Antimicrobial Peptide Epinecidin-1 Modulates MyD88 Protein Levels via the Proteasome Degradation Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
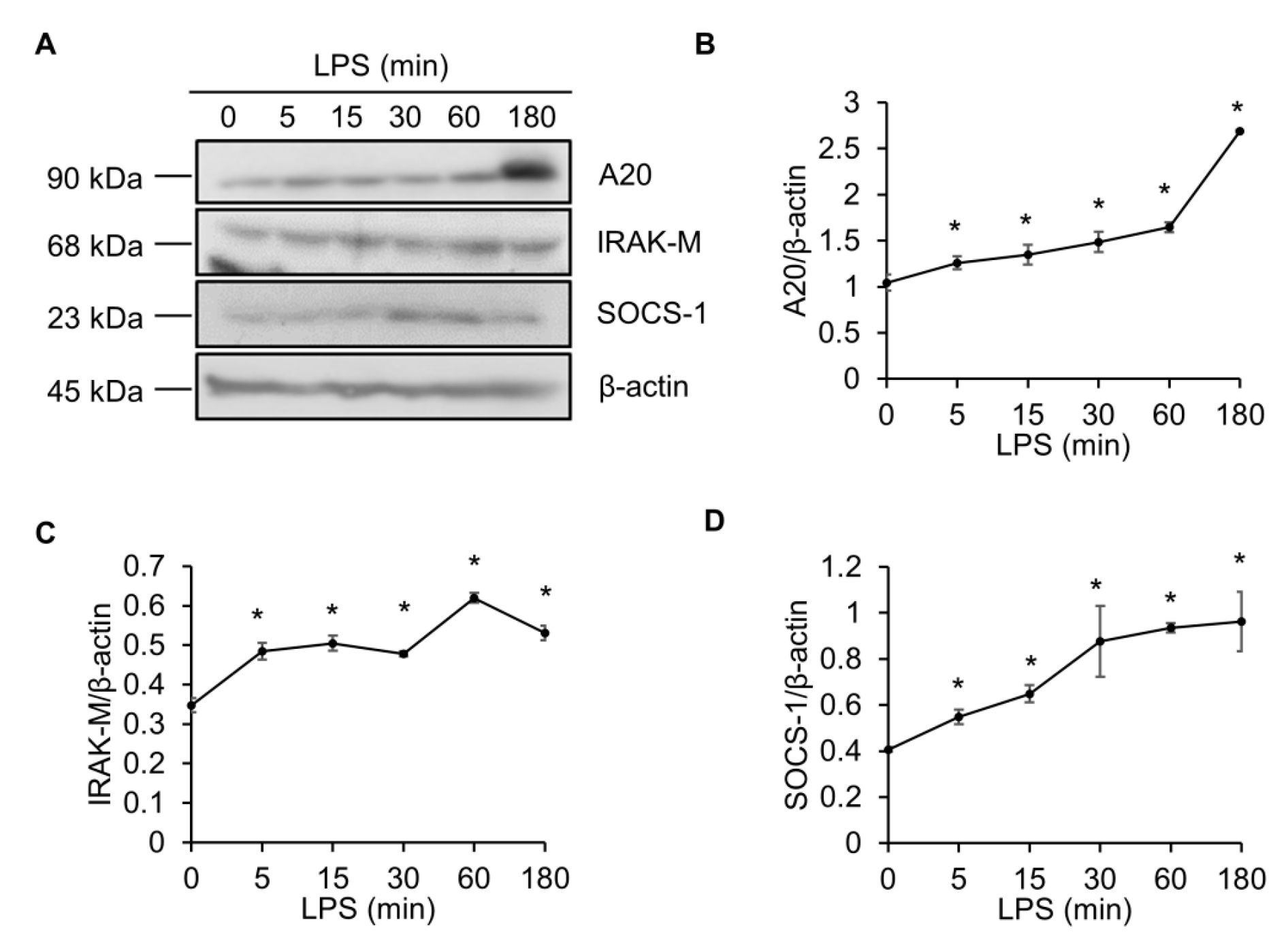
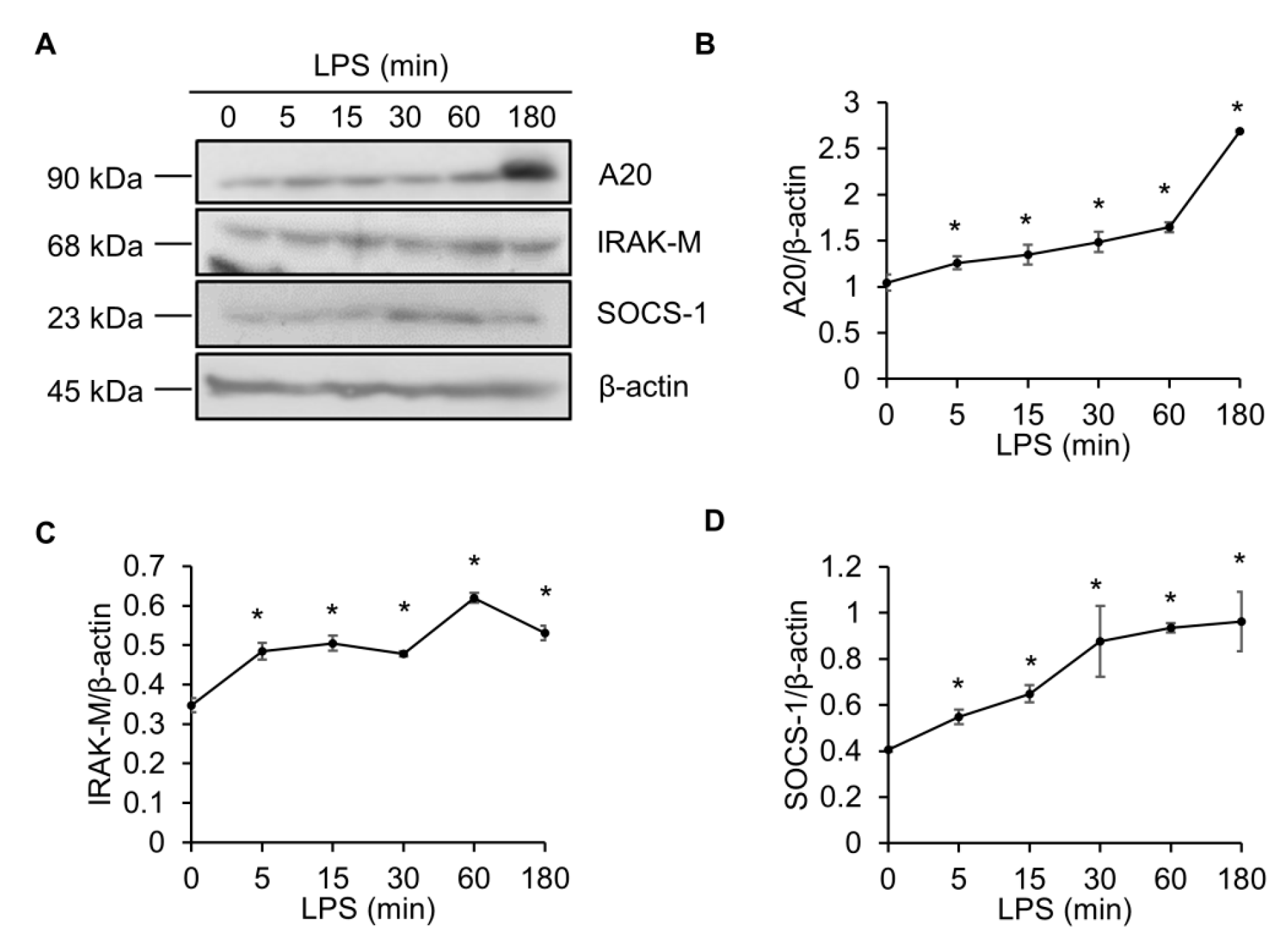
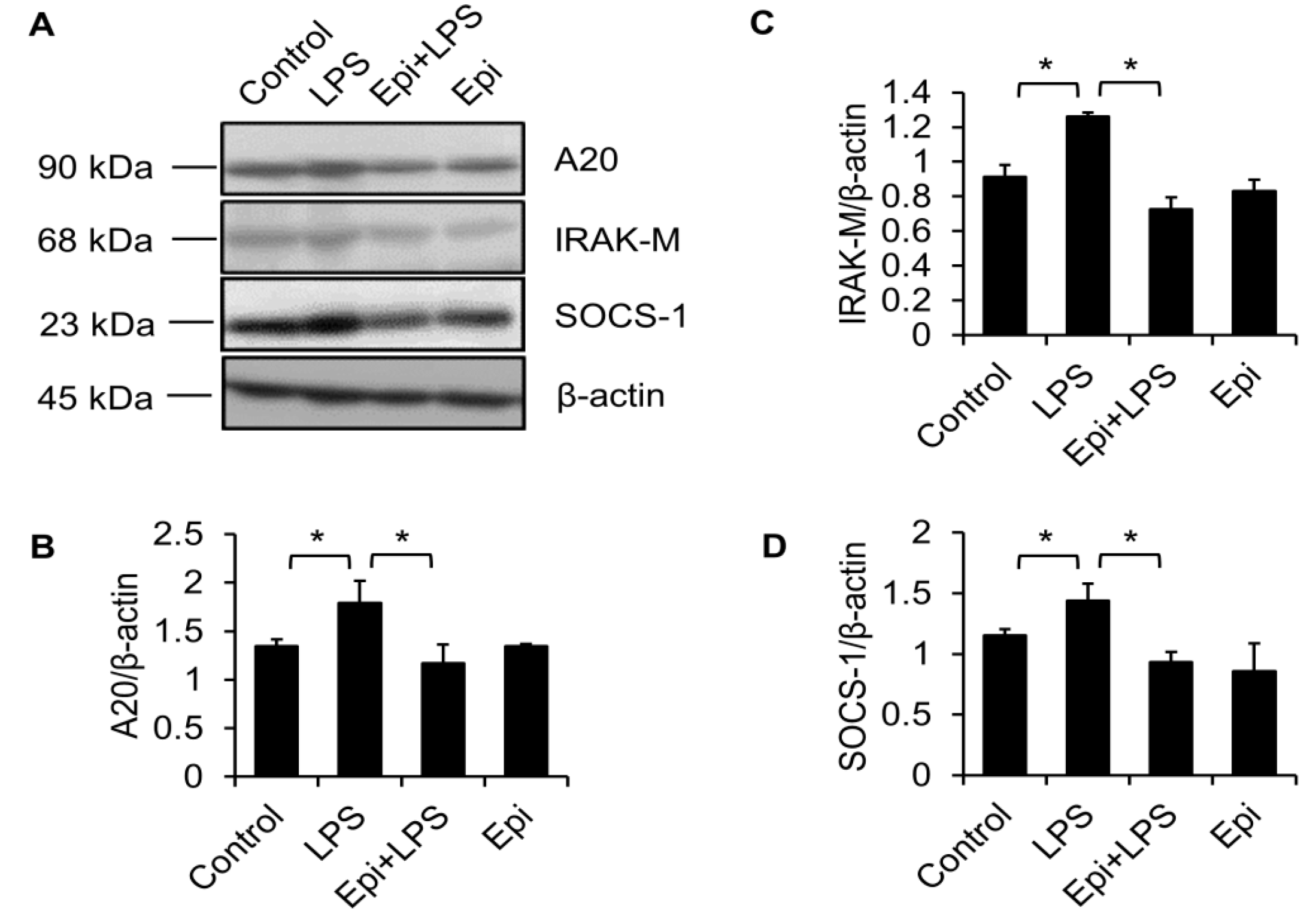
2.1. LPS Elevates the A20, IRAK-M and SOCS-1 Protein Levels in Raw264.7 Macrophages
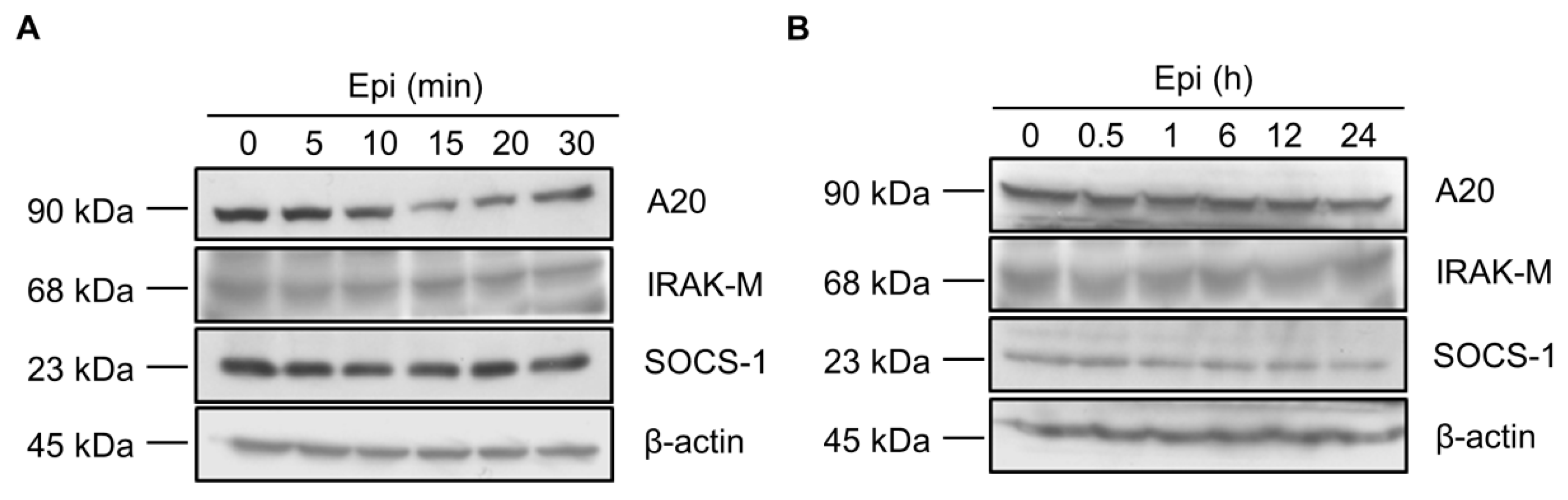
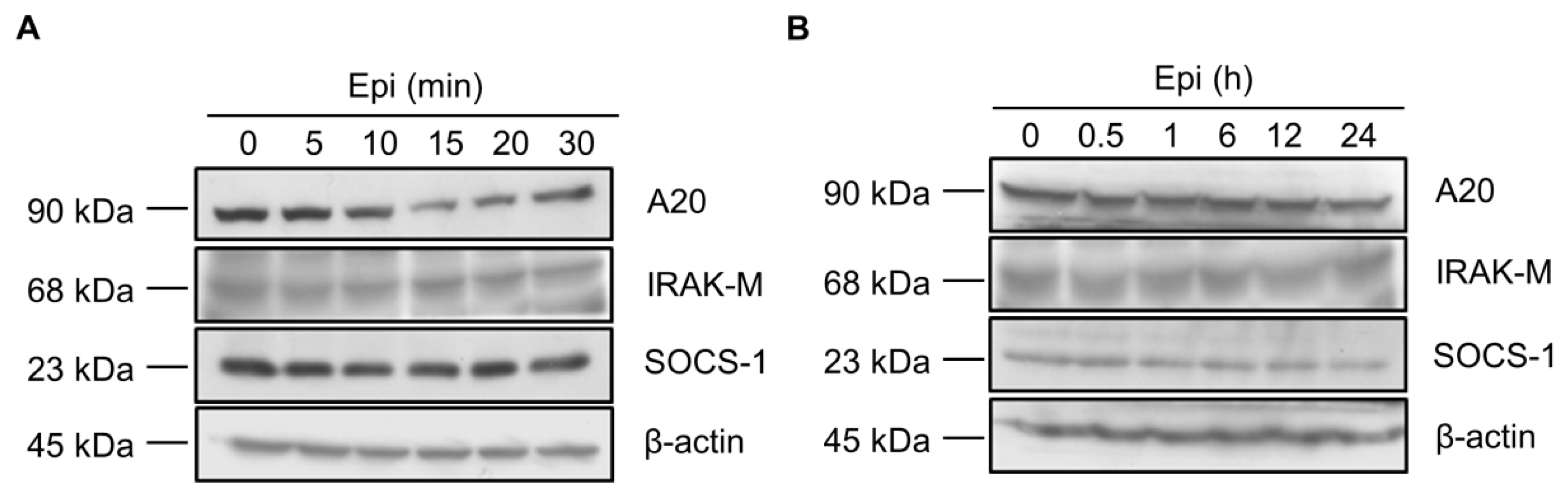
2.2. Epinecidin-1 Alone Does Not Affect the A20, IRAK-M and SOCS-1 Protein Levels
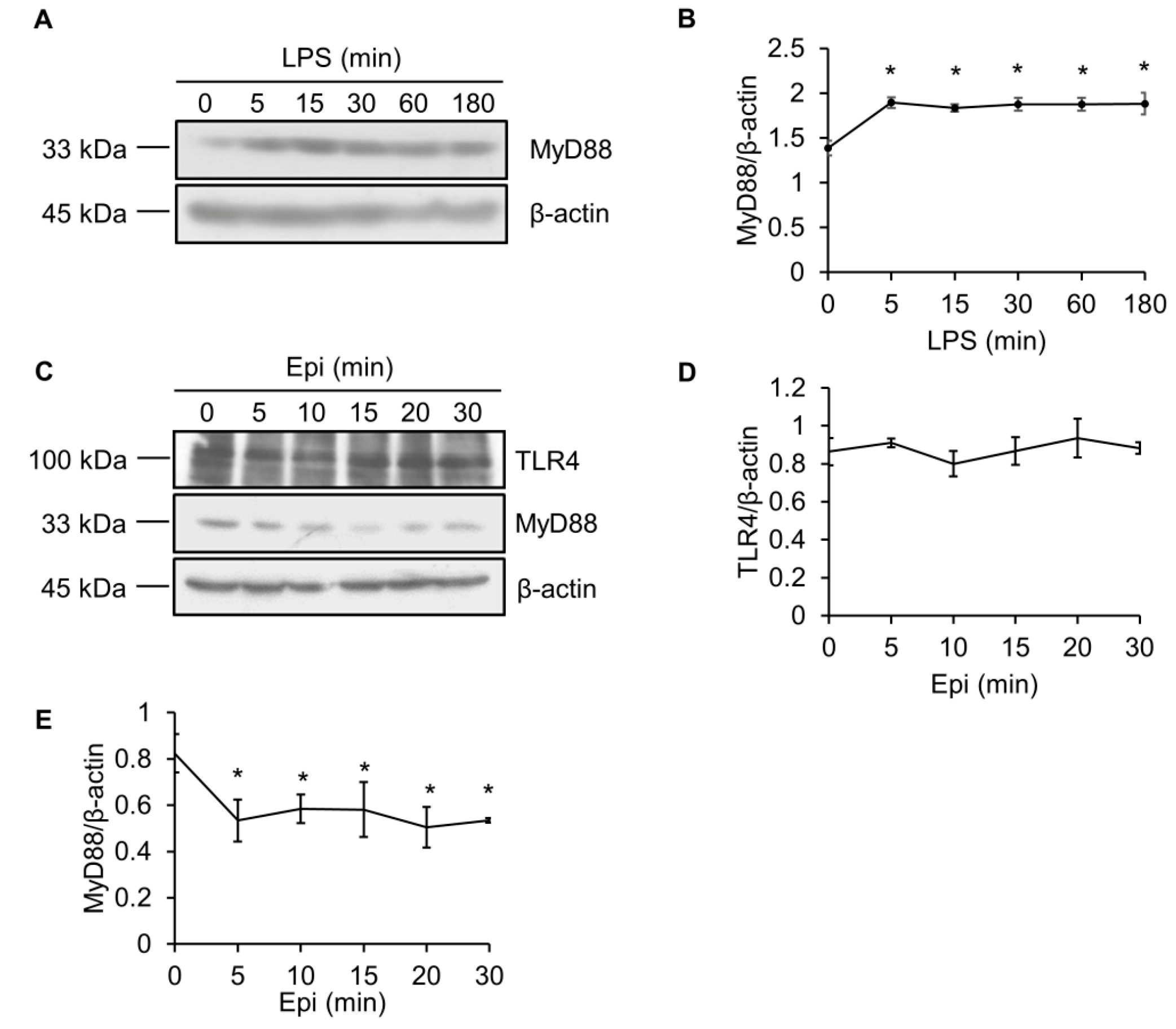
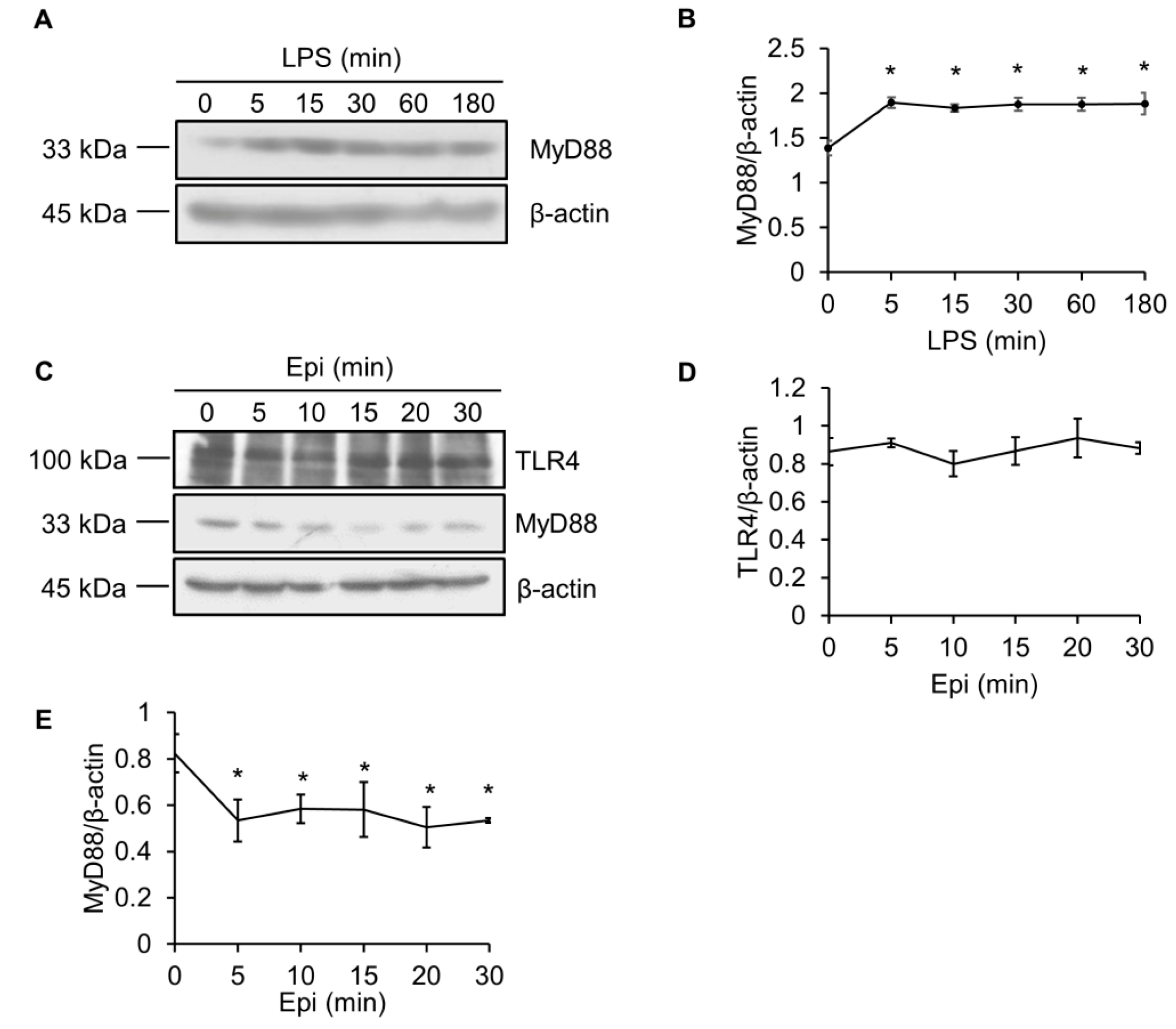
2.3. LPS Upregulates MyD88 Protein Level
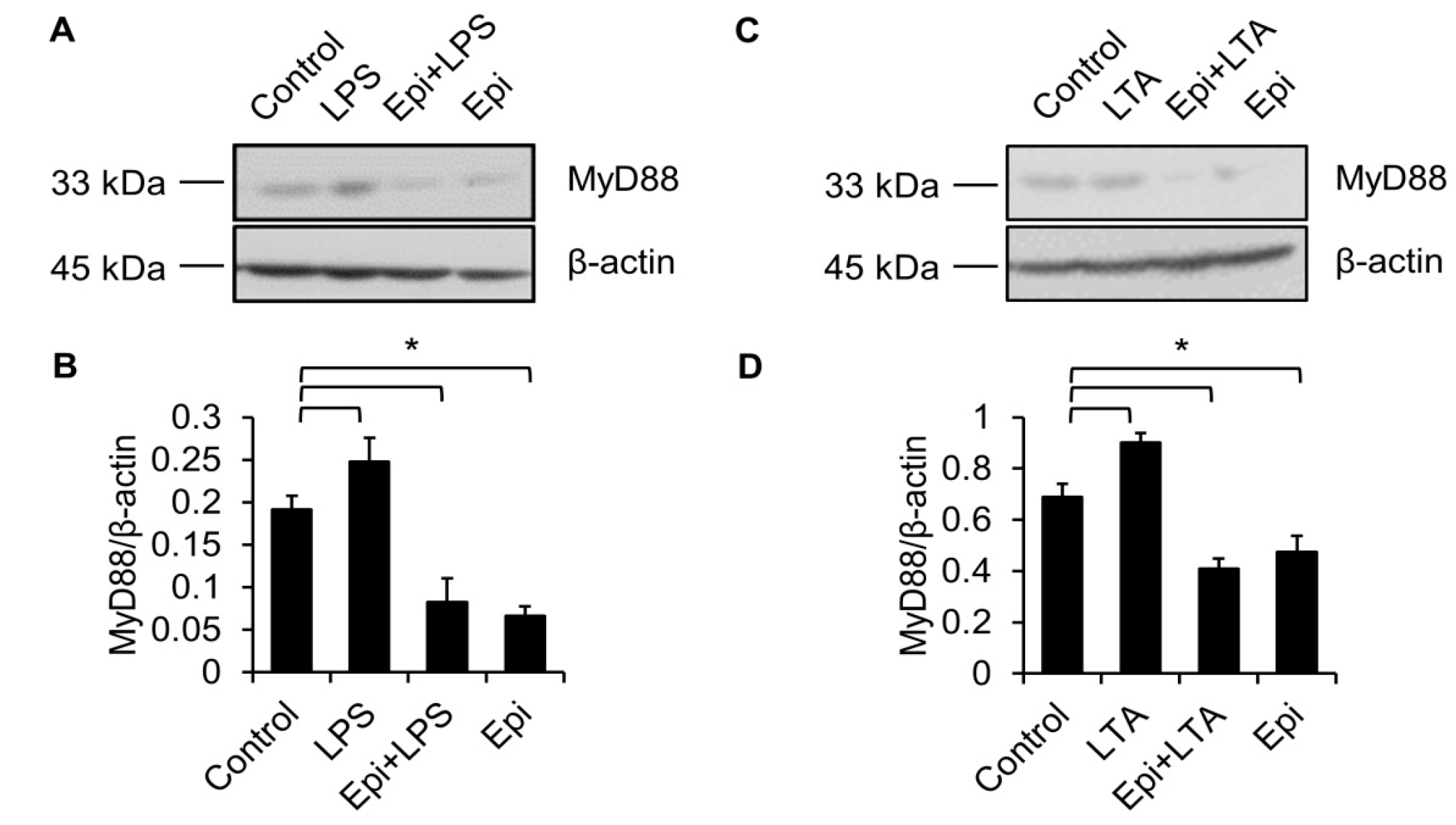
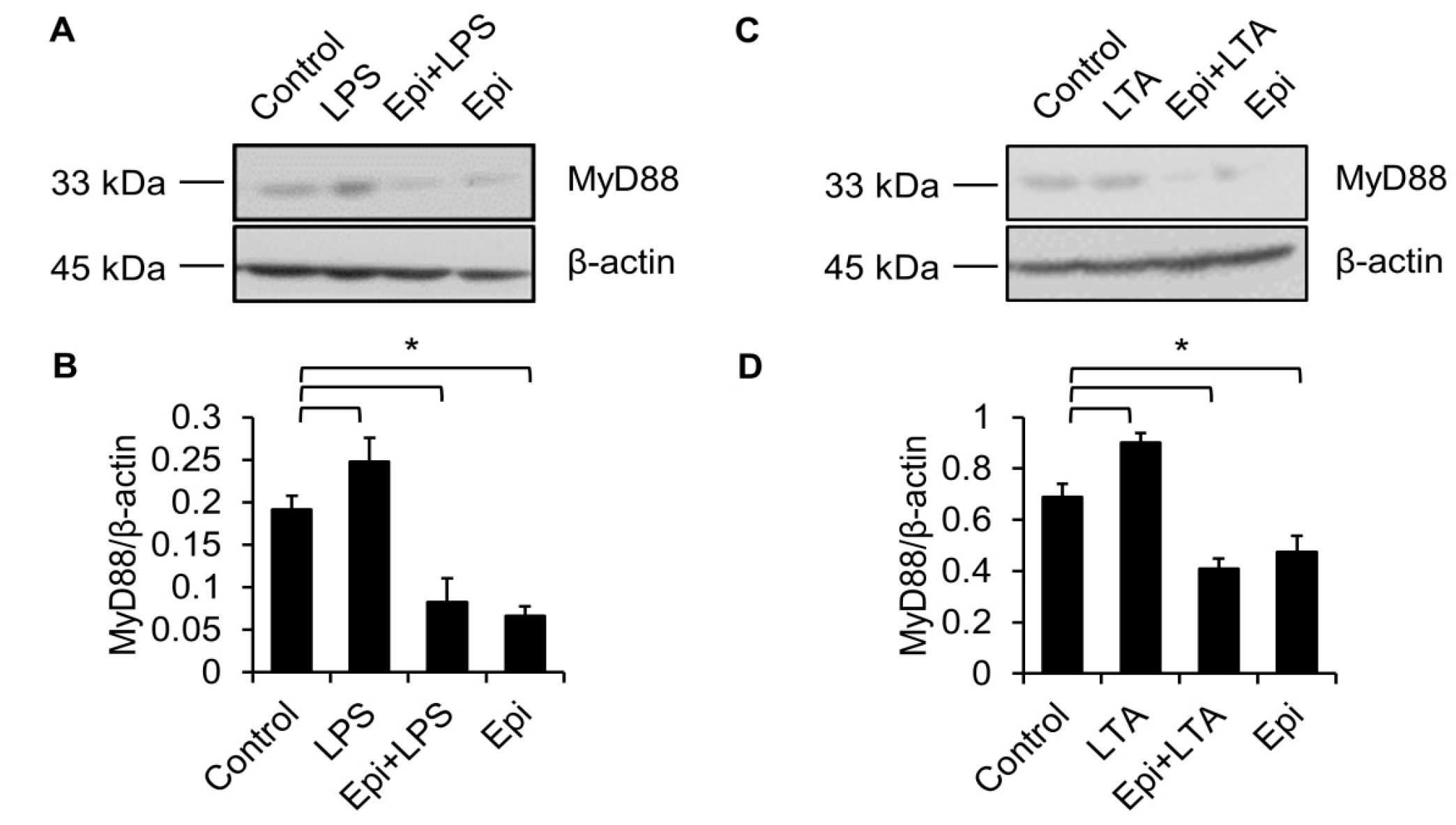
2.4. Epinecidin-1 Downregulates MyD88 Protein Level
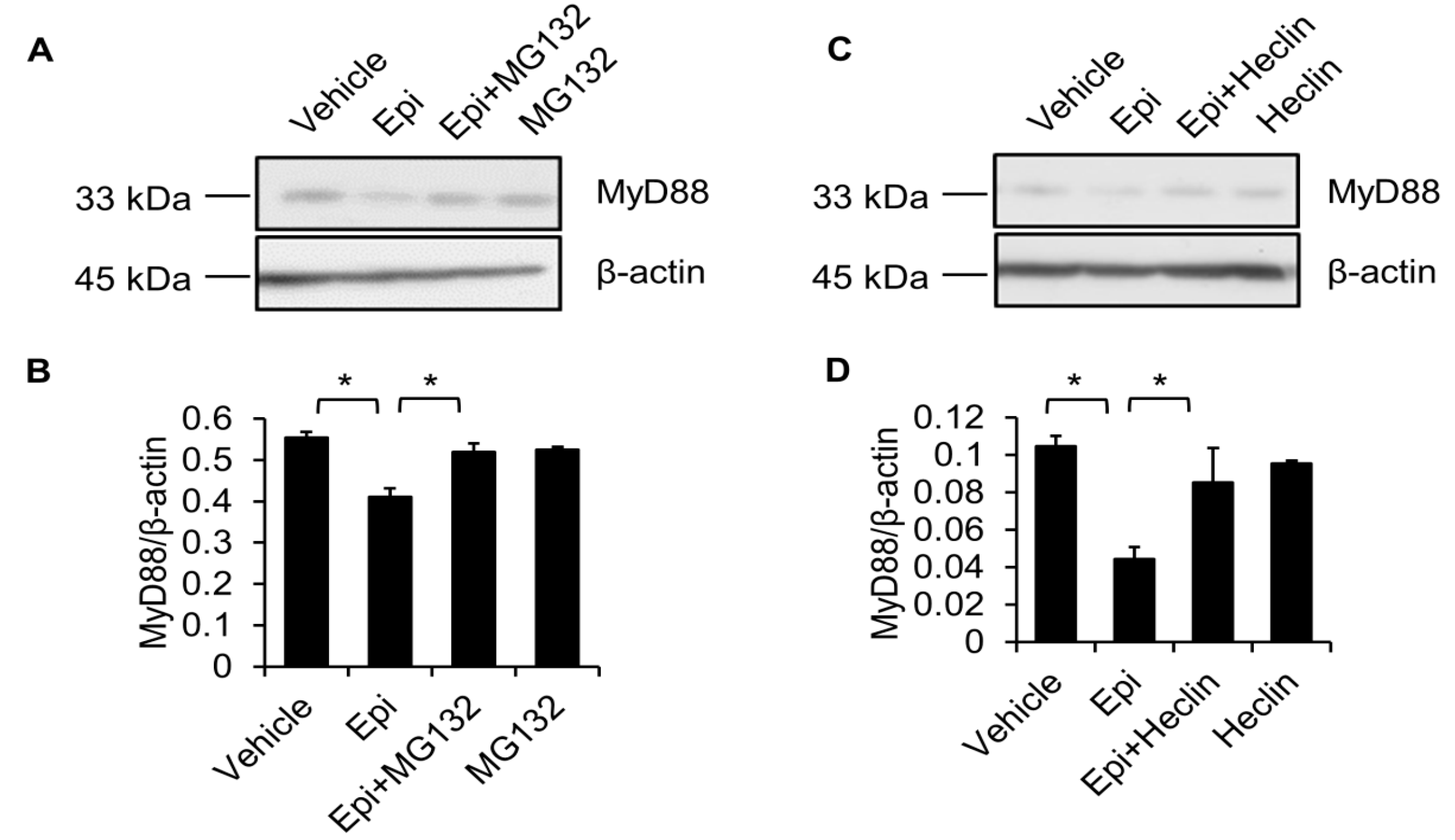
2.5. Proteasome Degradation Pathway Is Required for Epinecidin-1-Mediated MyD88 Downregulation
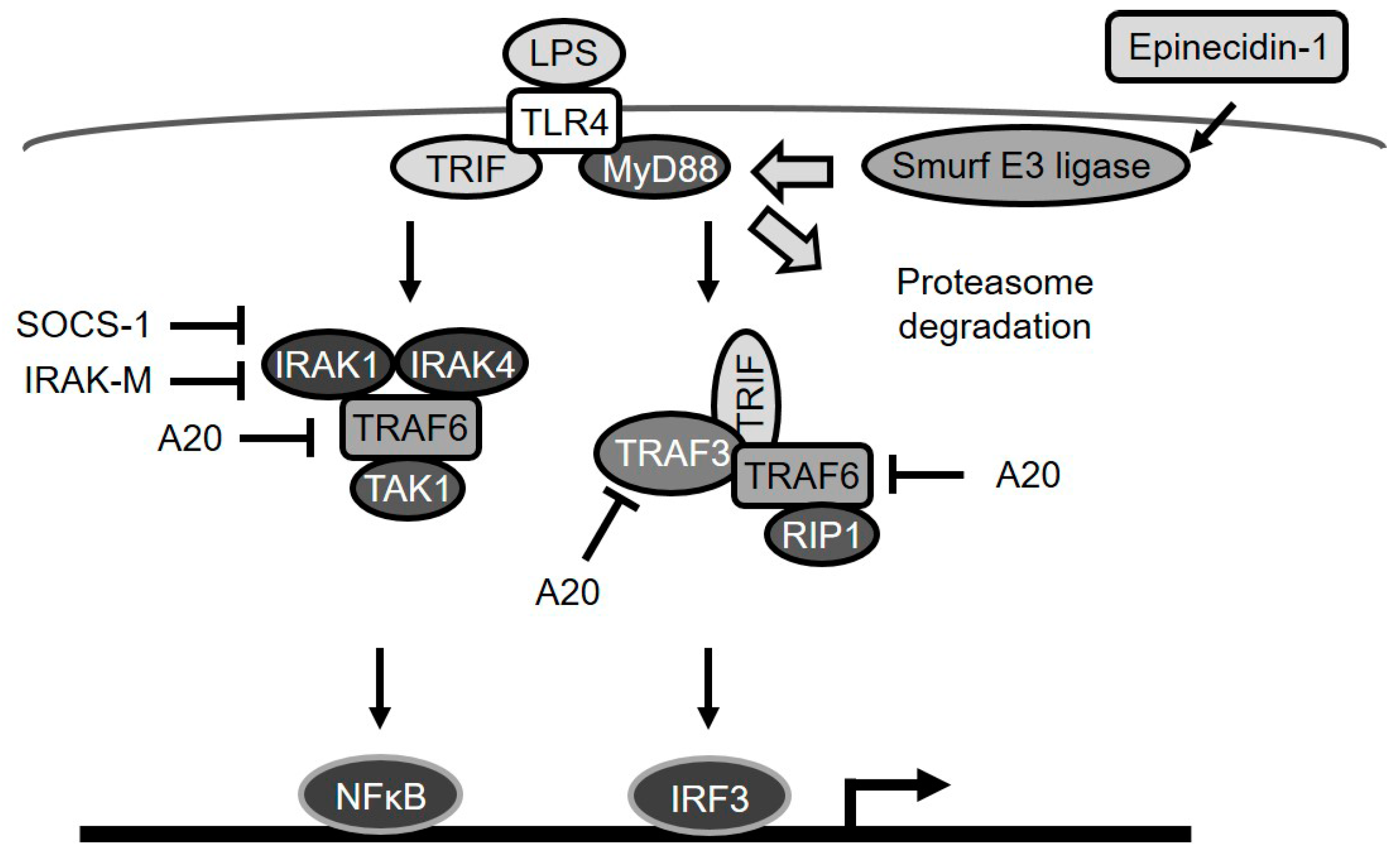
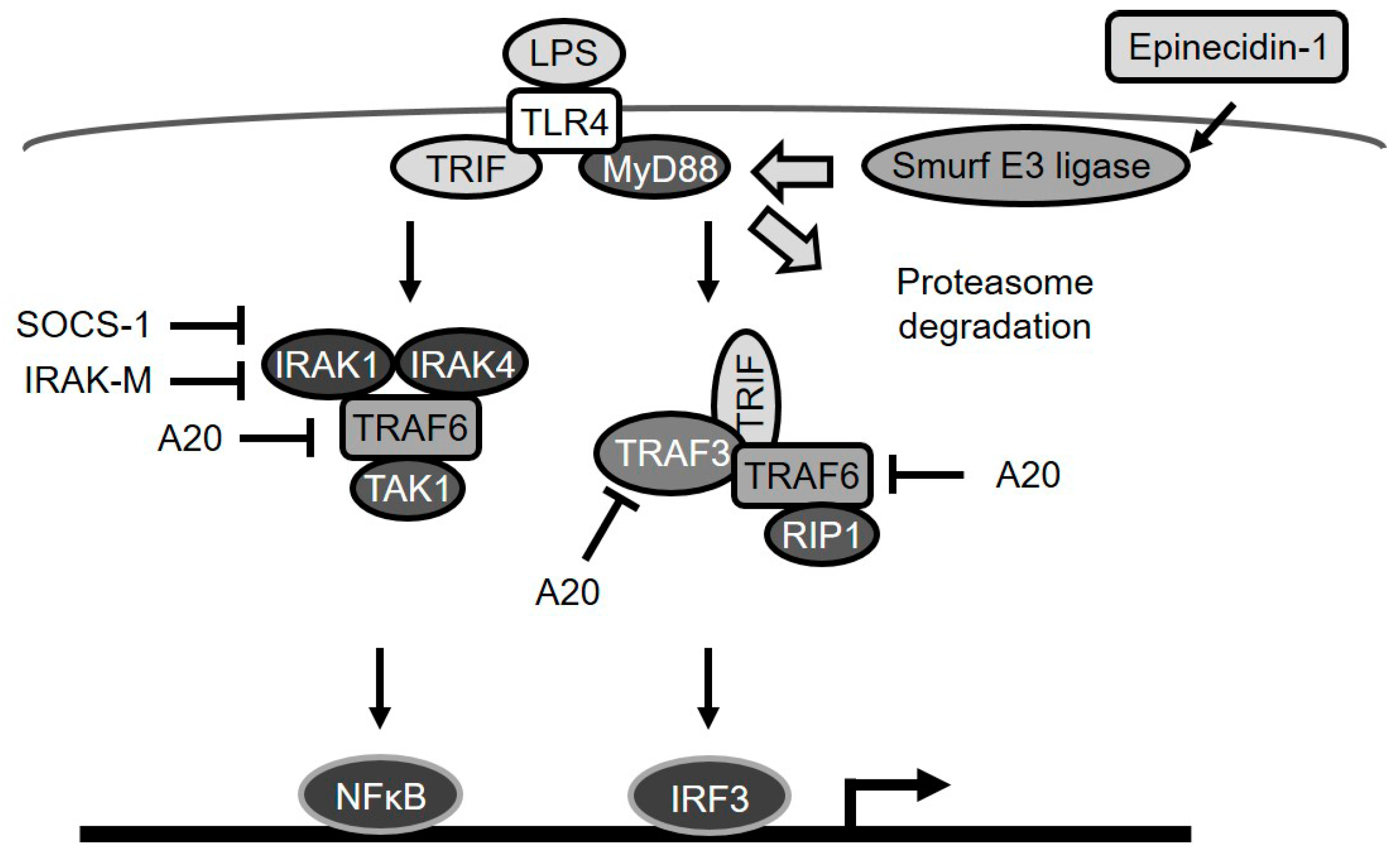
3. Discussion
4. Material and Methods
4.1. Reagents
4.2. Cell Culture and Treatment
4.3. Western Blotting Analysis
4.4. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pasare, C.; Medzhitov, R. Toll-like receptors: Linking innate and adaptive immunity. Adv. Exp. Med. Biol. 2005, 560, 11–18. [Google Scholar] [PubMed]
- Alexander, C.; Rietschel, E.T. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Van Lieshout, M.H.; van der Poll, T.; van’t Veer, C. Tlr4 inhibition impairs bacterial clearance in a therapeutic setting in murine abdominal sepsis. Inflamm. Res. 2014, 63, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, T.; Sato, S.; Jung, A.; Yamamoto, M.; Matsui, K.; Kato, H.; Uematsu, S.; Takeuchi, O.; Akira, S. Essential role of irak-4 protein and its kinase activity in toll-like receptor-mediated immune responses but not in tcr signaling. J. Exp. Med. 2007, 204, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; O’Neill, L.A. Signal transduction by the lipopolysaccharide receptor, toll-like receptor-4. Immunology 2004, 113, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor nf-kappab pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Rahman, F.Z.; Hayee, B.; Graham, S.J.; Marks, D.J.; Sewell, G.W.; Palmer, C.D.; Wilde, J.; Foxwell, B.M.; Gloger, I.S.; et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in crohn’s disease. J. Exp. Med. 2009, 206, 1883–1897. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.J.; Henry-Stanley, M.J.; Bendel, C.M.; Zhang, B.; Johnson, M.A.; Wells, C.L. Escherichia coli and tnf-alpha modulate macrophage phagocytosis of candida glabrata. J. Surg. Res. 2009, 155, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Geczy, C.L. Ifn-gamma and tnf regulate macrophage expression of the chemotactic s100 protein s100a8. J. Immunol. 2000, 164, 4916–4923. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Shibolet, O. Toll-like receptors in inflammatory bowel disease-stepping into uncharted territory. World J. Gastroenterol. 2008, 14, 5149–5153. [Google Scholar] [CrossRef] [PubMed]
- Drexler, S.K.; Foxwell, B.M. The role of toll-like receptors in chronic inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Molteni, M.; Gemma, S.; Rossetti, C. The role of toll-like receptor 4 in infectious and noninfectious inflammation. Mediat. Inflamm. 2016, 2016, 6978936. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhou, Y.; Feng, G.; Su, S.B. The critical role of toll-like receptor signaling pathways in the induction and progression of autoimmune diseases. Curr. Mol. Med. 2009, 9, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yu, M.; Fukuda, K.; Im, J.; Yao, P.; Cui, W.; Bulek, K.; Zepp, J.; Wan, Y.; Kim, T.W.; et al. Irak-m mediates toll-like receptor/il-1r-induced nfkappab activation and cytokine production. EMBO J. 2013, 32, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Mabilleau, G.; Chappard, D.; Sabokbar, A. Role of the a20-traf6 axis in lipopolysaccharide-mediated osteoclastogenesis. J. Biol.Chem. 2011, 286, 3242–3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, M.; Naka, T. Socs1, a negative regulator of cytokine signals and tlr responses, in human liver diseases. Gastroenterol. Res. Pract. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Reihill, J.A.; Malcomson, B.; Bertelsen, A.; Cheung, S.; Czerwiec, A.; Barsden, R.; Elborn, J.S.; Durkop, H.; Hirsch, B.; Ennis, M.; et al. Induction of the inflammatory regulator a20 by gibberellic acid in airway epithelial cells. Brit. J. Pharmacol. 2016, 173, 778–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, C.G.; Maccariello, E.R.; Wilson, S.W.; Putheti, P.; Daniel, S.; Damrauer, S.M.; Peterson, C.R.; Siracuse, J.J.; Kaczmarek, E.; Ferran, C. Hepatocyte growth factor preferentially activates the anti-inflammatory arm of nf-kappab signaling to induce a20 and protect renal proximal tubular epithelial cells from inflammation. J. Cell. Physiol. 2012, 227, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Le Morvan, C.; Troutaud, D.; Deschaux, P. Differential effects of temperature on specific and nonspecific immune defences in fish. J. Exp. Biol. 1998, 201, 165–168. [Google Scholar] [PubMed]
- Antoro, S.; Na-Nakorn, U.; Koedprang, W. Study of genetic diversity of orange-spotted grouper, epinephelus coioides, from thailand and indonesia using microsatellite markers. Mar. Biotechnol. 2006, 8, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.Y.; Chen, J.Y.; Cheng, Y.S.; Chen, C.Y.; Ni, I.H.; Sheen, J.F.; Pan, Y.L.; Kuo, C.M. Gene expression and localization of the epinecidin-1 antimicrobial peptide in the grouper (epinephelus coioides), and its role in protecting fish against pathogenic infection. DNA Cell Biol. 2007, 26, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Lin, W.J.; Wu, J.L.; Her, G.M.; Hui, C.F. Epinecidin-1 peptide induces apoptosis which enhances antitumor effects in human leukemia u937 cells. Peptides 2009, 30, 2365–2373. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.N.; Rajanbabu, V.; Pan, C.Y.; Chan, Y.L.; Wu, C.J.; Chen, J.Y. Use of the antimicrobial peptide epinecidin-1 to protect against mrsa infection in mice with skin injuries. Biomaterials 2013, 34, 10319–10327. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.Y.; Chen, J.C.; Sheen, J.F.; Lin, T.L.; Chen, J.Y. Epinecidin-1 has immunomodulatory effects, facilitating its therapeutic use in a mouse model of pseudomonas aeruginosa sepsis. Antimicrob. Agents Chemother. 2014, 58, 4264–4274. [Google Scholar] [CrossRef] [PubMed]
- Su, B.C.; Huang, H.N.; Lin, T.W.; Hsiao, C.D.; Chen, J.Y. Epinecidin-1 protects mice from lps-induced endotoxemia and cecal ligation and puncture-induced polymicrobial sepsis. Biochim. Biophys. Acta 2017, 1863, 3028–3037. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Song, D.; Wang, T.; Xia, X.; Hu, W.; Han, F.; Wang, Y. Lfp-20, a porcine lactoferrin peptide, ameliorates lps-induced inflammation via the myd88/nf-kappab and myd88/mapk signaling pathways. Dev. Comp. Immunol. 2015, 52, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Lee, C.W.; Tung, W.H.; Wang, S.W.; Lin, C.C.; Shu, J.C.; Yang, C.M. Cooperation of tlr2 with myd88, pi3k, and rac1 in lipoteichoic acid-induced cpla2/cox-2-dependent airway inflammatory responses. Am. J. Pathol. 2010, 176, 1671–1684. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.V.; Kim, E.R.; Ovchinnikov, L.P. Proteasome system of protein degradation and processing. Biochemistry 2009, 74, 1411–1442. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Park, J.S.; Kim, J.H.; Jung, S.M.; Lee, J.Y.; Kim, S.J.; Park, S.H. Smad6-specific recruitment of smurf e3 ligases mediates tgf-beta1-induced degradation of myd88 in tlr4 signalling. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yee, E.; Harlan, J.M.; Wong, F.; Karsan, A. Lipopolysaccharide induces the antiapoptotic molecules, a1 and a20, in microvascular endothelial cells. Blood. 1998, 92, 2759–2765. [Google Scholar] [PubMed]
- Gon, Y.; Asai, Y.; Hashimoto, S.; Mizumura, K.; Jibiki, I.; Machino, T.; Ra, C.; Horie, T. A20 inhibits toll-like receptor 2- and 4-mediated interleukin-8 synthesis in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.L.; Pei, D.A.; Yan, J.Z.; Xu, G.; Wu, P. A20 overexpression inhibits lipopolysaccharide-induced nf-kappab activation, traf6 and cd40 expression in rat peritoneal mesothelial cells. Int. J. Mol. Sci. 2014, 15, 6592–6608. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Seki, E. Toll-like receptors in liver fibrosis: Cellular crosstalk and mechanisms. Front. Physiol. 2012, 3, 138. [Google Scholar] [CrossRef] [PubMed]
- Catrysse, L.; Farhang Ghahremani, M.; Vereecke, L.; Youssef, S.A.; Mc Guire, C.; Sze, M.; Weber, A.; Heikenwalder, M.; de Bruin, A.; Beyaert, R.; et al. A20 prevents chronic liver inflammation and cancer by protecting hepatocytes from death. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Zhao, T.; Zhou, K.; Zhong, Q.; Wang, Y.; Xiong, X.; Wang, F.; Yang, Y.; Zhu, W.; Liu, J.; et al. A20 ameliorates intracerebral hemorrhage-induced inflammatory injury by regulating traf6 polyubiquitination. J. Immunol. 2017, 198, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Van’t Veer, C.; van den Pangaart, P.S.; van Zoelen, M.A.; de Kruif, M.; Birjmohun, R.S.; Stroes, E.S.; de Vos, A.F.; van der Poll, T. Induction of irak-m is associated with lipopolysaccharide tolerance in a human endotoxemia model. J. Immunol. 2007, 179, 7110–7120. [Google Scholar] [CrossRef]
- Im, J.; Baik, J.E.; Kim, K.W.; Kang, S.S.; Jeon, J.H.; Park, O.J.; Kim, H.Y.; Kum, K.Y.; Yun, C.H.; Han, S.H. Enterococcus faecalis lipoteichoic acid suppresses aggregatibacter actinomycetemcomitans lipopolysaccharide-induced il-8 expression in human periodontal ligament cells. Int. Immunol. 2015, 27, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, L.L.; Moore, B.B. Irak-m regulation and function in host defense and immune homeostasis. Infect. Dis. Rep. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Lee, J.Y.; Susuki-Miyata, S.; Wang, W.Y.; Xu, H.; Kai, H.; Kobayashi, K.S.; Flavell, R.A.; Li, J.D. Glucocorticoids suppress inflammation via the upregulation of negative regulator irak-m. Nat. Commun. 2015, 6, 6062. [Google Scholar] [CrossRef] [PubMed]
- Kinjyo, I.; Hanada, T.; Inagaki-Ohara, K.; Mori, H.; Aki, D.; Ohishi, M.; Yoshida, H.; Kubo, M.; Yoshimura, A. Socs1/jab is a negative regulator of lps-induced macrophage activation. Immunity 2002, 17, 583–591. [Google Scholar] [CrossRef]
- Baetz, A.; Frey, M.; Heeg, K.; Dalpke, A.H. Suppressor of cytokine signaling (socs) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J. Biol. Chem. 2004, 279, 54708–54715. [Google Scholar] [CrossRef] [PubMed]
- Strebovsky, J.; Walker, P.; Lang, R.; Dalpke, A.H. Suppressor of cytokine signaling 1 (socs1) limits nfkappab signaling by decreasing p65 stability within the cell nucleus. FASEB J. 2011, 25, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Adachi, O.; Ogawa, T.; Takeda, K.; Akira, S. Unresponsiveness of myd88-deficient mice to endotoxin. Immunity 1999, 11, 115–122. [Google Scholar] [CrossRef]
- Shotorbani, S.S.; Su, Z.L.; Xu, H.X. Toll-like receptors are potential therapeutic targets in rheumatoid arthritis. World J. Biol. Chem. 2011, 2, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.M.; Liu, J.H.; Xue, C.B.; Li, M.Q.; Xing, S.; Zhang, X.; He, W.T.; Jiang, F.C.; Lu, X.; Zhou, P. Pharmacological inhibition of myd88 homodimerization counteracts renal ischemia reperfusion-induced progressive renal injury in vivo and in vitro. Sci. Rep. 2016, 6, 26954. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ramke, M.; Chintakuntlawar, A.V.; Lee, J.Y.; Rajaiya, J.; Chodosh, J. Role of myd88 in adenovirus keratitis. Immunol. Cell Biol. 2017, 95, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Klekotka, P.A.; Yang, L.; Yokoyama, W.M. Contrasting roles of the il-1 and il-18 receptors in myd88-dependent contact hypersensitivity. J. Investig. Dermatol. 2010, 130, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Breglio, K.; Chen, A.; Vamadevan, A.S.; Goo, T.; Hsu, D.; Conduah, D.; Xu, R.; Abreu, M.T. The myeloid differentiation factor 88 (myd88) is required for cd4+ t cell effector function in a murine model of inflammatory bowel disease. J. Immunol. 2008, 180, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- Adachi, O.; Kawai, T.; Takeda, K.; Matsumoto, M.; Tsutsui, H.; Sakagami, M.; Nakanishi, K.; Akira, S. Targeted disruption of the myd88 gene results in loss of il-1- and il-18-mediated function. Immunity 1998, 9, 143–150. [Google Scholar] [CrossRef]
- Wang, J.; Kobayashi, M.; Han, M.; Choi, S.; Takano, M.; Hashino, S.; Tanaka, J.; Kondoh, T.; Kawamura, K.; Hosokawa, M. Myd88 is involved in the signalling pathway for taxol-induced apoptosis and tnf-alpha expression in human myelomonocytic cells. Br. J. Haematol. 2002, 118, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Ojiro, K.; Ebinuma, H.; Nakamoto, N.; Wakabayashi, K.; Mikami, Y.; Ono, Y.; Po-Sung, C.; Usui, S.; Umeda, R.; Takaishi, H.; et al. Myd88-dependent pathway accelerates the liver damage of concanavalin a-induced hepatitis. Biochem. Biophys. Res Commun. 2010, 399, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Danner, R.L.; Elin, R.J.; Hosseini, J.M.; Wesley, R.A.; Reilly, J.M.; Parillo, J.E. Endotoxemia in human septic shock. Chest 1991, 99, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lecker, S.; Post, M.J.; Hietaranta, A.J.; Li, J.; Volk, R.; Li, M.; Sato, K.; Saluja, A.K.; Steer, M.L.; et al. Inhibition of ubiquitin-proteasome pathway-mediated i kappa b alpha degradation by a naturally occurring antibacterial peptide. J. Clin. Investig. 2000, 106, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Niggemann, J.; Bozko, P.; Bruns, N.; Wodtke, A.; Gieseler, M.T.; Thomas, K.; Jahns, C.; Nimtz, M.; Reupke, I.; Bruser, T.; et al. Baceridin, a cyclic hexapeptide from an epiphytic bacillus strain, inhibits the proteasome. Chembiochem 2014, 15, 1021–1029. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, B.-C.; Chen, J.-Y. Antimicrobial Peptide Epinecidin-1 Modulates MyD88 Protein Levels via the Proteasome Degradation Pathway. Mar. Drugs 2017, 15, 362. https://doi.org/10.3390/md15110362
Su B-C, Chen J-Y. Antimicrobial Peptide Epinecidin-1 Modulates MyD88 Protein Levels via the Proteasome Degradation Pathway. Marine Drugs. 2017; 15(11):362. https://doi.org/10.3390/md15110362
Chicago/Turabian StyleSu, Bor-Chyuan, and Jyh-Yih Chen. 2017. "Antimicrobial Peptide Epinecidin-1 Modulates MyD88 Protein Levels via the Proteasome Degradation Pathway" Marine Drugs 15, no. 11: 362. https://doi.org/10.3390/md15110362
APA StyleSu, B.-C., & Chen, J.-Y. (2017). Antimicrobial Peptide Epinecidin-1 Modulates MyD88 Protein Levels via the Proteasome Degradation Pathway. Marine Drugs, 15(11), 362. https://doi.org/10.3390/md15110362