Guanidinium Toxins and Their Interactions with Voltage-Gated Sodium Ion Channels



Abstract
1. Introduction
2. Origin and Proximal Sources of Guanidinium Toxins
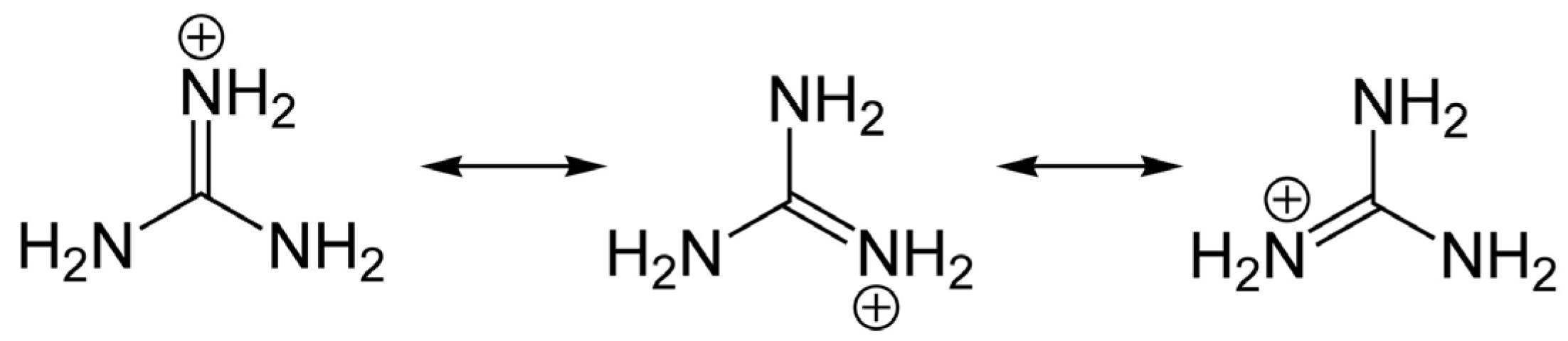
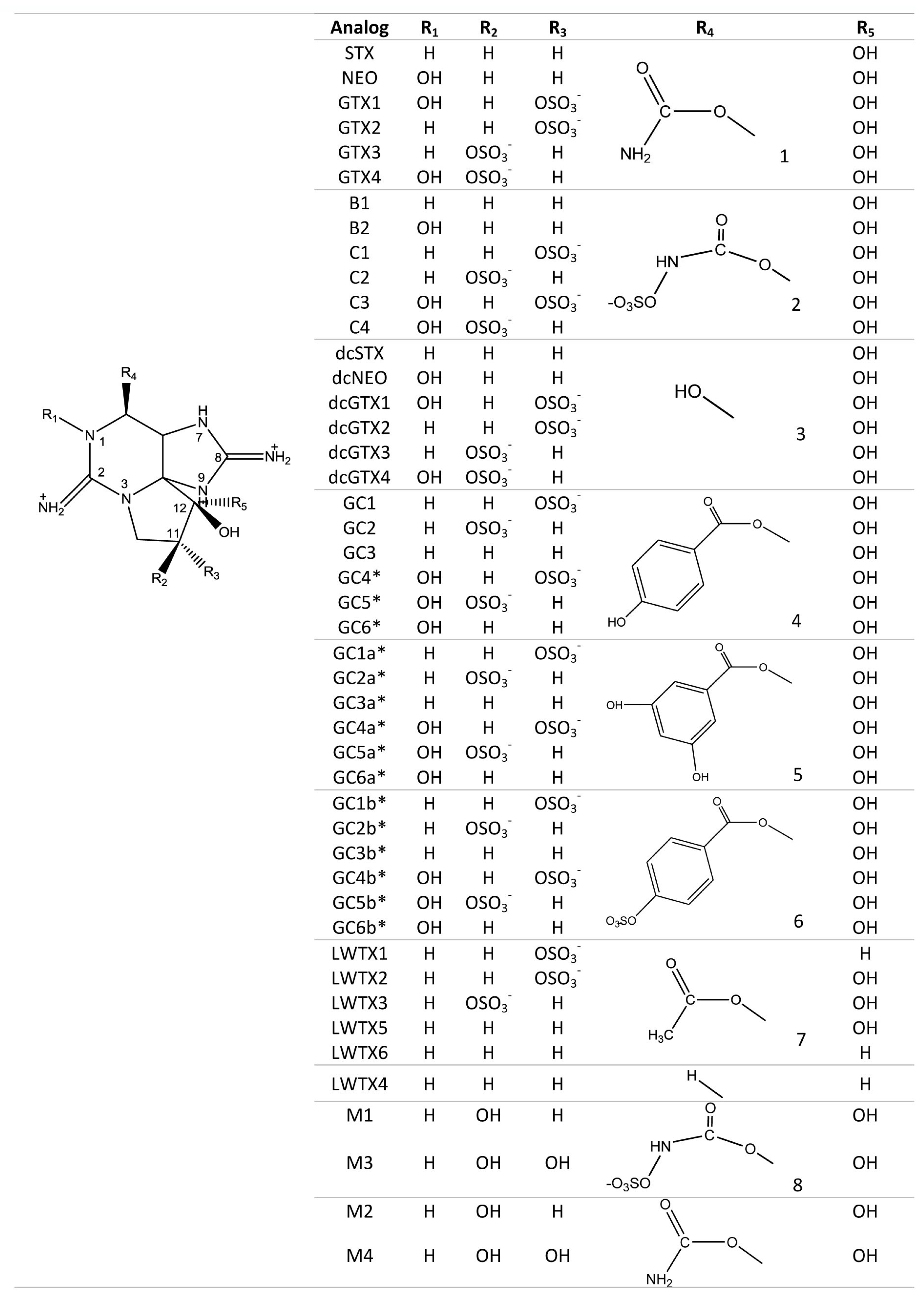
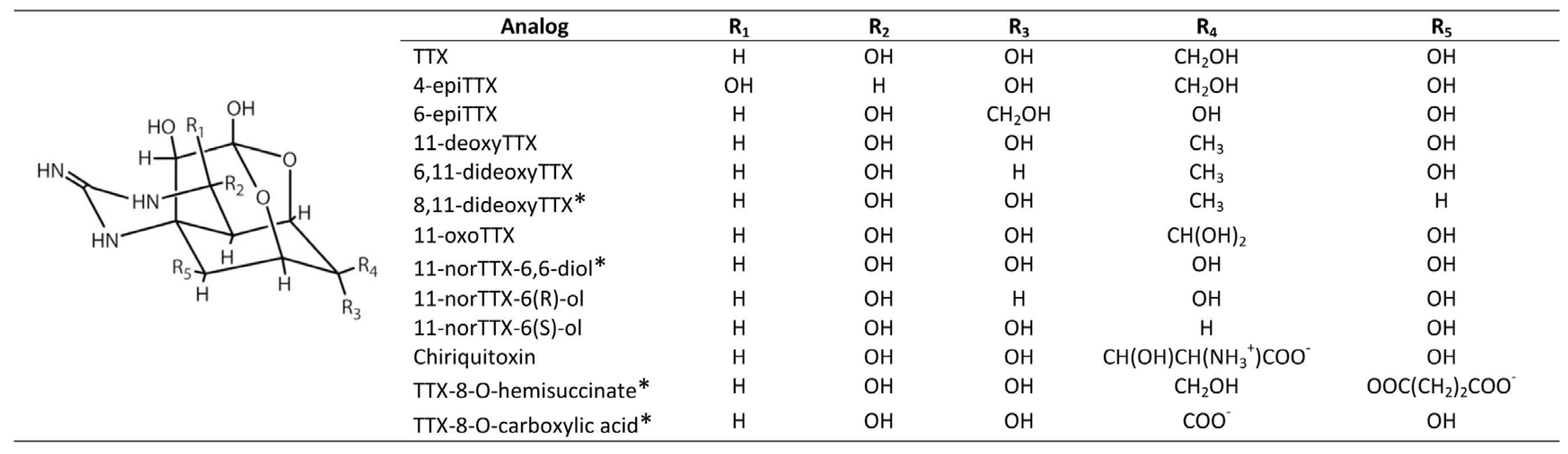
3. General Chemical and Toxicological Properties of Guanidinium Toxins
4. Symptomology and Etiology of Exposure to Guanidinium Toxins
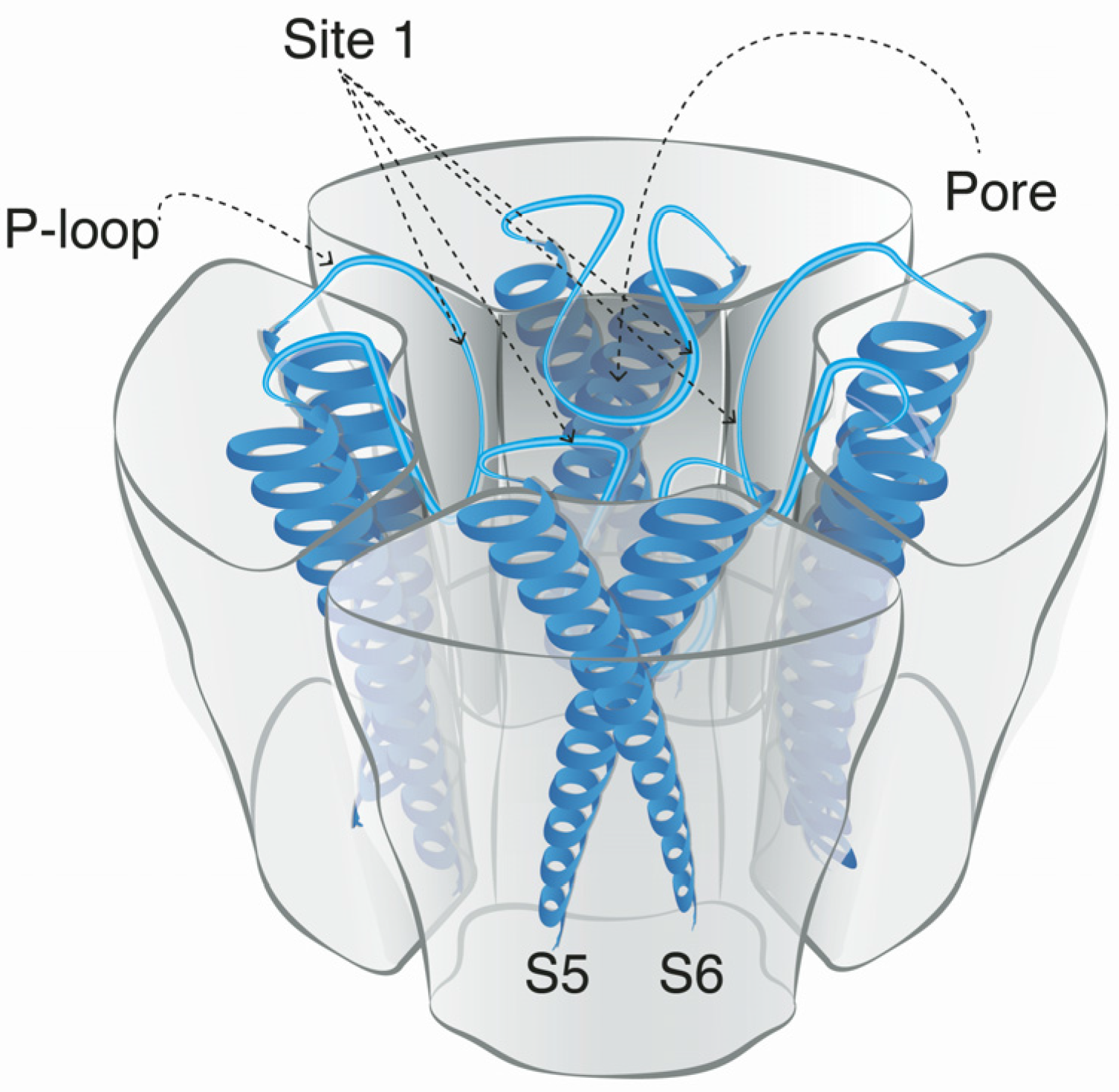
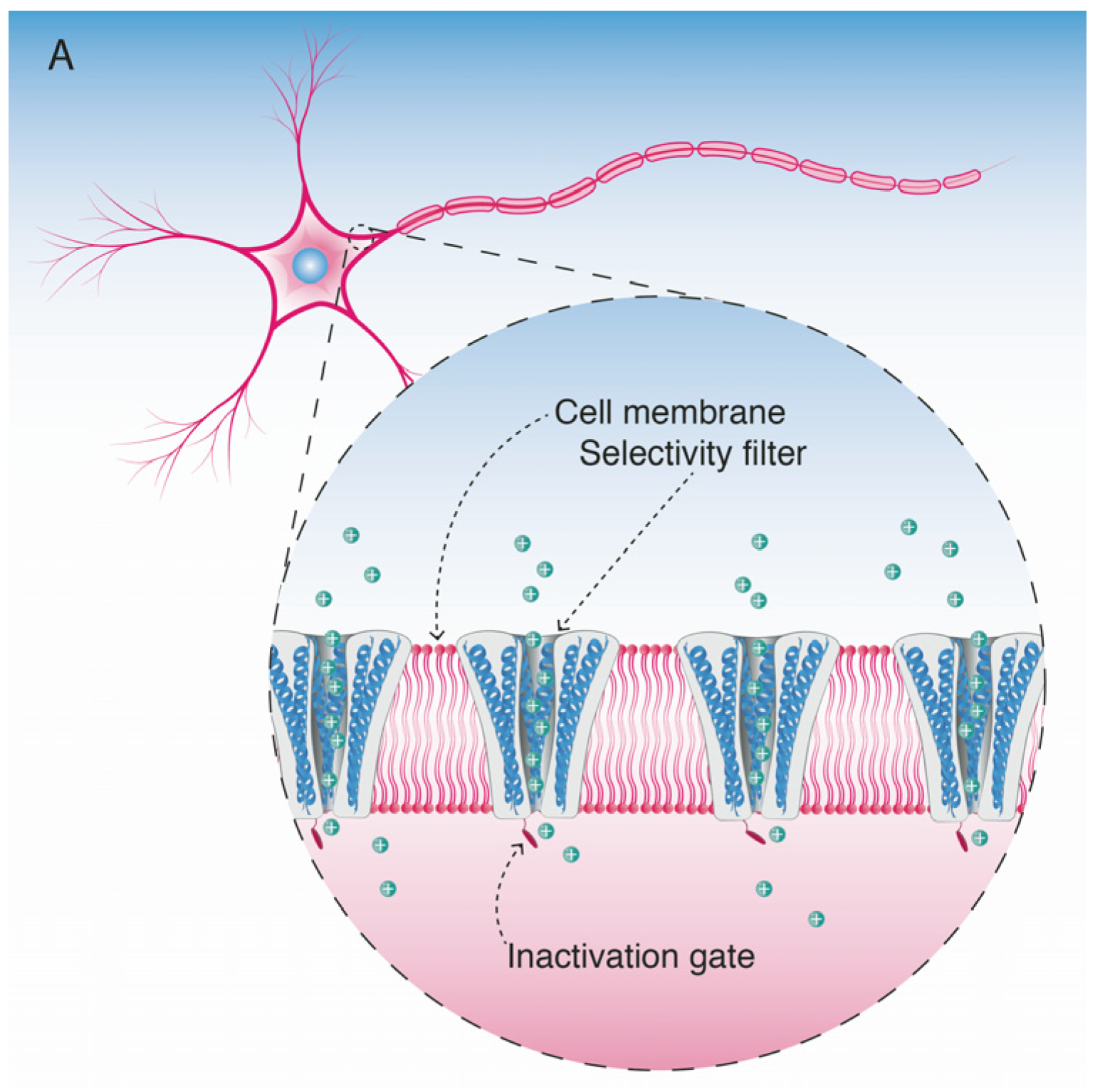
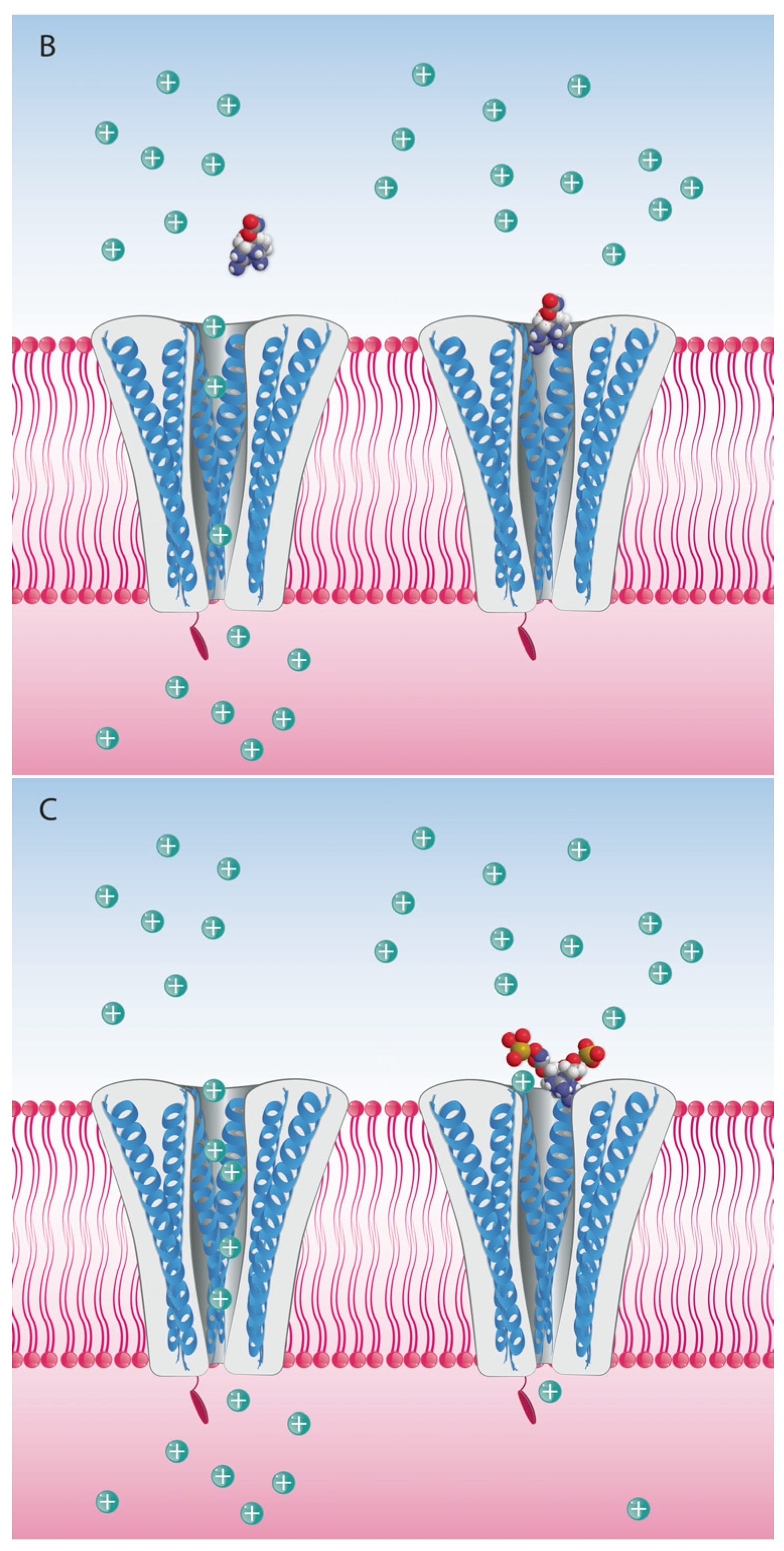
5. Mode of Action and Ion Channel Targets
6. Evolution of Voltage-Gated Na+ Ion Channels and Guanidinium Toxin Genes
7. Guanidinium Toxins as Ion Channel Research Subjects and Tools
7.1. Diagnostic Detection Assays for Guanidinium Toxins
7.2. Biomedical and Therapeutic Application of Guanidinium Toxins
7.3. Molecular Bioinformatics
8. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Yu, F.H.; Catterall, W.A. The VGL-chanome: A protein superfamily specialized for electrical signaling and ionic homeostasis. Sci. Signal. 2004, 253, re15. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.L.; Barchi, R.L.; Caldwell, J.H.; Hofmann, F.; Howe, J.R.; Hunter, J.C.; Kallen, R.G.; Mandel, G.; Meisler, M.H.; Netter, Y.B. Nomenclature of voltage-gated sodium channels. Neuron 2000, 28, 365–368. [Google Scholar] [CrossRef]
- Catterall, W.A.; Cestele, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.A.; Islam, Q.T.; Ekram, A.S. Puffer fish poisoning. TAJ 2008, 21, 199–202. [Google Scholar] [CrossRef]
- Davis, E.W. The ethnobiology of the Haitian zombi. J. Ethnopharmacol. 1983, 9, 85–104. [Google Scholar] [CrossRef]
- Schantz, E.J.; Ghazarossian, V.; Schnoes, H.K.; Strong, F.; Springer, J.; Pezzanite, J.O.; Clardy, J. Structure of saxitoxin. J. Am. Chem. Soc. 1975, 97, 1238–1239. [Google Scholar] [CrossRef] [PubMed]
- Field, J. Puffer fish poisoning. J. Accid Emerg. Med. 1998, 15, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Azevedo, J.; Rodriguez, P.; Alfonso, A.; Botana, L.M.; Vasconcelos, V. New gastropod vectors and tetrodotoxin potential expansion in temperate waters of the Atlantic Ocean. Mar. Drugs 2012, 10, 712–726. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.; Alfonso, A.; Vale, C.; Alfonso, C.; Vale, P.; Tellez, A.; Botana, L.M. First toxicity report of tetrodotoxin and 5, 6, 11-trideoxyTTX in the trumpet shell Charonia lampas lampas in Europe. Anal. Chem. 2008, 80, 5622–5629. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.; Powell, A.; Schofield, A.; Lees, D.; Baker-Austin, C. Detection of the pufferfish toxin tetrodotoxin in European bivalves, England, 2013 to 2014. Euro Surveill 2015, 20, 1–33. [Google Scholar] [CrossRef]
- Vlamis, A.; Katikou, P.; Rodriguez, I.; Rey, V.; Alfonso, A.; Papazachariou, A.; Zacharaki, T.; Botana, A.M.; Botana, L.M. First Detection of tetrodotoxin in Greek Shellfish by UPLC-MS/MS potentially linked to the presence of the dinoflagellate Prorocentrum minimum. Toxins 2015, 7, 1779–1807. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.S.; Rapoport, H. The pKa’s of saxitoxin. J. Am. Chem. Soc. 1980, 102, 7335–7339. [Google Scholar] [CrossRef]
- NCBI CID=11174599. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/11174599 (accessed on 31 August 2017).
- Fuhrman, F.A. Tetrodotoxin, tarichatoxin, and chiriquitoxin: Historical perspectives. Ann. NY Acad. Sci. 1986, 479, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Noguchi, T. Distribution and origin of tetrodotoxin. J. Toxicol. 2001, 20, 11–33. [Google Scholar] [CrossRef]
- Mosher, H.S.; Fuhrman, F.A. Occurrence and origin of tetrodotoxin. Seaf. Toxins 1984, 333–344. [Google Scholar] [CrossRef]
- Pires, O.R.; Sebben, A.; Schwartz, E.F.; Morales, R.A.; Bloch, C.; Schwartz, C.A. Further report of the occurrence of tetrodotoxin and new analogues in the Anuran family Brachycephalidae. Toxicon 2005, 45, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, F.A.; Fuhrman, G.J.; Mosher, H.S. Toxin from skin of frogs of the genus Atelopus: Differentiation from dendrobatid toxins. Science 1969, 165, 1376–1377. [Google Scholar] [CrossRef] [PubMed]
- Stokes, A.N.; Ducey, P.K.; Neuman-Lee, L.; Hanifin, C.T.; French, S.S.; Pfrender, M.E.; Brodie, E.D., III; Brodie, E.D., Jr. Confirmation and distribution of tetrodotoxin for the first time in terrestrial invertebrates: Two terrestrial flatworm species (Bipalium adventitium and Bipalium kewense). PLoS ONE 2014, 9, e100718. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-J.; Hwang, D.-F. Possible source of tetrodotoxin in the starfish Astropecten scoparius. Toxicon 2001, 39, 573–579. [Google Scholar] [CrossRef]
- Liu, F.-M.; Fu, Y.-M.; Shih, D.Y.-C. Occurrence of tetrodotoxin poisoning in Nassarius papillosus Alectrion and Nassarius gruneri Niotha. J. Food Drug Anal. 2004, 12, 189–192. [Google Scholar]
- Sato, K.-I.; Akai, S.; Shoji, H.; Sugita, N.; Yoshida, S.; Nagai, Y.; Suzuki, K.; Nakamura, Y.; Kajihara, Y.; Funabashi, M. Stereoselective and efficient total synthesis of optically active tetrodotoxin from D-glucose. J. Org. Chem. 2008, 73, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, T.; Yasumura, D.; Yotsu, M.; Michishita, T.; Endo, A.; Kotaki, Y. Bacterial production of tetrodotoxin and anhydrotetrodotoxin. Agr. Biol. Chem. Tokyo 1986, 50, 793–795. [Google Scholar]
- Bane, V.; Lehane, M.; Dikshit, M.; O’Riordan, A.; Furey, A. Tetrodotoxin: Chemistry, toxicity, source, distribution and detection. Toxins 2014, 6, 693–755. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Matsui, T.; Furukawa, K.; Yotsu-Yamashita, M.; Yamamori, K. Accumulation of tetrodotoxin and 4, 9-anhydrotetrodotoxin in cultured juvenile kusafugu Fugu niphobles by dietary administration of natural toxic komonfugu Fugu poecilonotus liver. Toxicon 2008, 51, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Taketsugu, S.; Sato, H.; Yamamori, K.; Kodama, K.; Ishii, A.; Hirose, H.; Shimizu, C. Toxification of cultured puffer fish by the administration of tetrodotoxin-producing bacteria. Nippon Suisan Gakk. 1990, 56, 705. [Google Scholar] [CrossRef]
- Matsumura, K. Production of tetrodotoxin in puffer fish embryos. Environ. Toxicol. Phar. 1998, 6, 217–219. [Google Scholar] [CrossRef]
- Hanifin, C.T.; Brodie, E.D. Tetrodotoxin levels of the rough-skin newt, Taricha granulosa, increase in long-term captivity. Toxicon 2002, 40, 1149–1153. [Google Scholar] [CrossRef]
- Shumway, S.E. A review of the effects of algal blooms on shellfish and aquaculture. J. World Aquacult. Soc. 1990, 21, 65–104. [Google Scholar] [CrossRef]
- Cembella, A. Ecophysiology and metabolism of paralytic shellfish toxins in marine microalgae. In Physiological Ecology of Harmful Algal Blooms; NATO-Advanced Study Institute Series; Anderson, D.M., Cembella, A.D., Hallegraeff, G.M., Eds.; Springer: Heidelberg, Germany, 1998; Volume 41, pp. 381–404. [Google Scholar]
- Hallegraeff, G.M. A review of harmful algal blooms and their apparent global increase. Phycologia 1993, 32, 79–99. [Google Scholar] [CrossRef]
- De Carvalho, M.; Jacinto, J.; Ramos, N.; de Oliveira, V.; Pinho e Melo, T.; de Sá, J. Paralytic shellfish poisoning: Clinical and electrophysiological observations. J. Neurol. 1998, 245, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Wacklin, P.; Hoffmann, L.; Komárek, J. Nomenclatural validation of the genetically revised cyanobacterial genus Dolichospermum (Ralfs ex Bornet et Flahault) comb. nova. Fottea 2009, 9, 59–64. [Google Scholar] [CrossRef]
- Carmichael, W.; Evans, W.; Yin, Q.; Bell, P.; Moczydlowski, E. Evidence for paralytic shellfish poisons in the freshwater cyanobacterium Lyngbya wollei (Farlow ex Gomont) comb. nov. Appl. Environ. Microb. 1997, 63, 3104–3110. [Google Scholar]
- Carmichael, W.W. Health effects of toxin-producing cyanobacteria: “The CyanoHABs”. Hum. Ecol. Risk Assess. 2001, 7, 1393–1407. [Google Scholar] [CrossRef]
- Negri, A.P.; Jones, G.J.; Hindmarsh, M. Sheep mortality associated with paralytic shellfish poisons from the cyanobacterium Anabaena circinalis. Toxicon 1995, 33, 1321–1329. [Google Scholar] [CrossRef]
- Wiese, M.; D’Agostino, P.M.; Mihali, T.K.; Moffitt, M.C.; Neilan, B.A. Neurotoxic alkaloids: Saxitoxin and its analogs. Mar. Drugs 2010, 8, 2185–2211. [Google Scholar] [CrossRef] [PubMed]
- Yotsu-Yamashita, M.; Sugimoto, A.; Takai, A.; Yasumoto, T. Effects of specific modifications of several hydroxyls of tetrodotoxin on its affinity to rat brain membrane. J. Pharmacol. Exp. Ther. 1999, 289, 1688–1696. [Google Scholar] [PubMed]
- Durán-Riveroll, L.; Cembella, A.; Band-Schmidt, C.; Bustillos-Guzmán, J.; Correa-Basurto, J. Docking simulation of the binding interactions of saxitoxin analogs produced by the marine dinoflagellate Gymnodinium catenatum to the voltage-gated sodium channel NaV1.4. Toxins 2016, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.J.; Wekell, M.M.; Kentala, L.L. Application of HPLC for the determination of PSP toxins in shellfish. J. Food Sci. 1985, 50, 26–29. [Google Scholar] [CrossRef]
- Oshima, Y. Post-column derivatization HPLC methods for paralytic shellfish poisons. In Manual on Harmful Marine Microalgae; Hallegraeff, G.M., Anderson, D.M., Cembella, A.D., Eds.; UNESCO: Paris, France, 1995; pp. 81–94. [Google Scholar]
- Munday, R.; Thomas, K.; Gibbs, R.; Murphy, C.; Quilliam, M.A. Acute toxicities of saxitoxin, neosaxitoxin, decarbamoyl saxitoxin and gonyautoxins 1&4 and 2&3 to mice by various routes of administration. Toxicon 2013, 76, 77–83. [Google Scholar] [PubMed]
- Llewellyn, L.E. Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat. Prod. Rep. 2006, 23, 200–222. [Google Scholar] [CrossRef] [PubMed]
- Islam, Q.; Razzak, M.; Islam, M.; Bari, M.; Basher, A.; Chowdhury, F.; Sayeduzzaman, A.; Ahasan, H.; Faiz, M.; Arakawa, O. Puffer fish poisoning in Bangladesh: Clinical and toxicological results from large outbreaks in 2008. Trans. R. Soc. Trop. Med. Hyg. 2011, 105, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Lago, J.; Rodriguez, L.P.; Blanco, L.; Vieites, J.M.; Cabado, A.G. Tetrodotoxin, an extremely potent marine neurotoxin: Distribution, toxicity, origin and therapeutical uses. Mar. Drugs 2015, 13, 6384–6406. [Google Scholar] [CrossRef] [PubMed]
- González-Cano, R.; Tejada, M.Á.; Artacho-Cordón, A.; Nieto, F.R.; Entrena, J.M.; Wood, J.N.; Cendán, C.M. Effects of tetrodotoxin in mouse models of visceral pain. Mar. Drugs 2017, 15, 188. [Google Scholar] [CrossRef] [PubMed]
- Abal, P.; Louzao, M.C.; Antelo, A.; Alvarez, M.; Cagide, E.; Vilariño, N.; Vieytes, M.R.; Botana, L.M. Acute oral toxicity of tetrodotoxin in mice: Determination of lethal dose 50 (LD50) and No Observed Adverse Effect Level (NOAEL). Toxins 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Nakamura, H.; Ohizumi, Y.; Kobayashi, J.I.; Hirata, Y. The amino acid sequences of homologous hydroxyproline-containing myotoxins from the marine snal Conus geographus venom. FEBS Lett. 1983, 155, 277–280. [Google Scholar] [CrossRef]
- Tubaro, A.; Sosa, S.; Hungerford, J. Toxicology and Diversity of Marine Toxins; Elsevier: New York, NY, USA, 2012; pp. 896–936. [Google Scholar]
- Wang, D.Z. Neurotoxins from marine dinoflagellates: A brief review. Mar. Drugs 2008, 6, 349–371. [Google Scholar] [CrossRef] [PubMed]
- How, C.-K.; Chern, C.-H.; Huang, Y.-C.; Wang, L.-M.; Lee, C.-H. Tetrodotoxin poisoning. Am. J. Emerg. Med. 2003, 21, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Garcı́a, C.; del Carmen Bravo, M.A.; Lagos, M.; Lagos, N. Paralytic shellfish poisoning: Post-mortem analysis of tissue and body fluid samples from human victims in the Patagonia fjords. Toxicon 2004, 43, 149–158. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, K.; Musgrave, I.F.; Humpage, A. Low dose extended exposure to saxitoxin and its potential neurodevelopmental effects: A review. Environ. Toxicol. Pharmacol. 2016, 48, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Baden, D.; Fleming, L.E.; Bean, J.A.; deWolf, F.A. (Eds.) Chapter: Marine Toxins. In Handbook of Clinical Neurology: Intoxications of the Nervous System Part H. Natural Toxins and Drugs; Elsevier: Amsterdam, The Nederland, 1995; pp. 141–175. [Google Scholar]
- Llewellyn, L.; Negri, A.; Robertson, A. Paralytic shellfish toxins in tropical oceans. Toxin Rev. 2006, 25, 159–196. [Google Scholar] [CrossRef]
- Chorus, I.; Bartram, J. (Eds.) Toxic cyanobacteria in water. A guide to their public health consequences, monitoring, and management. E & FN Spon: New York, NY, USA, 1999. [Google Scholar]
- Fishbein, I.; Segal, M. Miniature synaptic currents become neurotoxic to chronically silenced neurons. Cereb. Cortex 2007, 17, 1292–1306. [Google Scholar] [CrossRef] [PubMed]
- Brackenbury, W.J.; Calhoun, J.D.; Chen, C.; Miyazaki, H.; Nukina, N.; Oyama, F.; Ranscht, B.; Isom, L.L. Functional reciprocity between Na+ channel NaV1. 6 and β1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc. Natl. Acad. Sci. USA 2010, 107, 2283–2288. [Google Scholar] [CrossRef] [PubMed]
- Clemente, Z.; Busato, R.H.; Oliveira Ribeiro, C.A.; Cestari, M.M.; Ramsdorf, W.A.; Magalhaes, V.F.; Wosiack, A.C.; Silva de Assis, H.C. Analyses of paralytic shellfish toxins and biomarkers in a southern Brazilian reservoir. Toxicon 2010, 55, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Lam, P.K.; Hsieh, D.P. Interactions of paralytic shellfish toxins with xenobiotic-metabolizing and antioxidant enzymes in rodents. Toxicon 2003, 42, 425–431. [Google Scholar] [CrossRef]
- Da Silva, C.A.; Oba, E.T.; Ramsdorf, W.A.; Magalhães, V.F.; Cestari, M.M.; Ribeiro, C.A.O.; de Assis, H.C.S. First report about saxitoxins in freshwater fish Hoplias malabaricus through trophic exposure. Toxicon 2011, 57, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.B.; Diehl, F.; dos Santos, J.M.; Monserrat, J.M.; Yunes, J.S. Oxidative stress in rats induced by consumption of saxitoxin contaminated drink water. Harmful Algae 2014, 37, 68–74. [Google Scholar] [CrossRef]
- Gupta, R.C. Brain regional heterogeneity and toxicological mechanisms of organophosphates and carbamates. Toxicol. Mech. Method 2004, 14, 103–143. [Google Scholar] [CrossRef] [PubMed]
- Rein, K.S.; Borrone, J. Polyketides from dinoflagellates: Origins, pharmacology and biosynthesis. Comp. Biochem. Phys. B 1999, 124, 117–131. [Google Scholar] [CrossRef]
- Wang, J.; Salata, J.J.; Bennett, P.B. Saxitoxin is a gating modifier of HERG K+ channels. J. Gen. Physiol. 2003, 121, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Cusick, K.D.; Sayler, G.S. An overview on the marine neurotoxin, saxitoxin: Genetics, molecular targets, methods of detection and ecological functions. Mar. Drugs 2013, 11, 991–1018. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Sheets, M.; Ishida, H.; Li, F.; Barry, W.H. Saxitoxin blocks L-type ICa. J. Pharmacol. Exp. Ther. 2004, 308, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, L.E. Sodium channel inhibiting marine toxins. In Marine Toxins as Research Tools; Springer: Berlin, Germany, 2009; pp. 67–97. [Google Scholar]
- Hodgkin, A.L.; Huxley, A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 500. [Google Scholar] [CrossRef] [PubMed]
- Narahashi, T.; Moore, J.W.; Scott, W.R. Tetrodotoxin blockage of sodium conductance increase in lobster giant axons. J. Gen. Physiol. 1964, 47, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Kao, C. Comparison of the biological actions of tetrodotoxin and saxitoxin. In Animal Toxins: A Collection of Papers Presented at the First International Symposium on Animal Toxins; Elsevier: Atlantic City, NJ, USA, 1966; p. 109. [Google Scholar]
- Narahashi, T. Tetrodotoxin. P JPN Acad B 2008, 84, 147–154. [Google Scholar] [CrossRef]
- Thottumkara, A.P.; Parsons, W.H.; Du Bois, J. Saxitoxin. Angew. Chem. Int. Ed. Engl. 2014, 53, 5760–5784. [Google Scholar] [CrossRef] [PubMed]
- McGlothlin, J.W.; Chuckalovcak, J.P.; Janes, D.E.; Edwards, S.V.; Feldman, C.R.; Brodie, E.D., Jr.; Pfrender, M.E.; Brodie, E.D., III. Parallel evolution of tetrodotoxin resistance in three voltage-gated sodium channel genes in the garter snake Thamnophis sirtalis. Mol. Biol. Evol. 2014, 31, 2836–2846. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, N.; Yotsu-Yamashita, M. Selective blocking effects of 4, 9-anhydrotetrodotoxin, purified from a crude mixture of tetrodotoxin analogues, on NaV1. 6 channels and its chemical aspects. Mar. Drugs 2015, 13, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Cestèle, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Denac, H.; Mevissen, M.; Scholtysik, G. Structure, function and pharmacology of voltage-gated sodium channels. N-S Arch. Pharmakol. 2000, 362, 453–479. [Google Scholar] [CrossRef]
- Amir, R.; Argoff, C.E.; Bennett, G.J.; Cummins, T.R.; Durieux, M.E.; Gerner, P.; Gold, M.S.; Porreca, F.; Strichartz, G.R. The role of sodium channels in chronic inflammatory and neuropathic pain. J. Pain 2006, 7 (Suppl. 3), S1–S29. [Google Scholar] [CrossRef] [PubMed]
- Black, J.; Dib-Hajj, S.; McNabola, K.; Jeste, S.; Rizzo, M.; Kocsis, J.; Waxman, S. Spinal sensory neurons express multiple sodium channel α-subunit mRNAs. Mol. Brain Res. 1996, 43, 117–131. [Google Scholar] [CrossRef]
- Terlau, H.; Heinemann, S.H.; Stühmer, W.; Pusch, M.; Conti, F.; Imoto, K.; Numa, S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 1991, 293, 93–96. [Google Scholar] [CrossRef]
- Choudhary, G.; Shang, L.; Li, X.; Dudley, S.C. Energetic localization of saxitoxin in its channel binding site. Biophys. J. 2002, 83, 912–919. [Google Scholar] [CrossRef]
- Kirsch, G.; Alam, M.; Hartmann, H. Differential effects of sulfhydryl reagents on saxitoxin and tetrodotoxin block of voltage-dependent Na channels. Biophys. J. 1994, 67, 2305–2315. [Google Scholar] [CrossRef]
- Choudhary, G.; Yotsu-Yamashita, M.; Shang, L.; Yasumoto, T.; Dudley, S.C. Interactions of the C-11 hydroxyl of tetrodotoxin with the sodium channel outer vestibule. Biophys. J. 2003, 84, 287–294. [Google Scholar] [CrossRef]
- Zhorov, B.S.; Tikhonov, D.B. Potassium, sodium, calcium and glutamate-gated channels: Pore architecture and ligand action. J. Neurochem. 2004, 88, 782–799. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, S.; Kuyucak, S. Mechanism of ion permeation in mammalian voltage-gated sodium channels. PLoS ONE 2015, 10, e0133000. [Google Scholar] [CrossRef] [PubMed]
- Toledo, G.; Hanifin, C.; Geffeney, S.; Brodie, E. Convergent evolution of tetrodotoxin-resistant sodium channels in predators and prey. Curr. Top. Membr. 2016, 78, 87–113. [Google Scholar] [PubMed]
- Noda, M.; Suzuki, H.; Numa, S.; Stühmer, W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 1989, 259, 213–216. [Google Scholar] [CrossRef]
- Walker, J.R.; Novick, P.A.; Parsons, W.H.; McGregor, M.; Zablocki, J.; Pande, V.S.; Du Bois, J. Marked difference in saxitoxin and tetrodotoxin affinity for the human nociceptive voltage-gated sodium channel (NaV1. 7). Proc. Natl. Acad. Sci. USA 2012, 109, 18102–18107. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Ionic Channels of Excitable Membranes. Sinauer, Sunderland, MA. 1–426. Hondeghem, LM, and BG Katzung. 1977. Time and voltage dependent interaction of antiarrhythmic drugs with cardiac sodium channels. Biochim. Biophys. Acta 1984, 472, 373–398. [Google Scholar]
- Leys, S.P.; Mackie, G.O.; Meech, R. Impulse conduction in a sponge. J. Exp. Biol. 1999, 202, 1139–1150. [Google Scholar] [PubMed]
- Liebeskind, B.J.; Hillis, D.M.; Zakon, H.H. Evolution of sodium channels predates the origin of nervous systems in animals. Proc. Natl. Acad. Sci. USA 2011, 108, 9154–9159. [Google Scholar] [CrossRef] [PubMed]
- Zakon, H.H. Adaptive evolution of voltage-gated sodium channels: The first 800 million years. Proc. Natl. Acad. Sci. USA 2012, 109 (Suppl. 1), 10619–10625. [Google Scholar] [CrossRef] [PubMed]
- Twarog, B.M.; Hidaka, T.; Yamaguchi, H. Resistance to tetrodotoxin and saxitoxin in nerves of bivalve molluscs: A possible correlation with paralytic shellfish poisoning. Toxicon 1972, 10, 273–278. [Google Scholar] [CrossRef]
- Bricelj, V.M.; Connell, L.; Konoki, K.; MacQuarrie, S.P.; Scheuer, T.; Catterall, W.A.; Trainer, V.L. Sodium channel mutation leading to saxitoxin resistance in clams increases risk of PSP. Nature 2005, 434, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Feldman, C.R.; Brodie, E.D.; Pfrender, M.E. Constraint shapes convergence in tetrodotoxin-resistant sodium channels of snakes. Proc. Natl. Acad. Sci. USA 2012, 109, 4556–4561. [Google Scholar] [CrossRef] [PubMed]
- Kellmann, R.; Mihali, T.K.; Neilan, B.A. Identification of a saxitoxin biosynthesis gene with a history of frequent horizontal gene transfers. J. Mol. Evol. 2008, 67, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Kellmann, R.; Mihali, T.K.; Jeon, Y.J.; Pickford, R.; Pomati, F.; Neilan, B.A. Biosynthetic intermediate analysis and functional homology reveal a saxitoxin gene cluster in cyanobacteria. Appl. Environ. Microbiol. 2008, 74, 4044–4053. [Google Scholar] [CrossRef] [PubMed]
- Mihali, T.K.; Kellmann, R.; Neilan, B.A. Characterisation of the paralytic shellfish toxin biosynthesis gene clusters in Anabaena circinalis AWQC131C and Aphanizomenon sp. NH-5. BMC Biochem. 2009, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Soto-Liebe, K.; Murillo, A.A.; Krock, B.; Stucken, K.; Fuentes-Valdés, J.J.; Trefault, N.; Cembella, A.; Vásquez, M. Reassessment of the toxin profile of Cylindrospermopsis raciborskii T3 and function of putative sulfotransferases in synthesis of sulfated and sulfonated PSP toxins. Toxicon 2010, 56, 1350–1361. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Loram, J.E.; Hackett, J.D.; Anderson, D.M.; Plumley, F.G.; Bhattacharya, D. Origin of saxitoxin biosynthetic genes in cyanobacteria. PLoS ONE 2009, 4, e5758. [Google Scholar] [CrossRef] [PubMed]
- Stüken, A.; Orr, R.J.; Kellmann, R.; Murray, S.A.; Neilan, B.A.; Jakobsen, K.S. Discovery of nuclear-encoded genes for the neurotoxin saxitoxin in dinoflagellates. PLoS ONE 2011, 6, e20096. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.A.; Mihali, T.K.; Neilan, B.A. Extraordinary conservation, gene loss, and positive selection in the evolution of an ancient neurotoxin. J. Mol. Evol. 2010, 28, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Pomati, F.; Burns, B.P.; Neilan, B.A. Identification of an Na+-dependent transporter associated with saxitoxin-producing strains of the cyanobacterium Anabaena circinalis. Appl. Environ. Microbiol. 2004, 70, 4711–4719. [Google Scholar] [CrossRef] [PubMed]
- Pomati, F.; Rossetti, C.; Manarolla, G.; Burns, B.P.; Neilan, B.A. Interactions between intracellular Na+ levels and saxitoxin production in Cylindrospermopsis raciborskii T3. Microbiology 2004, 150, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 2016, 17, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Fensome, R.A. A classification of living and fossil dinoflagellates. Micropaleontol. Spec. Public. 1993, 7, 351. [Google Scholar]
- John, U.; Fensome, R.A.; Medlin, L.K. The application of a molecular clock based on molecular sequences and the fossil record to explain biogeographic distributions within the Alexandrium tamarense “species complex” (Dinophyceae). Mol. Biol. Evol. 2003, 20, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Orr, R.J.; Stuken, A.; Murray, S.A.; Jakobsen, K.S. Evolution and distribution of saxitoxin biosynthesis in dinoflagellates. Mar. Drugs 2013, 11, 2814–2828. [Google Scholar] [CrossRef] [PubMed]
- Orr, R.J.; Stüken, A.; Murray, S.A.; Jakobsen, K.S. Evolutionary acquisition and loss of saxitoxin biosynthesis in dinoflagellates: The second “core” gene, sxtG. Appl. Environ. Microbiol. 2013, 79, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Cembella, A.D. Chemical ecology of eukaryotic microalgae in marine ecosystems. Phycologia 2003, 42, 420–447. [Google Scholar] [CrossRef]
- AOAC. Official Method 959.08—Paralytic shellfish poison, biological method. In Official Methods of Analysis, 17th ed.; Chemists, A.O.O.A., Ed.; AOAC: Arlington, VA, USA, 1999; Volume 1. [Google Scholar]
- AOAC. Official Method 959.08. Paralytic Shellfish Poison. Biological Method, 18th ed.; AOAC International: Gaithersburg, MA, USA, 2005. [Google Scholar]
- Hallegraeff, G.M. 13 Seafood quality assurance for algal toxins. In Environmental Effects on Seafood Availability, Safety, and Quality; Taylor & Francis: New York, NY, USA, 2016; Volume 201. [Google Scholar]
- Hallegraeff, G.M.; Anderson, D.M.; Cembella, A.D.; Enevoldsen, H.O. Manual on Harmful Marine Microalgae; Unesco: Hong Kong, China, 2003. [Google Scholar]
- Vilariño, N.; Fraga, M.; Rodríguez, L.P. Functional and Receptor-Based Assays for Marine Toxins. In Seafood and Freshwater Toxins: Pharmacology, Physiology, and Detection; CRC Press: Boca Raton, FL, USA, 2014; Volume 333. [Google Scholar]
- McNabb, P.; Selwood, A.I.; Holland, P.T. Multiresidue method for determination of algal toxins in shellfish: Single-laboratory validation and interlaboratory study. J. AOAC Int. 2005, 88, 761–772. [Google Scholar] [PubMed]
- Catterall, W.A. Activation of the action potential Na+ ionophore of cultured neuroblastoma cells by veratridine and batrachotoxin. J. Biol. Chem. 1975, 250, 4053–4059. [Google Scholar] [PubMed]
- Catterall, W.A.; Risk, M. Toxin T446 from Ptychodiscus brevis (formerly Gymnodinium breve) enhances activation of voltage-sensitive sodium channels by veratridine. Mol. Pharmacol. 1981, 19, 345–348. [Google Scholar] [PubMed]
- Kimelberg, H. Active potassium transport and [Na+ K+] ATPase activity in cultured glioma and neuroblastoma cells. J. Neurochem. 1974, 22, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Kogure, K.; Tamplin, M.L.; Simidu, U.; Colwell, R.R. A tissue culture assay for tetrodotoxin, saxitoxin and related toxins. Toxicon 1988, 26, 191–197. [Google Scholar] [CrossRef]
- Jellett, J.F.; Marks, L.J.; Stewart, J.E.; Dorey, M.L.; Watson-Wright, W.; Lawrence, J.F. Paralytic shellfish poison (saxitoxin family) bioassays: Automated endpoint determination and standardization of the in vitro tissue culture bioassay, and comparison with the standard mouse bioassay. Toxicon 1992, 30, 1143–1156. [Google Scholar] [CrossRef]
- Humpage, A.R.; Ledreux, A.; Fanok, S.; Bernard, C.; Briand, J.F.; Eaglesham, G.; Papageorgiou, J.; Nicholson, B.; Steffensen, D. Application of the neuroblastoma assay for paralytic shellfish poisons to neurotoxic freshwater cyanobacteria: Interlaboratory calibration and comparison with other methods of analysis. Environ. Toxicol. Chem. 2007, 26, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Louzao, M.C.; Vieytes, M.R.; Cabado, A.G.; Vieites Baptista de Sousa, J.M.; Botana, L.M. A fluorimetric microplate assay for detection and quantitation of toxins causing paralytic shellfish poisoning. Chem. Res. Toxicol. 2003, 16, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Davio, S.R.; Fontelo, P.A. A competitive displacement assay to detect saxitoxin and tetrodotoxin. Anal. Biochem. 1984, 141, 199–204. [Google Scholar] [CrossRef]
- Vilarino, N.; Louzao, M.C.; Vieytes, M.R.; Botana, L.M. Biological methods for marine toxin detection. Anal. Bioanal. Chem. 2010, 397, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Usup, G.; Leaw, C.P.; Cheah, M.Y.; Ahmad, A.; Ng, B.K. Analysis of paralytic shellfish poisoning toxin congeners by a sodium channel receptor binding assay. Toxicon 2004, 44, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Ruberu, S.R.; Liu, Y.-G.; Wong, C.T.; Perera, S.K.; Langlois, G.W.; Doucette, G.J.; Powell, C.L. Receptor binding assay for paralytic shellfish poisoning toxins: Optimization and interlaboratory comparison. J. AOAC Int. 2003, 86, 737–745. [Google Scholar] [PubMed]
- Doucette, G.J.; Powell, C.L.; Do, E.U.; Byon, C.Y.; Cleves, F.; McClain, S.G. Evaluation of 11-[3 H]-tetrodotoxin use in a heterologous receptor binding assay for PSP toxins. Toxicon 2000, 38, 1465–1474. [Google Scholar] [CrossRef]
- Doucette, G.J.; Logan, M.M.; Ramsdell, J.S.; Van Dolah, F.M. Development and preliminary validation of a microtiter plate-based receptor binding assay for paralytic shellfish poisoning toxins. Toxicon 1997, 35, 625–636. [Google Scholar] [CrossRef]
- Vieytes, M.; Cabado, A.; Alfonso, A.; Louzao, M.; Botana, A.; Botana, L. Solid-phase radioreceptor assay for paralytic shellfish toxins. Anal. Biochem. 1993, 211, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Edlund, M.J.; Martin, B.C.; Russo, J.E.; Devries, A.; Braden, J.B.; Sullivan, M.D. The role of opioid prescription in incident opioid abuse and dependence among individuals with chronic non-cancer pain: The role of opioid prescription. Clin. J. Pain 2014, 30, 557. [Google Scholar] [CrossRef] [PubMed]
- Dowell, D.; Haegerich, T.M.; Chou, R. CDC guideline for prescribing opioids for chronic pain—United States, 2016. JAMA 2016, 315, 1624–1645. [Google Scholar] [CrossRef] [PubMed]
- Compton, W.M.; Volkow, N.D. Major increases in opioid analgesic abuse in the United States: Concerns and strategies. Drug Alcohol. Depend. 2006, 81, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Paulozzi, L.J.; Budnitz, D.S.; Xi, Y. Increasing deaths from opioid analgesics in the United States. Pharmacoepidemiol. Drug Saf. 2006, 15, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Berde, C.B.; Athiraman, U.; Yahalom, B.; Zurakowski, D.; Corfas, G.; Bognet, C. Tetrodotoxin-bupivacaine-epinephrine combinations for prolonged local anesthesia. Mar. Drugs 2011, 9, 2717–2728. [Google Scholar] [CrossRef] [PubMed]
- Kohane, D.S.; Yieh, J.; Lu, N.T.; Langer, R.; Strichartz, G.R.; Berde, C.B. A re-examination of tetrodotoxin for prolonged duration local anesthesia. Anesthesiology 1998, 89, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.; Duncan, K.G.; Fields, H.L.; Jones, M.R. Tetrodotoxin: Anesthetic activity in the de-epithelialized cornea. Graefes Arch. Clin. Exp. 1998, 236, 790–794. [Google Scholar] [CrossRef]
- Nieto, F.R.; Cobos, E.J.; Tejada, M.A.; Sanchez-Fernandez, C.; Gonzalez-Cano, R.; Cendan, C.M. Tetrodotoxin (TTX) as a therapeutic agent for pain. Mar. Drugs 2012, 10, 281–305. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.A.; Fisher, K.M.; Lapointe, B.; du Souich, P.; Chary, S.; Moulin, D.; Sellers, E.; Ngoc, A.H.; Group, C.T.S. An open-label, multi-dose efficacy and safety study of intramuscular tetrodotoxin in patients with severe cancer-related pain. J. Pain Symptom Mang. 2007, 34, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Garrido, R.; Lagos, N.; Lattes, K.; Abedrapo, M.; Bocic, G.; Cuneo, A.; Chiong, H.; Jensen, C.; Azolas, R.; Henriquez, A.; et al. Gonyautoxin: New treatment for healing acute and chronic anal fissures. Dis. Colon Rectum 2005, 48, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Garrido, R.; Lagos, N.; Lagos, M.; Rodriguez-NaVarro, A.J.; Garcia, C.; Truan, D.; Henriquez, A. Treatment of chronic anal fissure by gonyautoxin. Colorectal Dis. 2007, 9, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Lattes, K.; Venegas, P.; Lagos, N.; Lagos, M.; Pedraza, L.; Rodriguez-NaVarro, A.; Garcia, C. Local infiltration of gonyautoxin is safe and effective in treatment of chronic tension-type headache. Neurol. Res. 2009, 31, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Hinzpeter, J.; Barrientos, C.; Zamorano, A.; Martinez, A.; Palet, M.; Wulf, R.; Barahona, M.; Sepulveda, J.M.; Guerra, M.; Bustamante, T.; et al. Gonyautoxins: First evidence in pain management in total knee arthroplasty. Toxicon 2016, 119, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-NaVarro, A.J.; Lagos, N.; Lagos, M.; Braghetto, I.; Csendes, A.; Hamilton, J.; Berger, Z.; Wiedmaier, G.; Henriquez, A. Intrasphincteric neosaxitoxin injection: Evidence of lower esophageal sphincter relaxation in achalasia. Am. J. Gastroenterol. 2006, 101, 2667. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-NaVarro, A.J.; Lagos, N.; Lagos, M.; Braghetto, I.; Csendes, A.; Hamilton, J.; Figueroa, C.; Truan, D.; Garcia, C.; Rojas, A. Neosaxitoxin as a local anesthetic preliminary observations from a first human trial. Anesthesiology 2007, 106, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-NaVarro, A.J.; Lagos, M.; Figueroa, C.; Garcia, C.; Recabal, P.; Silva, P.; Iglesias, V.; Lagos, N. Potentiation of local anesthetic activity of neosaxitoxin with bupivacaine or epinephrine: Development of a long-acting pain blocker. Neurotox. Res. 2009, 16, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Manriquez, V.; Castro Caperan, D.; Guzman, R.; Naser, M.; Iglesia, V.; Lagos, N. First evidence of neosaxitoxin as a long-acting pain blocker in bladder pain syndrome. Int. Urogynecol. J. 2015, 26, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.W.; See, R.E. Dissociation of primary and secondary reward-relevant limbic nuclei in an animal model of relapse. Neuropsychopharmacology 2000, 22, 473–479. [Google Scholar] [CrossRef]
- Shi, J.; Liu, T.-T.; Wang, X.; Epstein, D.H.; Zhao, L.-Y.; Zhang, X.-L.; Lu, L. Tetrodotoxin reduces cue-induced drug craving and anxiety in abstinent heroin addicts. Pharmacol. Biochem. Behav. 2009, 92, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Rashid, M.H.; Mahdavi, S.; Kuyucak, S. Computational studies of marine toxins targeting ion channels. Mar. Drugs 2013, 11, 848–869. [Google Scholar] [CrossRef] [PubMed]
- Lipkind, G.M.; Fozzard, H.A. KcsA crystal structure as framework for a molecular model of the Na+ channel pore. Biochemistry 2000, 39, 8161–8170. [Google Scholar] [CrossRef] [PubMed]
- Tikhonov, D.B.; Zhorov, B.S. Modeling P-loops domain of sodium channel: Homology with potassium channels and interaction with ligands. Biophys. J. 2005, 88, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhou, Q.; Pan, X.; Li, Z.; Wu, J.; Yan, N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 2017, 355, eaal4326. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Du, Y.; Rinkevich, F.; Nomura, Y.; Xu, P.; Wang, L.; Silver, K.; Zhorov, B.S. Molecular biology of insect sodium channels and pyrethroid resistance. Insect Biochem. Mol. Biol. 2014, 50, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ching, B.; Woo, J.M.; Hiong, K.C.; Boo, M.V.; Wong, W.P.; Chew, S.F.; Ip, Y.K. Voltage-gated Na+ channel isoforms and their mRNA expression levels and protein abundance in three electric organs and the skeletal muscle of the electric eel Electrophorus electricus. PLoS ONE 2016, 11, e0167589. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Chen, R.; Chung, S.H. Computational methods of studying the binding of toxins from venomous animals to biological ion channels: Theory and applications. Physiol. Rev. 2013, 93, 767–802. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Andrews, C.W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S. A critical assessment of docking programs and scoring functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Bello, M.; Martínez-Archundia, M.; Correa-Basurto, J. Automated docking for novel drug discovery. Expert Opin. Drug Dis. 2013, 8, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Li, R.A.; Ennis, I.L.; French, R.J.; Dudley, S.C., Jr.; Tomaselli, G.F.; Marban, E. Clockwise domain arrangement of the sodium channel revealed by µ-conotoxin (GIIIA) docking orientation. J. Biol. Chem. 2001, 276, 11072–11077. [Google Scholar] [CrossRef] [PubMed]
- Fogh, R.H.; Kem, W.; Norton, R.S. Solution structure of neurotoxin I from the sea anemone Stichodactyla helianthus. A nuclear magnetic resonance, distance geometry, and restrained molecular dynamics study. J. Biol. Chem. 1990, 265, 13016–13028. [Google Scholar] [PubMed]
- Pietra, F. Docking and MD simulations of the interaction of the tarantula peptide psalmotoxin-1 with ASIC1a channels using a homology model. J. Chem. Inf. Model. 2009, 49, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Hasani, H.J.; Ganesan, A.; Houghton, M.; Barakat, K. Modeling the human NaV1. 5 sodium channel: Structural and mechanistic insights of ion permeation and drug blockade. Drug Des. Dev. Ther. 2017, 11, 2301. [Google Scholar] [CrossRef] [PubMed]
- Van Dolah, F.M. Marine algal toxins: Origins, health effects, and their increased occurrence. Environ. Health Persp. 2000, 108, 133. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin Analog | Relative Toxicity IP MBA | Relative Toxicity Voluntary Feeding/Gavage |
|---|---|---|
| STX | 1.0 | 1.0 |
| NEO | 1.0 | 2.5/1.7 |
| GTX1 | 1.0 | |
| GTX4 | 0.7 | |
| GTX1/4 | 0.9/0.7 | |
| GTX2 | 0.4 | |
| GTX3 | 0.6 | |
| GTX2/3 | 0.6/0.5 | |
| dcSTX | 1.0 | |
| dcGTX1 | 0.5 * | |
| dcGTX4 | 0.5 * | |
| dcGTX2 | 0.2 | |
| dcGTX3 | 0.4 | |
| dcGTX2/3 | 0.1/0.2 | |
| dcNEO | 0.4 | 0.2/0.2 |
| B1 | 0.1 | 0.06/0.05 |
| B2 | 0.1 | <0.02/0.04 |
| C1 | 0.01 ** | |
| C2 | 0.1 | |
| C1/2 | 0.04/0.03 | |
| C3 | 0.01 ** | |
| C4 | 0.1 |
| Animal Model | Toxin | Route of Administration | LD50 (nmol kg−1) | Reference |
|---|---|---|---|---|
| Pigeon | STX | O | 302 | [43] |
| Rabbit | i.v. | 1 | ||
| O | 601 | |||
| Rat | i.p. | 35 | ||
| i.m. * | 23 | |||
| O | 638 | |||
| Cat | O | 844 | ||
| Chicken | i.v. | 1 | ||
| Dog | O | 601 | ||
| Guinea pig | O | 449 | ||
| Mouse | i.v. | 11-28 | ||
| s.c. | 43 | |||
| i.p. | 26-33 | |||
| NEO | i.p. | 8.9 | [42] | |
| dcSTX | i.p. | 35.4 | ||
| GTX1/4 | i.p. | 14.6 | ||
| GTX1/3 | i.p. | 36.7 | ||
| STX | O/G | 1190 | ||
| O/F | 3200 | |||
| NEO | O/G | 700 | ||
| O/F | 1260 | |||
| dcSTX | O/G | 2600 | ||
| O/F | 8680 | |||
| GTX1/4 | O/G | 1610 | ||
| O/F | 3420 | |||
| GTX2/3 | O/G | 2230 | ||
| O/F | 5590 | |||
| TTX | i.v. | 28 | [44] | |
| i.p. | 34 | [45] | ||
| s.c. | 39-50 | [45,46] | ||
| i.g. | 1668 | [45] | ||
| O | 727 | [47] [47] | ||
| 11-deoxyTTX | i.p. | 231 | ||
| µ-conotoxin GIIIA | i.p. | 1066 | [48] | |
| µ-conotoxin GIIIB | i.p. | 345 |
| NaV Channel Isoform | Predominant Location | TTX IC50 [nM] | Sensitive/Resistant |
|---|---|---|---|
| NaV1.1 | CNS, PNS, Heart | 5.9 | Sensitive |
| NaV1.2 | CNS | 7.8 | Sensitive |
| NaV1.3 | CNS (embryonic) | 2.0 | Sensitive |
| NaV1.4 | Skeletal muscle | 4.5 | Sensitive |
| NaV1.5 | Heart, CNS | 1970 | Resistant |
| NaV1.6 | CNS, PNS, SMCs, DRG | 3.8 | Sensitive |
| NaV1.7 | PNS, DRG | 5.5 | Sensitive |
| NaV1.8 | PNS, DRG | 1330 | Resistant |
| NaV1.9 | PNS, DRG | 59,600 | Resistant |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durán-Riveroll, L.M.; Cembella, A.D. Guanidinium Toxins and Their Interactions with Voltage-Gated Sodium Ion Channels. Mar. Drugs 2017, 15, 303. https://doi.org/10.3390/md15100303
Durán-Riveroll LM, Cembella AD. Guanidinium Toxins and Their Interactions with Voltage-Gated Sodium Ion Channels. Marine Drugs. 2017; 15(10):303. https://doi.org/10.3390/md15100303
Chicago/Turabian StyleDurán-Riveroll, Lorena M., and Allan D. Cembella. 2017. "Guanidinium Toxins and Their Interactions with Voltage-Gated Sodium Ion Channels" Marine Drugs 15, no. 10: 303. https://doi.org/10.3390/md15100303
APA StyleDurán-Riveroll, L. M., & Cembella, A. D. (2017). Guanidinium Toxins and Their Interactions with Voltage-Gated Sodium Ion Channels. Marine Drugs, 15(10), 303. https://doi.org/10.3390/md15100303