 (br, 1H), 2.95–2.90 (m, 2H), 2.86–2.75 (m, 2H), 2.62 (t, J = 5.6 Hz, 2H), 2.16–2.06 (m, 2H), 1.91–1.84 (m, 2H), 1.50 (s, 3H), 1.47 (s, 3H), 1.06 (d, J = 6.4 Hz, 3H), 1.03 (d, J = 6.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 150.55, 149.68, 148.98, 107.39, 107.22, 99.93, 99.01, 98.31, 82.66, 73.96, 72.39, 69.21, 61.70, 51.59, 47.58, 47.42, 46.05, 35.51, 35.37, 23.02, 22.75, 18.76, 18.37, 16.01, 15.91. HR-EI-MS m/z 475.2563; calculated for C26H37O7N1: 475.2565.
Compound 20. The title compound was obtained from xyloketal B and aniline with a procedure similar to that for compound 16. Yield 93%; white solid; m. p. 68–69 °C. 1H NMR (400 MHz, CDCl3) δ 7.25 (t, J = 8.8 Hz, 2H), 7.10 (d, J = 8.8 Hz, 2H), 6.88 (t, J = 8.0 Hz, 1H), 5.35–5.25 (m, 2H), 4.46 (s, 2H), 4.14 (dd, J = 17.2, 8.0 Hz, 2H), 3.49 (dd, J = 17.2, 8.0 Hz, 2H), 2.79 (t, J = 17.2 Hz, 2H), 2.65–2.58 (m, 2H), 2.11–2.05 (m, 2H), 1.88–1.83 (m, 2H), 1.48 (s, 3H), 1.46 (s, 3H), 1.04 (d, J = 6.4 Hz, 3H), 1.01 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 101 MHz) δ 151.05, 149.86, 148.38, 148.38, 129.03, 120.59, 117.51, 107.46, 107.29, 101.23, 99.05, 96.66, 78.84, 73.99, 47.59, 47.46, 46.34, 35.41, 23.04, 22.75, 18.81, 18.73, 18.50, 16.06, 15.97. HR-EI-MS m/z 463.2352; calculated for C28H33O5N1: 463.2353.
Compound 21. The title compound was obtained from xyloketal B and p-methoxyaniline with a procedure similar to that for compound 16. Yield 89%; white solid; m. p. 89–90 °C. 1H NMR (400 MHz, CDCl3) δ 7.01 (d, J = 9.2 Hz, 2H), 6.78 (d, J = 9.2 Hz, 2H), 5.25–5.17 (m, 2H), 4.38 (s, 2H), 4.13 (dd, J = 17.2, 8.0 Hz, 2H), 3.73 (s, 3H), 3.49 (dd, J = 17.2, 8.0 Hz, 2H), 2.78 (t, J = 17.2 Hz, 2H), 2.64–2.56 (m, 2H), 2.10–2.06 (m, 2H), 1.87–1.81 (m, 2H), 1.51 (s, 3H), 1.46 (s, 3H), 1.03 (d, J = 6.8 Hz, 3H), 1.00 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 101 MHz) δ 154.33, 150.96, 149.83, 148.38, 142.55, 119.87, 114.35, 107.43, 107.28, 101.14, 98.99, 98.62, 80.10, 73.99, 55.53, 47.57, 47.49, 47.036, 35.432, 23.06, 22.79, 18.74, 18.47, 16.08, 15.98. HR-EI-MS m/z 493.2458; calculated for C29H35O6N1: 493.2459.
Compound 22. The title compound was obtained from xyloketal B and 4-aminophenol with a procedure similar to that for compound 16. Yield 85%; white solid; m. p. 102–103 °C. 1H NMR (400 MHz, CDCl3) δ 6.99 (d, J = 8.8 Hz, 2H), 6.74 (d, J = 8.8 Hz, 2H), 5.25–5.16 (m, 2H), 4.37 (s, 2H), 4.14 (dd, J = 17.2, 8.0 Hz, 2H), 3.51 (dd, J = 17.2, 8.0 Hz, 2H), 2.79 (t, J = 17.2 Hz, 2H), 2.66–2.58 (m, 2H), 2.12–2.04 (m, 2H), 1.90–1.84 (m, 2H), 1.47 (s, 6H), 1.04 (d, J = 6.4 Hz, 3H), 1.01 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 101 MHz) δ 150.91, 150.78, 149.73, 148.24, 142.18, 120.16, 115.77, 109.64, 107.53, 107.36, 101.20, 99.00, 98.76, 80.23, 73.98, 47.53, 47.47, 47.07, 35.39, 23.08, 22.84, 18.69, 18.42, 16.29, 15.98. HR-EI-MS m/z 479.2308; calculated for C28H33O6N1: 479.2309.
Compound 23. The title compound was obtained from xyloketal B and p-aminobenzoic with a procedure similar to that for compound 16. Yield 88%; white solid; m. p. 128–129 °C. 1H NMR (400 MHz, CDCl3) δ 12.58 (s, 1H), 8.01 (d, J = 8.8 Hz, 2H), 7.08 (d, J = 8.8 Hz, 2H), 5.42–5.32 (m, 2H), 4.54 (s, 2H), 4.16 (dd, J = 17.2, 8.0 Hz, 2H), 3.55–3.49 (m, 2H), 2.81 (t, J = 17.2 Hz, 2H), 2.68–2.60 (m, 2H), 2. 11–2.06 (m, 2H), 1.90–1.86 (m, 2H), 1.51 (s, 3H), 1.48 (s, 3H), 1.05 (d, J = 6.8 Hz, 3H), 1.03 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 101 MHz) δ 171.51, 152.21, 150.86, 149.97, 148.25, 131.82, 120.05, 114.94, 107.54, 107.40, 100.82, 99.38, 98.68, 73.97, 47.39, 47.21, 45.53, 35.29, 22.96, 22.71, 18.71, 18.39, 16.01, 15.95. HR-EI-MS m/z 507.2253; calculated for C29H33O7N1: 507.2252.
Compound 24. The title compound was obtained from xyloketal B and paranitroaniline with a procedure similar to that for compound 16. Yield 82%; white solid; m. p. 70–71 °C. 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 9.2 Hz, 2H), 7.06 (d, J = 9.2 Hz, 2H), 5.42–5.33 (m, 2H), 4.56 (s, 2H), 4.17 (dd, J = 17.2, 8.0 Hz, 2H), 3.55–3.50 (m, 2H), 2.82 (t, J = 17.2 Hz, 2H), 2.68–2.60 (m, 2H), 2.13–2.04 (m, 2H), 1.92–1.88 (m, 2H), 1.51 (s, 3H), 1.49 (s, 3H), 1.06 (d, J = 6.8 Hz, 3H), 1.04 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 101 MHz) δ 153.00, 150.90, 150.47, 150.31, 148.56, 139.93, 125.74, 114.46, 107.81, 107.64, 100.47, 99.98, 99.85, 98.99, 76.57, 73.97, 47.57, 47.37, 45.49, 35.46, 35.34, 23.02, 22.78, 18.63, 18.34, 15.99, 15.86. HR-EI-MS m/z 508.2205; calculated for C28H32O7N2: 508.2204.
Compound 25. The title compound was obtained from xyloketal B and p-fluoro aniline with a procedure similar to that for compound 16. Yield 87%; white solid; m. p. 79–80 °C. 1H NMR (400 MHz, CDCl3) δ 7.07 (d, J = 9.2 Hz, 2H), 6.95 (d, J = 9.2 Hz, 1H), 6.93 (d, J = 9.2 Hz, 1H), 5.24 (s, 2H), 4.42 (s, 2H), 4.15 (dd, J = 17.2, 8.0 Hz, 2H), 3.49 (dd, J = 17.2, 8.0 Hz, 2H), 2.79 (t, J = 17.2 Hz, 2H), 2.67–2.58 (m, 2H), 2.14–2.03 (m, 2H), 1.90–1.84 (m, 2H), 1.48 (s, 6H), 1.05 (d, J = 6.8 Hz, 3H), 1.02 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 159.24, 156.05, 150.85, 149.89, 148.28, 144.96, 144.817, 119.78, 119.55, 115.62, 115.34, 107.46, 107.28, 100.84, 99.14, 98.64, 79.52, 73.94, 70.38, 47.55, 46.99, 35.50, 35.39, 23.04, 22.77, 18.76, 18.41, 16.02. HR-EI-MS m/z 481.2259; calculated for C28H32O5N1F1: 481.2259.
Compound 26. The title compound was obtained from xyloketal B and parachloroaniline with a procedure similar to that for compound 16. Yield 78%; white solid; m. p. 82–83 °C. 1H NMR (400 MHz, CDCl3) δ 7.20 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.4 Hz, 2H), 5.31–5.22 (m, 2H), 4.43 (s, 2H), 4.15 (dd, J = 17.2, 8.4 Hz, 2H), 3.52 (dd, J = 17.2, 8.0 Hz, 2H), 2.79 (t, J = 17.2 Hz, 2H), 2.66–2.58 (m, 2H), 2.13–2.03 (m, 2H), 1.90–1.85 (m, 2H), 1.48 (s, 6H), 1.47 (s, 3H), 1.05 (d, J = 6.8 Hz, 3H), 1.02 (d, J = 6.8 Hz, 3H); 13C NMR (101MHz, CDCl3) δ 150.84, 149.96, 149.81, 148.34, 147.08, 128.90, 125.55, 118.85, 107.52, 107.32, 100..82, 99.24, 98.67, 78.73, 73.98, 47.56, 47.40, 46.54, 35.40, 23.05, 22.78, 18.70, 18.43, 16.04. HR-EI-MS m/z 497.1965; calculated for C28H32O5N1Cl1: 497.1964.
Compound 27. The title compound was obtained from xyloketal B and parabromoaniline with a procedure similar to that for compound 16. Yield 88%; white solid; m. p. 83–84 °C. 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 9.2 Hz, 2H), 6.98 (d, J = 9.2 Hz, 2H), 5.31–5.22 (m, 2H), 4.43 (s, 2H), 4.15 (dd, J = 17.2, 8.0 Hz, 2H), 3.51 (dd, J = 17.2, 8.0 Hz, 2H), 2.79 (t, J = 17.2 Hz, 2H), 2.64–2.56 (m, 2H), 2.10–2.01 (m, 2H), 1.88–1.83 (m, 2H), 1.48 (s, 6H), 1.05 (d, J = 6.8 Hz, 3H), 1.02 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 150.82, 149.96, 149.80, 148.33, 147.51, 131.81, 119.21, 112.89, 107.51, 107.31, 100.79, 99.24, 98.66, 78.56, 73.97, 47.55, 47.40, 46.44, 35.50, 35.39, 23.04, 22.79, 18.70, 18.46, 16.04, 15.93. HR-EI-MS m/z 541.1455; calculated for C28H32O5N1Br1: 541.1458.
Compound 28. The title compound was obtained from xyloketal B and paraiodoaniline with a procedure similar to that for compound 16. Yield 86%; white solid; m. p. 85–86 °C. 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 5.29–5.20 (m, 2H), 4.41 (s, 2H), 4.13 (dd, J = 17.2, 8.4 Hz, 2H), 3.50 (dd, J = 17.2, 8.4 Hz, 2H), 2.77 (t, J = 17.2 Hz, 2H), 2.63–2.59 (m, 2H), 2.9–2.01 (m, 2H), 1.87–1.82 (m, 2H), 1.46 (s, 6H), 1.03 (d, J = 6.8 Hz, 3H), 1.01 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 150.82, 149.97, 149.82, 148.34, 148.14, 137.76, 119.62, 107.57, 107.32, 100.82, 99.25, 98.67, 82.75, 78.36, 73.99, 47.55, 47.39, 46.32, 35.39, 23.06, 22.77, 18.69, 18.42, 16.05, 15.94. HR-EI-MS m/z 589.1312; calculated for C28H32O5N1I1: 589.1320.
Compound 29. First, 50 mg (0.14 mmol) of xyloketal B [16] and 24 mg (0.28 mmol) of morpholine were dissolved in 10 mL of DMF and stirred, followed by adding 0.05 mL (0.28 mmol) of 40% formaldehyde solution, stirred at room temperature for 1 h. The reaction was quenched with water. The aqueous layer was extracted with ethyl acetate, 3 × 25 mL. The combined organic extracts were washed with saturated ammonium chloride and brine, dried over anhydrous MgSO4 and concentrated in vacuo. Purification by flash chromatography (petroleum ether: ethyl acetate = 5:1~2:1) gave the title compound. Yield 81%; white solid; m. p. 80–81 °C. 1H NMR (400 MHz, CDCl3) δ 4.12–4.03 (m, 2H), 3.77–3.67 (m, 4H), 3.71 (s, 2H), 3.44 (q, J = 8.8 Hz, 2H), 2.77 (t, J = 17.2 Hz, 2H), 2.66–2.62 (m, 2H), 2.61–2.52 (m, 4H), 2.11–1.98 (m, 2H), 1.89–1.79 (m, 2H), 1.47 (s, 3H), 1.43 (s, 3H), 1.03 (d, J = 8.4 Hz, 3H), 0.99 (d, J = 8.4 Hz, 3H); 13C NMR (CDCl3, 101 MHz) δ 155.18, 150.84, 150.03, 108.00, 107.77, 100.04, 98.83, 98.59, 74.26, 67.16, 54.62, 53.11, 48.19, 47.97, 36.16, 35.82, 23.52, 23.16, 19.35, 19.03, 16.54, 16.44. HR-EI-MS m/z 445.2458; calculated for C25H35O6N1: 445.2459.
Compound 30. The title compound was obtained from xyloketal B and diethylamine with a procedure similar to that for compound 29. Yield 81%; white solid; m. p. 94–95 °C. 1H NMR (400 MHz, CDCl3) δ 4.15 (dd, J = 17.2, 8.4 Hz, 2H), 3.78 (q, J = 11.2 Hz, 2H), 3.49 (dd, J = 17.2, 8.4 Hz, 2H), 2.86–2.80 (m, 2H), 2.70–2.64 (m, 2H), 2.64–2.58 (m, 4H), 2.12–2.03 (m, 2H), 1.92–1.83 (m, 2H), 1.50 (s, 6H), 1.09 (t, 6H), 1.06 (d, J = 6.8 Hz, 3H), 1.03 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 101 MHz) δ 155.76, 150.29, 149.37, 107.45, 107.32, 100.76, 98.42, 97.50, 73.93, 49.36, 47.62, 46.31, 35.71, 35.46, 23.13, 22.83, 18.70, 16.07, 11.21. HR-EI-MS m/z 431.2668; calculated for C25H37O5N1: 431.2666.
3.3. Biological Evaluation
3.3.1. Pharmacological Assays
Xyloketals derivatives were obtained using the above synthetic method; DMEM (High Glucose) and FBS were purchased from Gibco BRL (Grand Island, NY, USA); JC-1 probe was purchased from Beyotime Institute of Biotechnology (Haimen, China); H2O2 was purchased from Guangzhou Chemical Reagent Factory (GCRF, Guangzhou, China) and was freshly prepared for each experiment from a 30% stock solution. All other reagents were purchased from Sigma (St. Louis, MO, USA).
The HUVECs cell line was provided by the Pharmaceutical Biotechnology Centre of Jinan University (Guangdong, China). The cells were cultured in a DMEM medium (High Glucose) (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA), penicillin (100 U/mL) and streptomycin (100 U/mL) at 37 °C in a 5% CO2 humidified incubator. Endothelial cells appear as “cobblestone” mosaic after reaching confluence under a microscope.
HUVECs were harvested during the logarithmic growth phase and seeded in 96-well plates at a density of 6 × 104/mL and cultured at 37 °C in a 5% CO2 humidified incubator for 24 h. The cell viability was assessed using the mitochondrial tetrazolium assay (MTT) in HUVECs. The cells were pre-incubated with xyloketals at different concentrations (10 μM, 1 μM) for 30 min, followed by exposure to H2O2 at a concentration of 600 μM and additional incubation for 20 h. MTT solution (15 μL/well, 5 μg/mL) was added and processed to examine the cell viability. The optical density was read at λ = 570 nm using a Thermo Multiskan FC plate reader. At the tested concentration, all of the xyloketals showed no significant effects on cell viability.
3.3.2. Construction and Validation of the QSAR Model
The three-dimensional structures of the compounds were constructed using the SYBYL programming package (version 7.3.5, Tripos, St. Louis, MO, USA). The MMFF94 force field and MMFF94 partial atomic charges were applied to these compounds. In addition, the compounds were minimized using a non-bond cut-off of 8 angstroms and the Powell conjugate-gradient algorithm. The convergence criterion was set to 0.05 kcal/mol. The activities of the compounds at 10 μM were expressed using the LOGIT transformation shown in the following formula to give a value in proportion to energy:
The training set and test set were randomly divided out of a total of 35 molecules. A training set of 30 molecules was used to construct the QSAR model. In addition, a training set of 5 molecules was used to validate it. All of the molecules were aligned using the most active compound, 22, as the template. Each compound was mapped onto a 3D lattice with grid points 2.0 Å apart. The mapped region was created automatically by the program with an attenuation factor of 0.3. The electrostatic, hydrophobic, donor and acceptor columns were used to construct the model. The model was constructed using the partial-least-squares (PLS) analysis without any column filtering.
The robustness of the model was addressed based on the internal cross-validation using the leave-one-out (LOO) procedure and the external validation of the test set. All of the statistical parameters are listed in Table 3.
3.3.3. Mitochondrial Membrane Potentials Assay
The JC-1 probe was used to measure the mitochondrial depolarization in HUVECs. Briefly, HUVECs were cultured in six-well plates at a density of 2.5 × 105/mL, incubated with 23 and 24 at different concentrations (25 μM and10 μM) for 30 min, then exposed to 400 μM H2O2 for 20 h. According to the instructions for the test kits, all cells were collected into 1.5-mL tubes, incubated with JC-1 for 20 min at 37 °C and rinsed twice with PBS. The mitochondrial membrane potentials (ΔΨm) were monitored by determining the relative amounts of dual emissions from JC-1 monomers or aggregates using a BD FACS Aria flow cytometry. The red/green fluorescence was calculated using the Flowjo (v7.6.5, Ashland, OR, USA).
3.3.4. Statistics
Results are presented as mean ± S.E.M. Comparisons between multiple groups were performed using the Excel by t-test. Differences were considered to be significant at p ≤ 0.05.
4. Conclusions
The water insolubility of the xyloketal compounds from marine fungus may be challenging for further clinical development. Therefore, a new series of derivatives with the introduction of amino groups at the C-12 and C-13 positions of xyloketal B were designed and synthesized to improve the solubility and biological activity. All 28 new derivatives and seven known compounds (14, 15, 31–35) were evaluated for their protection against H2O2-induced HUVEC injury. The results indicated that some compounds exhibited strong anti-oxidative activities, especially compounds 23 and 24, which displayed the best excellent protective activities out of all of the derivatives. Then, a CoMSIA was constructed using the SYBYL programming package (version 7.3.5) to explain the structural activity relationship of the xyloketal derivatives. A 3D QSAR model generated using the CoMSIA was analyzed and provided good advice to modify the molecules for better activity in the future. Compounds 23 and 24, which had the most remarkable anti-oxidative activities, were further examined in the JC-1 mitochondrial membrane potential (MMP) assay of HUVECs. The results showed that compound 23 and 24 would significantly inhibit H2O2-induced the decrease in the cell mitochondrial membrane potential (ΔΨm) at 25 μM. In conclusion, we designed and synthesized a new series of xyloketal derivatives to improve solubility and biological activity. Among them, compounds 23 and 24 effectively protected HUVECs against oxidative damage and further mitochondrial membrane integrity impairment. These derivatives will be new candidates for the treatment of CVD.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21172271, 81202454), the Natural Science Foundation of Guangdong Province, China (Grant No. S2011020001231) and Major Scientific and Technological Special Project of Administration of Ocean and Fisheries of Guangdong Province (A201301C08). We are indebted to Haibin Luo, School of Pharmaceutical Sciences, Sun Yat-sen University for providing the SYBYL programming package (version 7.3.5).
Author Contributions
Conceived and designed the experiments: S. Liu, R. Luo, J. Pang; Performed the experiments: S. Liu, R. Luo, Q. Xiang, X. Xu; Analyzed the data: S. Liu, R. Luo, L. Qiu, J. Pang; Wrote the paper: S. Liu, R. Luo, J. Pang.
Abbreviations
| 3D-QSAR | three-dimensional quantitative structure-activity relationship |
| As | atherosclerosis |
| BOP | (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate |
| CoMSIA | a comparative molecular similarity indices analysis |
| CVD | cardiovascular disease |
| DCM | dichloromethane |
| DIEA | N,N-diisopropylethylamine |
| DMF | N,N-dimethylformamide |
| DMSO | dimethyl sulfoxide |
| FACs | fluorescence activating cell sorter |
| FCM | flow cytometry |
| H2DCFDA | 2′,7′-dihydrodichlorofluorescein diacetate |
| H2O2 | hydrogen peroxide |
| HCHO | formaldehyde |
| HUVECs | human umbilical vein endothelial cells |
| LOO | leave-one-out |
| MAPK | mitogen-activated protein kinase |
| MMP | mitochondrial membrane potential |
| MPP+ | 1-methyl-4-phenylpyridinium |
| NMR | nuclear magnetic resonance |
| OGD | oxygen-glucose deprivation |
| OxLDL | oxidized low density lipoprotein |
| PLS | partial-least-squares |
| ROS | reactive oxygen species |
| SAR | structure-activity relationship |
| THF | tetrahydrofuran |
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yang, B.H.; Oo, T.N.; Rizzo, V. Lipid rafts mediate H2O2 prosurvival effects in cultured endothelial cells. FASEB J. 2006, 20, 1501–1503. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gu, L.; Ma, Q.; Zhu, D.; Huang, X. Resveratrol attenuates hydrogen peroxide-induced apoptosis in human umbilical vein endothelial cells. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 88–94. [Google Scholar] [PubMed]
- Kamata, H; Hirata, H. Redox regulation of cellular signalling. Cell. Signal. 1999, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Favero, T.G.; Zable, A.C.; Abramson, J.J. Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1995, 270, 25557–25563. [Google Scholar] [CrossRef] [PubMed]
- Suhara, T.; Fukuo, K.; Sugimoto, T.; Morimoto, S.; Nakahashi, T.; Hata, S.; Shimizu, M.; Ogihara, T. Hydrogen peroxide induces up-regulation of Fas in human endothelial cells. J. Immunol. 1998, 160, 4042–4047. [Google Scholar] [PubMed]
- Sabri, A.; Byron, K.L.; Samarel, A.M.; Bell, J.; Lucchesi, P.A. Hydrogen peroxide activates mitogen-activated protein kinases and Na+-H+ exchange in neonatal rat cardiac myocytes. Circ. Res. 1998, 82, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Cyrne, L.; Marques, V.O.; Marinho, H.S.; Antunes, F. H2O2 in the induction of NF-κB-dependent selective gene expression. Methods Enzymol. 2013, 528, 173–188. [Google Scholar] [PubMed]
- Xia, Z.Y.; Liu, M.; Wu, Y.; Sharma, V.; Luo, T.; Ouyang, J.P.; McNeill, J.H. N-acetylcysteine attenuates TNF-alpha-induced human vascular endothelial cell apoptosis and restores eNOS expression. Eur. J. Pharmacol. 2006, 550, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Martinou, J.C.; Green, D.R. Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol. 2001, 2, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Guido, K.; Bruno, D.; Michele, R.R. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998, 60, 619–642. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Wu, X.Y.; Feng, S.; Jiang, G.; Luo, J.H.; Zhou, S.N.; Vrijmoed, L.L.P.; Jones, E.B.G.; Krohn, K.; Steingrover, K.; et al. Five unique compounds: Xyloketals from mangrove fungus Xylaria sp. from the South China Sea coast. J. Org. Chem. 2001, 66, 6252–6256. [Google Scholar]
- Wu, X.Y.; Liu, X.H.; Lin, Y.C.; Luo, J.H.; She, Z.G.; Li, H.J.; Chan, W.L.; Antus, S.; Kurtan, T.; Elsasser, B.; et al. Xyloketal F: A strong l-calcium channel blocker from the mangrove fungus Xylaria sp. (#2508) from the South China Sea coast. Eur. J. Org. Chem. 2005, 12, 4061–4064. [Google Scholar] [CrossRef]
- Chen, W.L.; Qian, Y.; Meng, W.F.; Pang, J.Y.; Lin, Y.C.; Guan, Y.Y.; Chen, S.P.; Liu, J.; Pei, Z.; Wang, G.L. A novel marine compound xyloketal B protects against oxidized LDL-induced cell injury in vitro. Biochem. Pharmacol. 2009, 78, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, L.; Ling, C.; Li, J.; Pang, J.Y.; Lin, Y.C.; Liu, J.; Huang, R.X.; Wang, G.L.; Pei, Z.; et al. Marine compound Xyloketal B protects PC12 cells against OGD-induced cell damage. Brain Res. 2009, 1302, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.L.; Yao, X.L.; Liu, Z.Y.; Zhang, H.; Li, W.; Li, Z.X.; Wang, G.L.; Pang, J.Y.; Lin, Y.C.; Xu, Z.L.; et al. Protective effects of xyloketal B against MPP+-induced neurotoxicity in Caenorhabditiselegans and PC12 cells. Brain Res. 2010, 1332, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, Y.; Xiang, Q.; Pei, Z.; Liu, X.; Lu, B.; Chen, L.; Wang, G.; Pang, J.; Lin, Y. Design and synthesis of novel xyloketal derivatives and their vasorelaxing activities in rat thoracic aorta and angiogenic activities in zebrafish angiogenesis screen. J. Med. Chem. 2010, 53, 4642–4653. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.X.; Chen, J.W.; Feng, Y.; Huang, Y.Y.; Zhao, L.Y.; Li, J.; Su, H.X.; Liu, J.; Pang, J.Y.; Lin, Y.C.; et al. Xyloketal B exhibits its antioxidant activity through induction of HO-1 in vascular endothelial cells and zebrafish. Mar. Drugs 2013, 11, 504–522. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Shen, C.Z.; Guo, W.Y.; Zhang, X.F.; Liu, S.X.; Liang, F.Y.; Xu, Z.L.; Pei, Z.; Song, H.C.; Qiu, L.Q.; et al. Synthesis and neuroprotective action of xyloketal derivatives in Parkinson’s Disease models. Mar. Drugs 2013, 11, 5159–5189. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.Q.; Zhang, W.N.; Ji, H.T.; Zhang, M.; Song, Y.L.; Xu, H.; Zhu, J.; Miao, Z.Y.; Jiang, Q.F.; Yao, J.Z.; et al. Structure-based optimization of azole antifungal agents by CoMFA, CoMSIA, and molecular docking. J. Med. Chem. 2006, 49, 2512–2525. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.P.; Moroni, E.; Yan, B.; Colombo, G.; Blagg, B.S.J. 3D-QSAR-assisted design, synthesis, and evaluation of novobiocin analogues. Med. Chem. Lett. 2013, 4, 57–62. [Google Scholar] [CrossRef]
- Fleury, C.; Mignotte, B.; Vayssière, J.L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 8, 131–141. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, J.D.; Bexrud, J.A.; Freeman, R.P.; Wilson, P.D. Total synthesis of (+/−)-xyloketal D and model studies towards the total synthesis of (−)-xyloketal A. Heterocycles 2004, 62, 445–452. [Google Scholar] [CrossRef]
- Rodriguez, R.; Adlington, R.M.; Moses, J.E.; Cowley, A.; Baldwin, J.E. A new and efficient method for o-ouinone methide intermediate generation: application to the biomimetic synthesis of (±)-alboatrin. Org. Lett. 2004, 6, 3617–3619. [Google Scholar] [CrossRef] [PubMed]
- Krohn, K.; Riaz, M. Total synthesis of (+)-xyloketal D, a secondary metabolite from the mangrove fungus Xylaria sp. Tetrahedron Lett. 2004, 45, 293–294. [Google Scholar] [CrossRef]
- Pettigrew, J.D.; Freeman, R.P.; Wilson, P.D. Total synthesis of (−)-xyloketal D and its enantiomer—Confirmation of absolute stereochemistry. Can. J. Chem. 2004, 82, 1640–1648. [Google Scholar] [CrossRef]
- Pettigrew, J.D.; Wilson, P.D. Synthesis of xyloketal A, B, C, D and G analogues. J. Org. Chem. 2006, 71, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, J.D.; Wilson, P.D. Total synthesis of (−)-xyloketal A. Org. Lett. 2006, 8, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Krohn, K.; Riaz, M.; Flörke, U. Synthesis of xyloketals, natural products from the mangrove fungus Xylaria sp. Eur. J. Org. Chem. 2004, 1261–1270. [Google Scholar] [CrossRef]
- Xu, Z.L.; Li, Y.Y.; Lu, B.T.; Pang, J.Y.; Lin, Y.C. An expedient approach to the benzopyran core: application to synthesis of the natural products (±)-xyloketals and (±)-alboatrin. Chin. J. Chem. 2010, 28, 2441–2446. [Google Scholar] [CrossRef]
- Gong, G.H.; Qin, Y.; Huang, W.; Zhou, S.; Yang, X.H.; Li, D. Rutin inhibits hydrogen peroxide-induced apoptosis through regulating reactive oxygen species mediated mitochondrial dysfunction pathway in human umbilical vein endothelial cells. Eur. J. Pharmacol. 2010, 628, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Bresgen, N.; Karlhuber, G.; Krizbai, I.; Bauer, H.; Bauer, H.C.; Eckl, P.M. Oxidative stress in cultured cerebral endothelial cells induces chromosomal aberrations, micronuclei, and apoptosis. J. Neurosci. Res. 2003, 72, 327–333. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).