
Activation of RAF1 (c-RAF) by the Marine Alkaloid Lasonolide A Induces Rapid Premature Chromosome Condensation

Abstract
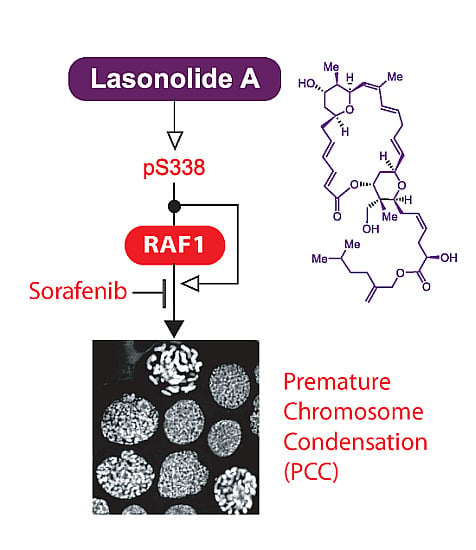
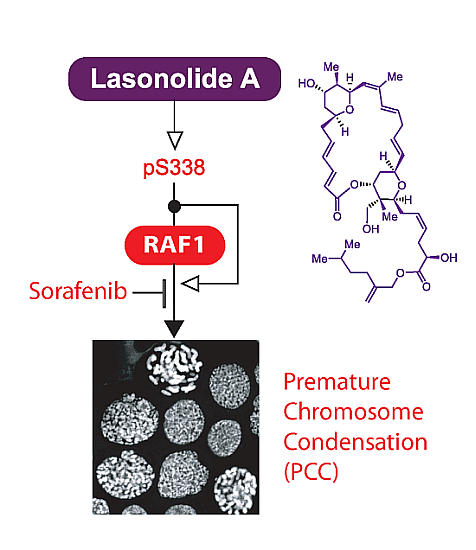
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Results and Discussion
2.1. A Kinome shRNA Screen Identified Candidate LSA Molecular Targets and Pathways
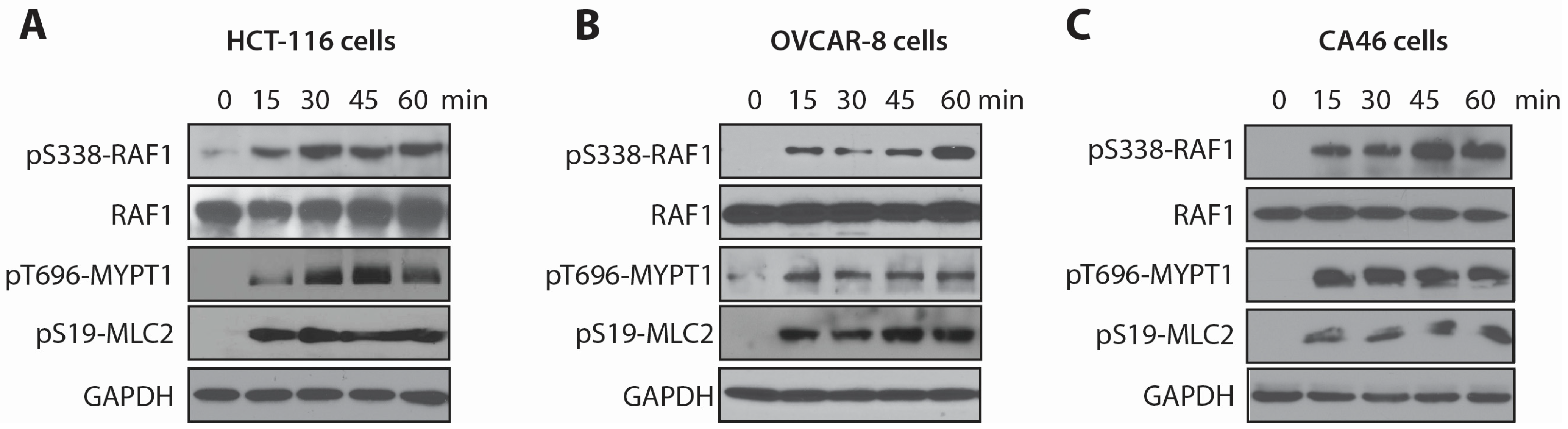
2.2. LSA Rapidly Activates RAF1 by Inducing Its Phosphorylation on Ser338

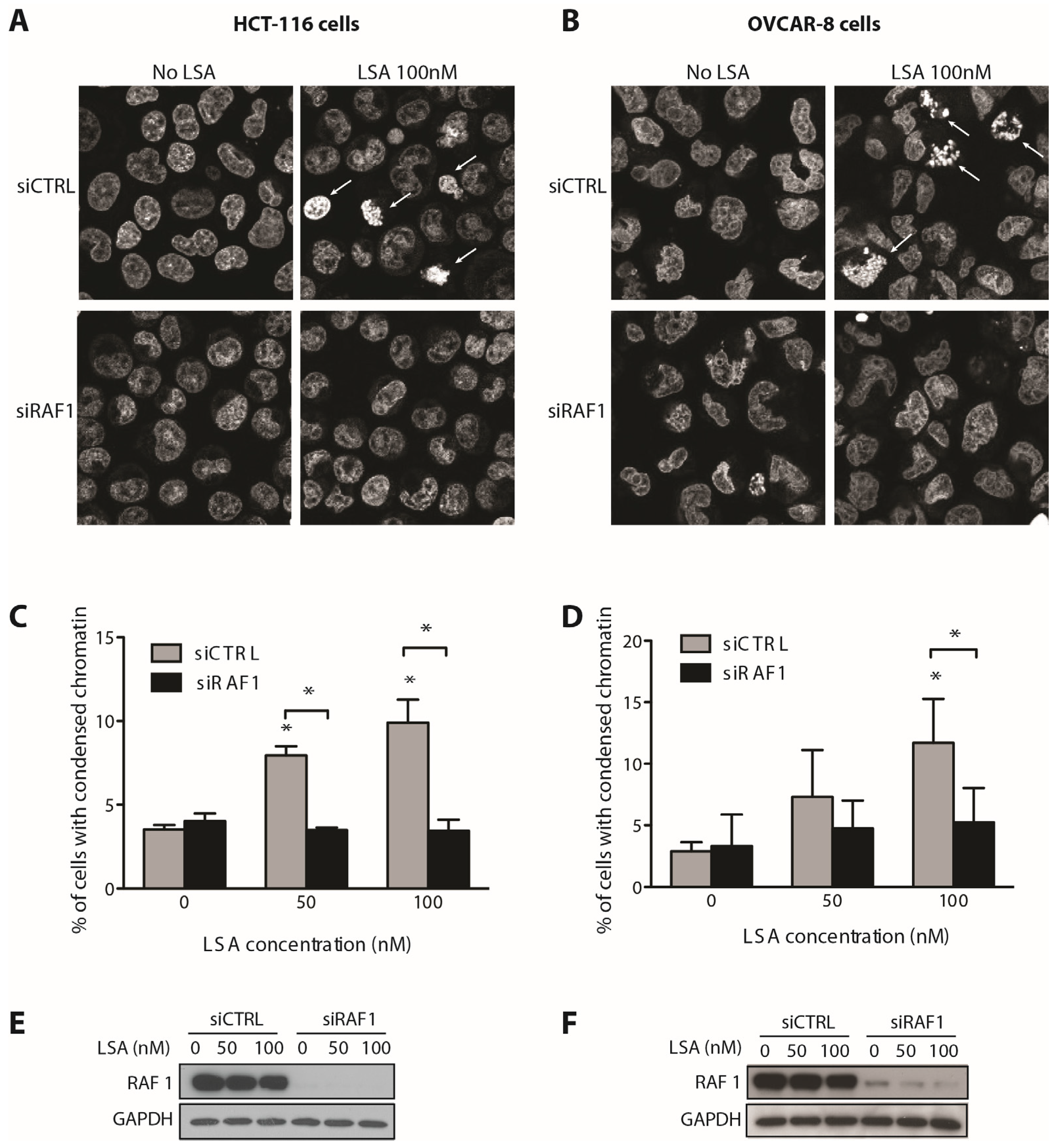
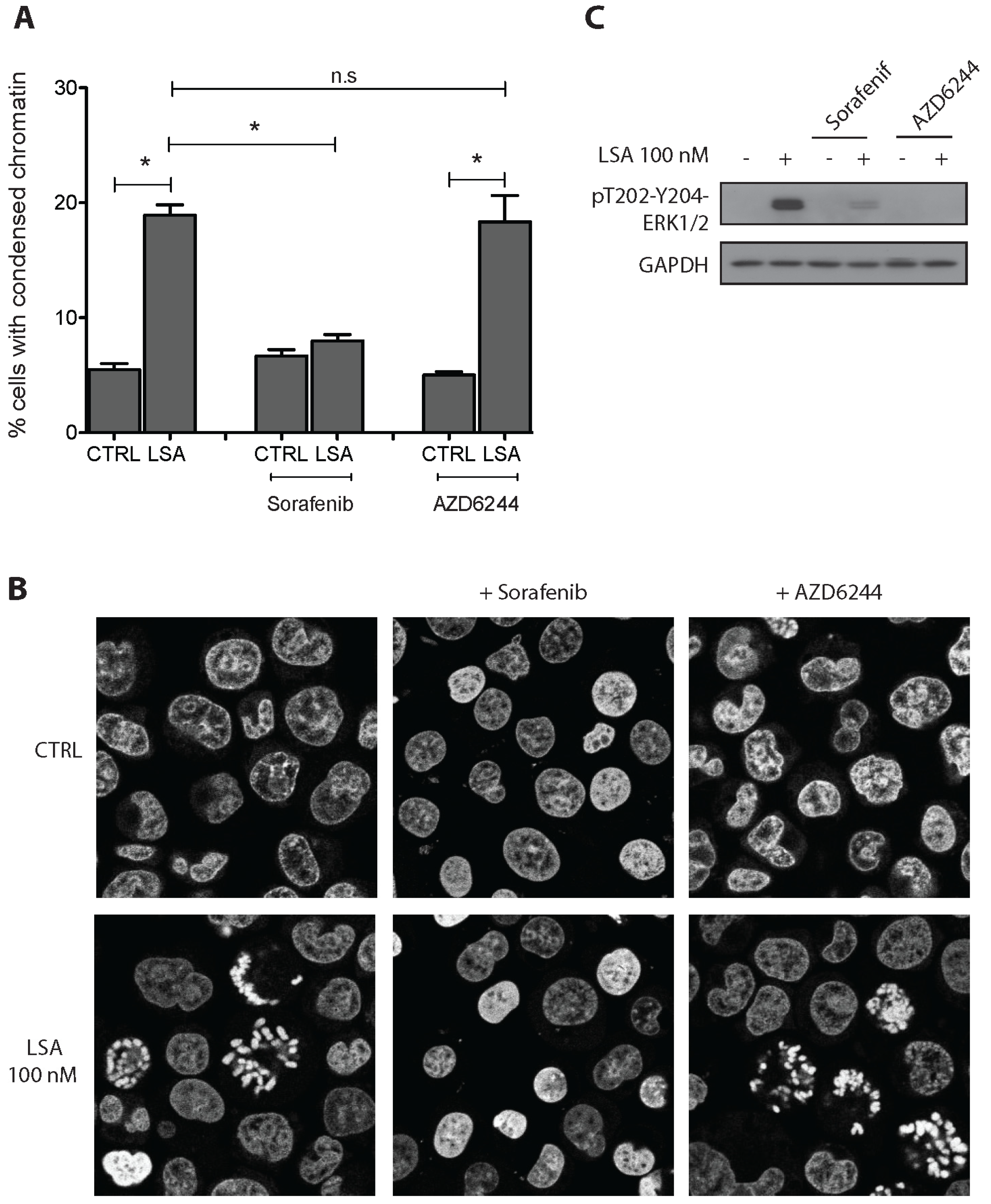
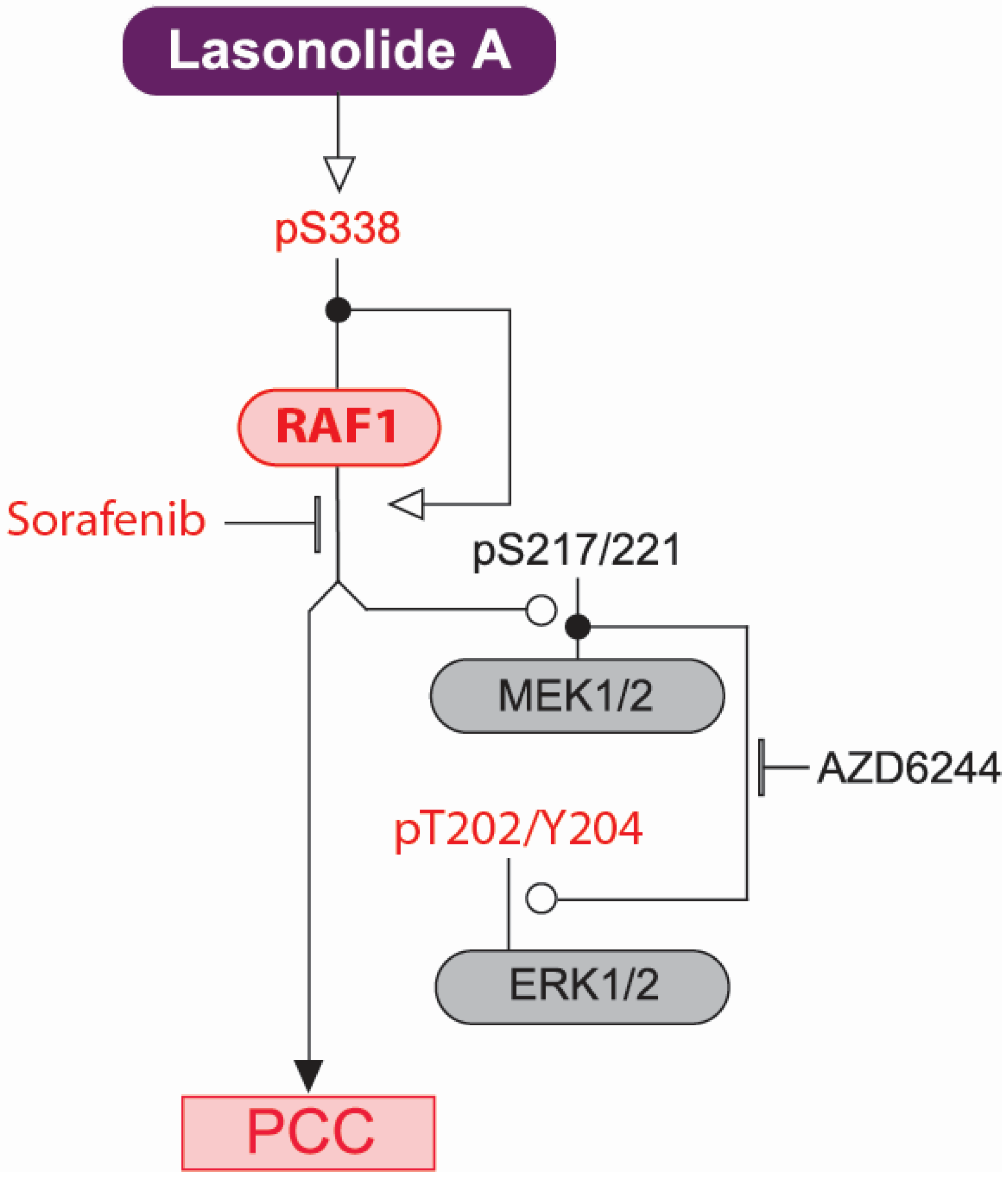
2.3. RAF1 Kinase Activity Is Involved in LSA-Induced PCC



2.4. Discussion

3. Materials and Methods
3.1. Drugs and Antibodies
3.2. Cell Culture and Treatments
3.3. shRNA Screening
3.4. Complementation of Raf−/− MEFs
3.5. siRNA Transfection
3.6. Protein Extracts and Western Blotting
3.7. Chromosome Morphology
3.8. Statistical Analyses
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bailly, C. Lamellarins, from A to Z: A family of anticancer marine pyrrole alkaloids. Curr. Med. Chem. Anticancer Agents 2004, 4, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar. Drugs 2014, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Kelkel, M.; Dicato, M.; Diederich, M. Gold from the sea: Marine compounds as inhibitors of the hallmarks of cancer. Biotechnol. Adv. 2011, 29, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Radjasa, O.K.; Vaske, Y.M.; Navarro, G.; Vervoort, H.C.; Tenney, K.; Linington, R.G.; Crews, P. Highlights of marine invertebrate-derived biosynthetic products: Their biomedical potential and possible production by microbial associants. Bioorg. Med. Chem. 2011, 19, 6658–6674. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Marchand, C. Interfacial inhibitors: Targeting macromolecular complexes. Nat. Rev. Drug Discov. 2012, 11, 25–36. [Google Scholar] [CrossRef]
- Changeux, J.-P. 50th anniversary of the word “allosteric”. Protein Sci. 2011, 20, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.A.; Koehn, F.E.; Longley, R.E.; McConnell, O.J. Lasonolide A, a new cytotoxic macrolide from the marine sponge Forcepia sp. J. Am. Chem. Soc. 1994, 116, 6015–6016. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Gong, G. Total synthesis of potent antitumor agent (−)-lasonolide A: A cycloaddition-based strategy. Chem. Asian J. 2008, 3, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Ren, G.B. Stereoselective synthesis of both tetrahydropyran rings of the antitumor macrolide, (−)-lasonolide A. J. Org. Chem. 2012, 77, 2559–2565. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Stivala, C.E.; Hull, K.L.; Huang, A.; Fandrick, D.R. A concise synthesis of (−)-lasonolide A. J. Am. Chem. Soc. 2014, 136, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Isbrucker, R.A.; Guzman, E.A.; Pitts, T.P.; Wright, A.E. Early effects of lasonolide A on pancreatic cancer cells. J. Pharmacol. Exp. Ther. 2009, 331, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Ghosh, A.K.; Pommier, Y. Lasonolide A, a potent and reversible inducer of chromosome condensation. Cell Cycle 2012, 11, 4424–4435. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, E.; Durante, M. Chromosome condensation outside of mitosis: Mechanisms and new tools. J. Cell Physiol. 2006, 209, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.S.; Springer, C.J.; Cooper, R.G.; Superti-Furga, G.; Marshall, C.J.; Marais, R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not b-Raf activation. EMBO J. 1999, 18, 2137–2148. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; von Kriegsheim, A.; Grindlay, J.; Kolch, W. Phosphatase and feedback regulation of Raf-1 signaling. Cell Cycle 2007, 6, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Laird, A.D.; Morrison, D.K.; Shalloway, D. Characterization of Raf-1 activation in mitosis. J. Biol. Chem. 1999, 274, 4430–4439. [Google Scholar] [CrossRef] [PubMed]
- Ziogas, A.; Lorenz, I.C.; Moelling, K.; Radziwill, G. Mitotic Raf-1 is stimulated independently of ras and is active in the cytoplasm. J. Biol. Chem. 1998, 273, 24108–24114. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Gong, J.; Luo, L.; Zhou, J.; Xiang, X.; Huang, W.; Huang, Q.; Luo, X.; Olbrot, M.; Peng, Y.; et al. Characterization of ser338 phosphorylation for Raf-1 activation. J. Biol. Chem. 2008, 283, 31429–31437. [Google Scholar] [CrossRef] [PubMed]
- Achiwa, H.; Lazo, J.S. Prl-1 tyrosine phosphatase regulates c-src levels, adherence, and invasion in human lung cancer cells. Cancer Res. 2007, 67, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.W.; Bugde, A.; Luby-Phelps, K.; Cobb, M.H. Wnk1 is required for mitosis and abscission. Proc. Natl. Acad. Sci. USA 2011, 108, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kirby, C.E.; Herbst, R. The tyrosine phosphatase prl-1 localizes to the endoplasmic reticulum and the mitotic spindle and is required for normal mitosis. J. Biol. Chem. 2002, 277, 46659–46668. [Google Scholar] [CrossRef] [PubMed]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Paavilainen, V.O.; Bertling, E.; Falck, S.; Lappalainen, P. Regulation of cytoskeletal dynamics by actin-monomer-binding proteins. Trends Cell Biol. 2004, 14, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Abaan, O.D.; Polley, E.C.; Davis, S.R.; Zhu, Y.J.; Bilke, S.; Walker, R.L.; Pineda, M.; Gindin, Y.; Jiang, Y.; Reinhold, W.C.; et al. The exomes of the nci-60 panel: A genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013, 73, 4372–4382. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Sahasrabudhe, R.M.; Luo, L.Z.; Lewis, D.W.; Gollin, S.M.; Saunders, W.S. Deficiency in myosin light-chain phosphorylation causes cytokinesis failure and multipolarity in cancer cells. Oncogene 2010, 29, 4183–4193. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.A.; Borman, M.A.; Muranyi, A.; Somlyo, A.V.; Hartshorne, D.J.; Haystead, T.A. Identification of the endogenous smooth muscle myosin phosphatase-associated kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 2419–2424. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Stites, E.C.; Yu, H.; Germino, E.A.; Meharena, H.S.; Stork, P.J.; Kornev, A.P.; Taylor, S.S.; Shaw, A.S. Allosteric activation of functionally asymmetric Raf kinase dimers. Cell 2013, 154, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.K.; Ritt, D.A.; Morrison, D.K. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol. Cell 2013, 49, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Reardon, D.B.; Morrison, D.K.; Sturgill, T.W. Regulation of Raf-1 and Raf-1 mutants by Ras-dependent and Ras-independent mechanisms in vitro. Mol. Cell Biol. 1995, 15, 4125–4135. [Google Scholar] [PubMed]
- Beeram, M.; Patnaik, A.; Rowinsky, E.K. Raf: A strategic target for therapeutic development against cancer. J. Clin. Oncol. 2005, 23, 6771–6790. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. Cot drives resistance to Raf inhibition through map kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Mielgo, A.; Seguin, L.; Huang, M.; Camargo, M.F.; Anand, S.; Franovic, A.; Weis, S.M.; Advani, S.J.; Murphy, E.A.; Cheresh, D.A. A mek-independent role for cRaf in mitosis and tumor progression. Nat. Med. 2011, 17, 1641–1645. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Sun, H.; Diaz, B.; Barnard, D.; Miao, W.; Bagrodia, S.; Marshall, M.S. The protein kinase pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature 1998, 396, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Salzano, M.; Rusciano, M.R.; Russo, E.; Bifulco, M.; Postiglione, L.; Vitale, M. Calcium/calmodulin-dependent protein kinase II (CaMKII) phosphorylates Raf-1 at serine 338 and mediates Ras-stimulated Raf-1 activation. Cell Cycle 2012, 11, 2100–2106. [Google Scholar] [CrossRef] [PubMed]
- Kohn, K.W.; Aladjem, M.I.; Weinstein, J.N.; Pommier, Y. Molecular interaction maps of bioregulatory networks: A general rubric for systems biology. Mol. Biol. Cell 2006, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Niault, T.; Sobczak, I.; Meissl, K.; Weitsman, G.; Piazzolla, D.; Maurer, G.; Kern, F.; Ehrenreiter, K.; Hamerl, M.; Moarefi, I.; et al. From autoinhibition to inhibition in trans: The Raf-1 regulatory domain inhibits Rok-alpha kinase activity. J. Cell Biol. 2009, 187, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreiter, K.; Piazzolla, D.; Velamoor, V.; Sobczak, I.; Small, J.V.; Takeda, J.; Leung, T.; Baccarini, M. Raf-1 regulates rho signaling and cell migration. J. Cell Biol. 2005, 168, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Schlabach, M.R.; Luo, J.; Solimini, N.L.; Hu, G.; Xu, Q.; Li, M.Z.; Zhao, Z.; Smogorzewska, A.; Sowa, M.E.; Ang, X.L.; et al. Cancer proliferation gene discovery through functional genomics. Science 2008, 319, 620–624. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jossé, R.; Zhang, Y.-W.; Giroux, V.; Ghosh, A.K.; Luo, J.; Pommier, Y. Activation of RAF1 (c-RAF) by the Marine Alkaloid Lasonolide A Induces Rapid Premature Chromosome Condensation. Mar. Drugs 2015, 13, 3625-3639. https://doi.org/10.3390/md13063625
Jossé R, Zhang Y-W, Giroux V, Ghosh AK, Luo J, Pommier Y. Activation of RAF1 (c-RAF) by the Marine Alkaloid Lasonolide A Induces Rapid Premature Chromosome Condensation. Marine Drugs. 2015; 13(6):3625-3639. https://doi.org/10.3390/md13063625
Chicago/Turabian StyleJossé, Rozenn, Yong-Wei Zhang, Valentin Giroux, Arun K. Ghosh, Ji Luo, and Yves Pommier. 2015. "Activation of RAF1 (c-RAF) by the Marine Alkaloid Lasonolide A Induces Rapid Premature Chromosome Condensation" Marine Drugs 13, no. 6: 3625-3639. https://doi.org/10.3390/md13063625
APA StyleJossé, R., Zhang, Y.-W., Giroux, V., Ghosh, A. K., Luo, J., & Pommier, Y. (2015). Activation of RAF1 (c-RAF) by the Marine Alkaloid Lasonolide A Induces Rapid Premature Chromosome Condensation. Marine Drugs, 13(6), 3625-3639. https://doi.org/10.3390/md13063625