Abstract
Red algae of Laurencia continue to provide wide structural diversity and complexity of halogenated C15 acetogenin medium-ring ethers. Here, we described the isolation of three new C15 acetogenins (3–5), and one truncated derivative (6) from Laurencia viridis collected on the Canary Islands. These compounds are interesting variations on the pinnatifidenyne structure that included the first examples of ethynyl oxirane derivatives (3–4). The structures were elucidated by extensive study of NMR (Nuclear Magnetic Resonance) data, J-based configuration analysis and DFT (Density Functional Theory) calculations. Their antiproliferative activity against six human solid tumor cell lines was evaluated.
1. Introduction
Medium-ring haloethers of Laurencia, a significant subset of biologically active marine natural products [1,2], continue to challenge innovative efforts as attractive targets for the synthesis of stereochemically rich medium-sized oxacyclic compounds [3,4] or to explore biogenetic hypothesis [5,6].
Z-Pinnatifidenyne and E-pinnatifidenyne (1–2) are representative and were reported in 1982 after isolation from the red algae Laurencia pinnatifida collected at Canary Islands [7]. Their structures were established by espectroscopic methods, X-ray diffraction analysis and chemical correlations, although, in 1991, the absolute configurations were reassigned based on a later X-ray analysis [8]. Recent synthetic approaches and biosynthetic studies of these eight-membered compounds have been reported [9,10].
As part of our continuing interest on the chemistry of the genus Laurencia [11,12,13,14] and during the course of our anticancer drug discovery program, we report the isolation of three new C15 acetogenins and one truncated derivative from Laurencia viridis (Figure 1). The structures were elucidated based on detailed analysis of 1D and 2D NMR data and revealed that these compounds are variations on the pinnatifidenyne structure with biogenetic relevance. In addition, a detailed study of NMR chemical shifts by DFT calculations analysis was also undertaken and their antiproliferative activity against human cancer cell lines A549 (lung), HBL-100 (breast), HeLa (cervix), SW1573 (lung), T-47D (breast) and WiDr (colon) was evaluated.
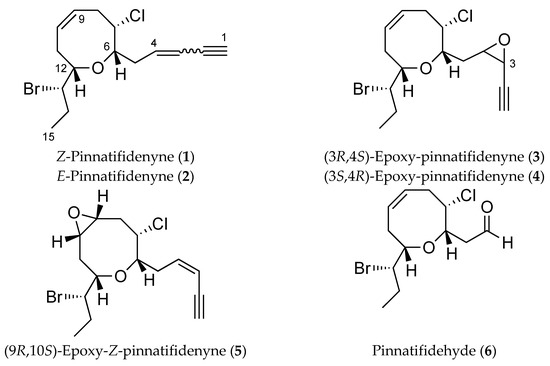
Figure 1.
Structures of pinnatifidenynes and new metabolites.
2. Results and Discussion
(3R,4S)-Epoxy-pinnatifidenyne (3) was isolated with yellow amorphous solid appearance. A molecular formula of C15H20BrClO2 was deduced by the presence of three pseudomolecular [M + Na]+ peaks in the HR-ESI-MS (High Resolution Electro Spray Ionization Mass Spectroscopy) spectrum at m/z 369.0227, 371.0216 and 373.0201 (ratio: 76:100:24, calcd. 369.0233, 371.0203, 371.0212 and 373.0183).
The 1H NMR spectrum revealed the presence of signals for two olefinic protons (δH 5.93 and 5.72), six deshielded methines (δH 4.12, 3.97, 3.95, 3.65, 3.52 and 3.25), one acetylenic proton (δH 2.39), four methylenes (ranging from δH 1.64 to 2.96) and a terminal methyl group (δH 1.08) (Table 1).

Table 1.
1H NMR data for compounds 3 and 4 (600 MHz, CDCl3).
According to the molecular formula, the 13C NMR data (Table 2) along with the analysis of the edited HSQC (Heteronuclear Single Quantum Correlation) spectrum confirmed the presence of two olefinic carbons (δC 131.0 and 128.9), six heteroatom-bearing methynes (δC 83.1, 77.5, 65.8, 61.1, 55.3 and 45.6), four methylenes (δC 34.5, 34.3, 30.0 and 27.1) and one methyl (δC 12.8). Careful examination of the homonuclear and heteronuclear NMR correlations exhibited in the COSY (Correlation SpectroscopY) and HSQC spectra allowed to observe a single 1H-1H spin system, C-3→C-15, containing a double bond between C-9 and C-10 together with heteroatoms located on carbons C-3, C-4, C-6, C-7, C-12 and C-13. Moreover, HMBC (Heteronuclear Multiple Bond Correlation) cross-peaks from H-6 (δH 4.12) to C-12 (δC 83.1) established an ether linkage between C-6 and C-12, thus indicating the presence of a 3,4,7,8-tetrahydro-2H-oxocin heterocycle. The presence of a terminal epoxy alkyne was evident by the characteristic 1H and 13C signals (δH/δC, 3.25, ddd, J = 3.8, 4.0, 8.4 Hz/55.3 (CH-4), 3.52, dd, J = 1.7, 4.0 Hz/45.6 (CH-3), 78.6 (C-2), and 2.39, d, J = 1.7 Hz/74.5 (CH-1)) and by IR (Infrared) absorption at 3023 (it is the strongest signal) and 2134 cm−1.
Table 2.
13C NMR data for compounds 3–6 (150 MHz, CDCl3).
The relative configuration of 3 was determined by a combination of NOESY (Nuclear Overhauser Spectroscopy) data and J-based configuration approach. 1D-NOE (Nuclear Overhauser Effect) correlations observed between H-6 and H-7/H-12 located all these protons on the same face of the heterocycle (Figure 2), consistent with the orientation previously observed in the pinnatifidenynes. The relationship between the configurations of C-12 and C-13 was established comparing chemical shifts and coupling constants of 3 with those of pinnatifidenynes sharing the same C-15–C-6 fragment [7]. To complete the structural determination, the relative configuration of the epoxide as well as its stereochemical relationship with C-6, the homo- and heteronuclear J couplings were measured [15,16]. The nJC,H values were accurately obtained using HSQC-HECADE (Heteronuclear Couplings from ASSCI (American Standard Code for Information Interchange)-Domain experiments with E.COSY-type cross peaks) experiment. The value of the coupling constants observed for 3JH3-H4 = 4.0 Hz, confirmed by the observation of a strong dipolar correlation between H-3 and H-4, ensure the cis configuration for the epoxide (Figure 2c). Finally, the relative configurations at C-6 and C-4 can be conveniently started from the large coupling constant displayed by the protons H-6 and H-5a (3JH6-H5a = 9.7 Hz) and the observed value of 2JC6-H5b = −1.5 Hz that suggested an anti relationship for H-6 and H-5a (δH 2.35) and a gauche orientation between H-6 and H-5b (δH 1.64), confirmed by the dipolar correlation between H-5b and H-7. Similarly, it was established a threo-configuration between H-4 and H-5a explained by the observed values of 3JH4-H5b = 8.4 Hz, 3JC3-H5a = 1.2 Hz and 2JC4-H5a = −2.2 Hz. Further support was obtained from additional long-range heteronuclear coupling constants shown in Figure 3.
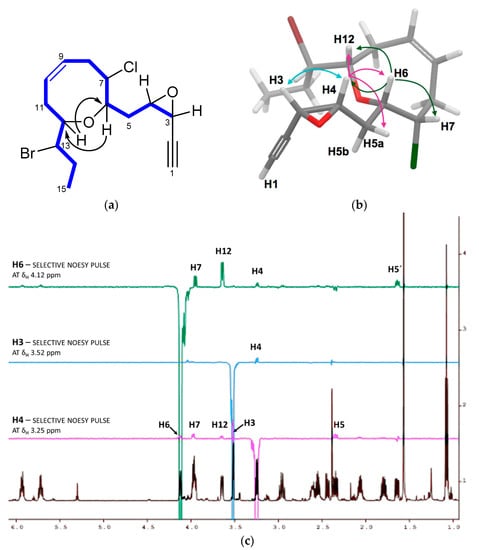
Figure 2.
(a) C-3–C-15 spin system, correlated by 1H-1H COSY experiment and selected HMBC correlations of 3; (b) key NOE correlations used to determine the relative configuration; and (c) 1D-NOE spectra.
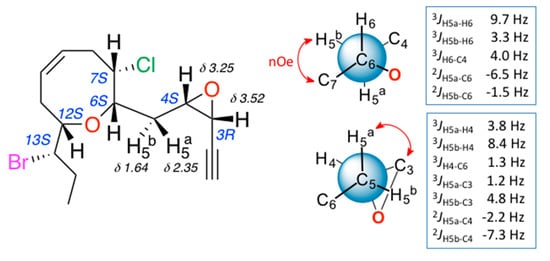
Figure 3.
J-based configuration analysis for C-4/C-6 fragment of (3R,4S)-epoxy-pinnatifidenyne (3).
Compound 4 has the same molecular formula as 3 together with a close structural relationship. Comparison of their 1H and 13C NMR chemical shifts and the analysis of their 2D NMR data allowed us to establish an identical planar structure for both compounds (see Table 1 and Table 2 and Experimental Section).
The stereochemical relationships between the different stereogenic centres, including those of epoxide, were performed using the above-described methods. The results of the NMR configurational analysis are shown in Figure 4. The conclusion was that compounds 3 and 4 share the relative configuration at centers C-6, C-7, C-12 and C-13, as well as the cis configuration of the epoxide, while the oxirane rings present an opposite relative configuration.
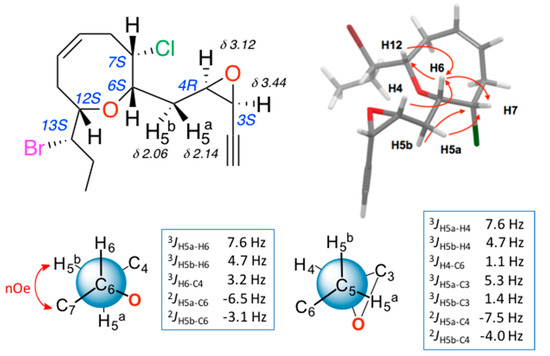
Figure 4.
Configuration analysis for (3S,4R)-epoxy-pinnatifidenyne (4).
Although a J-based configurational analysis on 3 and 4 was done to determine its relative configuration, a complementary computational study was undertaken. Chemical shift calculations of carbon atoms attached to halogens are not accurate or difficult to calculate due to spin–orbit contributions. However, Kutateladze et al. [17] have recently reported the use of parametric corrections to calculate 13C chemical shifts by DFT using inexpensive computations at the B3LYP/6-31G(d) level for structure optimization and using the ωB97xD/6-31G(d) level for the chemical shift calculations. Using this methodology, we built all possible stereoisomers for compound 3 followed by the corresponding conformational searches, structure optimizations and chemical shift calculations for each calculated conformer. As a result, it turned out that the 3R,4S,6S,7S,12S,13S isomer showed the lowest RMSD (Root Mean Square Deviation) for compounds 3 and 4 (see Supplementary Materials, Table S1). This result was coincident with our NMR-based proposal for the relative configuration of all stereogenic centers within the oxirane ring. However, it was clear that it was unable to solve the uncertainty within the oxirane ring. Taking into account that the previously mentioned methodology uses only 13C data, we decided to use chemical shift calculations of 1H for a second comparison as it has been shown that the combination of both chemical shifts yields better results [12,13,18,19,20]. Thus, models of the two possible diastereoisomers, 3S,4R,6S,7S,12S,13S (3a) and 3R,4S,6S,7S,12S,13S (3b), were built and conformational searches on each one, consisting on 5000 steps of a hybrid MCMM (Monte Carlo Multiple Minimum), Low-Mode sampling using the MMFF94 (Merck Molecular Force Field) force field, were completed. Redundant conformers within a 12 kJ/mol energy window of the global minimum found were eliminated using an RMSD cutoff of 1.0 Å. Next, all the resulting structures (seven conformers for 3a and five conformers for 3b) were geometrically optimized using DFT calculations [21] at the B3LYP/6-31G** level of theory with the LACVP basis set in gas phase [22]. NMR shielding constants (σ) were calculated according to the calculated relative Boltzmann populations for each conformer. Finally, NMR chemical shifts were obtained scaling the calculated values by linear regression analysis of experimental and computed data. In this analysis, the carbon nucleus with the attached bromine atom and the acetylenic proton were not included due to their high deviations. Correlation coefficients were almost identical for both stereoisomers (0.9885 for 3a vs. 0.9881 for 3b) using the 13C NMR data but clearly better for isomer 3b using 1H-NMR data (0.9867 vs. 0.9696) (Figure 5 and Supplementary Materials, Table S2). Calculation of the DP4 parameter [23] showed that the 3R,4S,6S,7S,12S,13S diastereoisomer (3b) is the most likely solution, with a probability 99.9% using both 13C and 1H data, therefore supporting our NMR-based proposal. Moreover, the same procedure was applied to fit the data of compound 4 to the calculated values for diasteroisomers 3a and 3b (Figure 6 and Supplementary Materials, Table S2). In this case, it turned out that the experimental data of 4 fitted better with calculated data for the 3S,4R,6S,7S,12S,13S diasteroisomer (3a), confirming that compounds 3 and 4 are the two-possible cis oxirane stereoisomers.
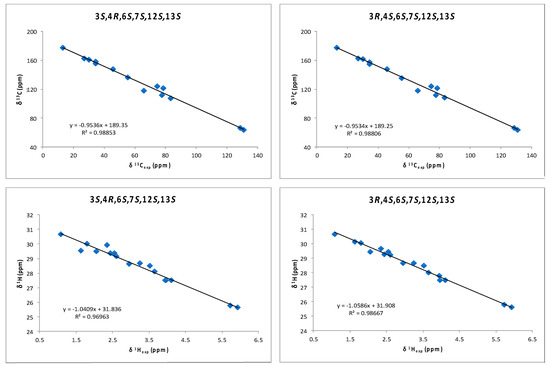
Figure 5.
1H and 13C correlations between calculated isotropic shieldings for the two simulated diastereoisomers of (3,4)-epoxy-pinnatifidenyne and experimentally observed chemical shifts for compound 3. Fitting parameters are indicated.
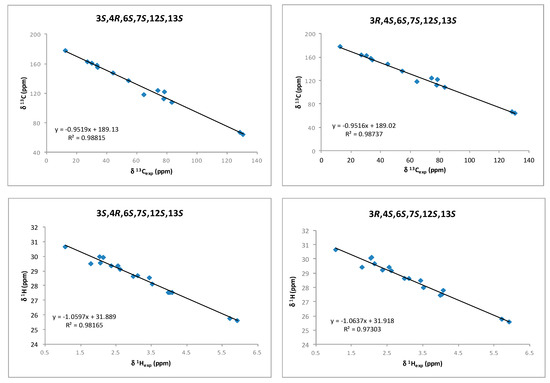
Figure 6.
1H and 13C correlations between calculated isotropic shieldings for the two simulated diastereoisomers of (3,4)-epoxy-pinnatifidenyne and experimentally observed chemical shifts for compound 4. Fitting parameters are indicated.
Compound 5 was isolated as an amorphous solid that was determined to have the molecular formula C15H20BrClO2 by HR-ESI-MS analysis. The presence in 5 of a terminal acetylene group conjugated with a double bond was evident from UV (Ultra Violet) [λmax (log ε) 225 (3.59) nm] and IR [νmax 3289 and 2300 cm−1] spectra. The 1H NMR experiment showed the characteristic shift signals at δH 3.17 (1H, br.s), 5.63 (1H, br.d, J = 10.8 Hz) and 6.05 (1H, ddd, J = 7.6, 7.7, 10.8 Hz) of the Z-enyne unit clearly related to Z-pinnatifidenyne (1). An extensive assessment of 1D and 2D NMR data (Table 2 and Table 3) compared to reported data for 1 [7] indicated that compound 5 differs to 1 containing an extra disubstituted epoxide situated at C-9–C-10. The relative configuration of 5 was determined by observation of dipolar correlations. Accordingly, the observed NOE enhancements from H-10 to H-9 and H-12, and from H-6 to H-7 and H-12 located all these protons on the same face of the molecule (Supplementary Materials, Figure S23). The above data and the coupling constant for 3JH9-H10 = 4.0 Hz provide evidence of the cis-orientation. This structure corresponds to a previously reported synthetic epoxide obtained by epoxidation of 1 and published by our group in 1982 [7]. However, the data published at that time and those obtained for our natural product did not correlate well. Therefore, to confirm the structure of 5 as (9R,10S)-epoxy-Z-pinnatifidenyne, we decided to repeat the selective epoxidation of 1. A product identical to 5 in all respects was obtained (see Experimental part).
Table 3.
1H NMR data for compounds 5 and 6 (600 MHz, CDCl3).
Pinnatifidehyde 6 is a yellow amorphous solid that shows the molecular formula C12H18BrClO2, evidenced by the presence of three pseudomolecular [M + Na]+ ions in the HR-ESI-MS spectrum at m/z 331.0086, 333.0056 and 335.0033 (ratio 74:100:36, calcd. 331.0076, 333.0047, 335.0026).
1H and 13C NMR spectra (Table 2 and Table 3) were reminiscent of the corresponding partial spectral signals of pinnatifidenynes 1–2 or compounds 3–4. All those compounds share identical oxacyclic and bromopropyl terminal chain (C-6→C-15 moiety), whereas the only notable differences were fixed going towards C-4 were the structure appears truncated. Thus, the deshielded signals of a methylene at δH 3.10 (br.dd, J = 8.0, 18.7 Hz) and 2.71 (dd, J = 4.0, 18.7 Hz), as well as the methine at δH 4.46 (ddd, J = 2.9, 4.0, 8.0 Hz), equivalent respectively to H2-5 and H-6 in pinnatifidenynes, showed correlations in the HMBC with a carbonyl signal at δC 200.2 corresponding to one aldehyde group. The structure of compound 6 possesses a truncated C12 carbon skeleton that may be derived from compounds 1 or 2 based on a probable oxidative process. Finally, analysis of the ROESY experiment confirmed the relative configuration of all stereogenic centers presented in the molecule as equivalent to those observed in the pinnatifidenynes (1, 2). Pinnatifidehyde 6 has been a synthetic intermediate target in the total synthesis of compounds 1 and 2 [10,24]. The spectroscopic data of synthesized intermediate by the Snyder group [10] was in agreement with the spectroscopic data of the natural product 6.
As far as we know, pinnatifidehyde (6) is the third example of C12 acetogenins; the other two, okamuragenin (7) and desepilaurallene (8) (Figure 7), have been isolated from Laurencia okamurai [25,26]. It has to be noted that the new compounds with the ethynyl oxirane unit (3 and 4) could be considered biogenetic precursors of pinnatifidehyde.
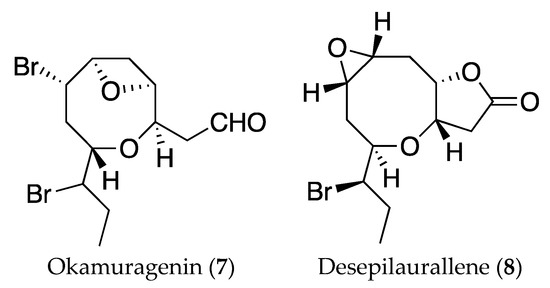
Figure 7.
Structures of C12 metabolites isolated from Laurencia okamurai, okamuragenin (7) and desepilaurallene (8).
The antiproliferative activity against six representative human solid tumor cell lines was evaluated for compounds 3–6 [27]. The results showed that compound 5 was the most potent compound of the series, with a modest activity against four of the cell lines tested (GI50 13–48 μM). Compound 4 was active against two of the cell lines (GI50 33–45 μM), whilst the remaining compounds were inactive (GI50 > 50 μM) (see Supplementary Materials, Table S15).
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured at room temperature in CHCl3 on a PelkinElmer-241 polarimeter (Waltham, MA, USA) by using a sodium lamp. IR spectra were recorded on a Bruker IFS55 spectrophotometer (Ettlingen, Germany) using methanolic solutions over NaCl disk. NMR spectra were recorded on a Bruker Avance 600 instrument (Karlsruhe, Germany) equipped with a 5-mm TCI (Triple Resonance CryoProbe) inverse detection cryo-probe. 1H and 13C NMR chemical shifts were referenced either to the CDCl3 or C6D6 solvent peaks at 300 K (CDCl3: δH 7.26, δC 77.0). COSY, HSQC, HMBC and ROESY experiments were performed using standard pulse sequences. 3JH,H values were measured from 1D 1H NMR. The HSQC-HECADE pulse sequence was used to measure long-range heteronuclear coupling constants. All experiments were performed in the phase-sensitive mode (States-TPPI (Time-Proportional Phase-Incrementation frequency discrimination) or echo-antiecho for quadrature detection in F1) and used gradient coherence selection. The HSQC-HECADE experiment was recorded using DIPSI (Decoupling in the Presence of Scalar Interactions) during the 40 ms of the isotropic mixing period using a bandwidth of 10 kHz, and a J-scale factor of 1 was used. Prior to Fourier transformation, zero filling was performed to expand the data to at least double the number of acquired data points. HR-ESI-MS data were obtained on a LCT Premier XE Micromass spectrometer (Waters, Milford, CT, USA). HPLC (High performance liquid chromatography) separations were carried out with a LKB 2248 system (Bromma, Sweden) equipped with a photodiode array detector. TLC (Thin layer chromatography) (Merck, Darmstadt, Germany) was visualized by spraying with phosphomolybdic acid reagent (10% in EtOH) and heating.
3.2. Computational Methods
Conformational searches were undertaken using the Macromodel software (version 8.5, Schrödinger Inc., San Diego, CA, USA) and the MMFF94 force field. Solvation effects of CHCl3 were simulated using the generalized Born/surface area (GBSA) solvation model. Extended non-bonded cutoff distances (a van der Waals cutoff of 8.0 Å and an electrostatic cutoff of 20.0 Å) were used. Local minima within 10 kJ of the global minimum were saved and analysis of the results was undertaken using Maestro software. Quantum mechanical calculations were carried out with the Jaguar package (Jaguar; Schrödinger LLC, New York, NY, USA). Single-point energy calculations were performed at the DFT theoretical level in the gas phase. The B3LYP hybrid functional with the LACVP ** basis set was used. Chemical shifts were calculated using the gauge-including atomic orbital (GIAO) method. Chemical shifts were calculated from their shielding constants that were first averaged according to their relative Boltzmann populations using a Schrödinger Inc. python script. Proton chemical shifts for each methyl group were averaged due to their conformational freedom.
3.3. Biological Material
Specimens of Laurencia viridis were collected by hand in the intertidal zone at Callao Salvaje, Tenerife, Canary Islands, Spain (28°07′12′′ N, 16°46′45′′ W) in April 2013. A voucher specimen was deposited at the Department of Biología Vegetal, Botánica, University of La Laguna, Tenerife (TFC Phyc 7180, Herbarium Code of University of La Laguna).
3.4. Extraction and Isolation
Fresh specimens were extracted at room temperature using CHCl3: CH3OH (1:1, v/v). The resulting extract (77.5 g) was separated in Silicagel 0.2–0.5 mm (Sigma-Aldrich, St. Louis, MO, USA) using a gradient of n-hexane:ethyl acetate. The fraction corresponding at 10% of n-hexane was chromatographed in Sephadex LH-20 (Sigma, St. Louis, MO, USA) (n-hexane:CHCl3:CH3OH, 2:1:1). The first fraction was treated in Sephadex LH-20 in the same conditions, yielding eight fractions. Selected fractions that exhibits similar TLC profiles were rechromatographed on normal phase open column (Silicagel, 0.2–0.5 mm), using a gradient of n-hexane:acetone from 49:1 to 1:1, yielding compound 6 (3.2 mg) in the second fraction. The compounds 3–5 were isolated and purified from the first fraction by HPLC using µ-Porasil™ HPLC column (Waters, Milford, CT, USA), 10 µm, 19 × 150 mm, and n-hexane:ethyl acetate (49:1) as mobile phase (Supplementary Materials, Scheme S1).
(3R,4S)-Epoxy-pinnatifidenyne (3): yellow, amorphous substance; −17 (c 0.09, CHCl3); IR νmax (CHCl3) 3023, 2788, 2379, 2134, 1748, 1698, 1647, 1571, 1500, 1401, 1311, 1189, 1098 cm−1; 1H and 13C NMR data (CDCl3), see Table 1 and Table 2; HR-ESI-MS m/z 369.0227, 371.0216 and 373.0201 [M + Na]+ (76:100:24) (calcd. for C15H20O235Cl79BrNa, 369.0233; C15H20O237Cl79BrNa, 371.0203; C15H20O235Cl81BrNa, 371.0212; C15H20O237Cl81BrNa, 373.0183).
(3S,4R)-Epoxy-pinnatifidenyne (4): yellow, amorphous solid; +30 (c 0.1, CHCl3); IR νmax (CHCl3) 3289, 2935, 2117, 1743, 1454, 1384, 1103 cm−1; 1H and 13C NMR data (CDCl3), see Table 1 and Table 2; HR-ESI-MS m/z 369.0222, 371.0204 and 373.0195 [M + Na]+ (79:100:31) (calcd. for C15H20O235Cl79BrNa, 369.0233; C15H20O237Cl79BrNa, 371.0203; C15H20O237Cl81BrNa, 371.0212; C15H20O237Cl81BrNa, 373.0183).
(9R,10S)-Epoxy-Z-pinnatifidenyne (5): yellow, amorphous solid; +8 (c 0.24, CHCl3); IR νmax (CHCl3) 3289, 2935, 2300, 2015, 1743, 1454, 1379, 1103 cm−1; 1H and 13C NMR data (CDCl3), see Table 1 and Table 2; HR-ESI-MS m/z 369.0219, 371.0214 and 373.0197 [M + Na]+ (84:100:25) (calcd. for C15H20O235Cl79BrNa, 369.0233; C15H20O237Cl79BrNa, 371.0203; C15H20O235Cl81BrNa, 371.0212; C15H20O237Cl81BrNa, 373.0183).
Pinnatifidehyde (6): yellow, amorphous substance; +33 (c 0.3, CHCl3); IR νmax (CHCl3) 3021, 2971, 2881, 2181, 2157, 2010, 1965, 1751, 1700, 1645, 1610, 1500, 1312, 1193, 1099 cm−1; 1H and 13C NMR data (CDCl3), see Table 1 and Table 2; HR-ESI-MS m/z 331.0086, 333.0056 and 335.0033 [M + Na]+ (74:100:36) (calcd. for C12H18O235Cl79BrNa, 331.0076; C12H18O237Cl79BrNa, 333.0047; C12H18O235Cl81BrNa, 333.0047; C12H18O237Cl81BrNa, 335.0026).
3.5. Chemical Conversion of Z-Pinnatifidenyne (1) to (9R,10S)-Epoxy-Z-Pinnatifidenyne (5)
m-Chloroperbenzoic acid (26 mg) in dry benzene (1.2 mL) was added dropwise to a solution of Z-pinnatifidenyne (23 mg) in benzene (0.84 mL) with stirring [7]. The reaction was led overnight and in the final step calcium hydroxide was added to remove the excess of peracid. The solution was then filtered and evaporated. The residue obtained was applied in to a silica gel (230–400 mesh, 60 Å) column eluted whit ethyl acetate:n-hexane gradient (2:8 to 3:7) yielding (9R,10S)-epoxy-Z-pinnatifidenyne (2.4 mg).
3.6. Antiproliferative Activity
Human solid tumor cell lines A549, HBL-100, HeLa, SW1573, T-47D and WiDr were a kind gift from Prof. G. J. Peters (VU Medical Center, Amsterdam, The Netherlands). The cell lines were cultured in RPMI 1640 medium (Flow Laboratories, Irvine, UK), supplemented with 5% fetal calf serum (FCS, Gibco, Grand Island, NY, USA), 2 mM l-glutamine (Merck, Darmstadt, Germany), 100 U/mL of penicillin G and 0.1 mg/mL of streptomycin (Sigma, St. Louis, MO, USA) at 37 °C in a 95% humidified with 5% CO2 atmosphere. The in vitro antiproliferative activity was evaluated using the sulforhodamine B (SRB, Sigma, St. Louis, MO, USA) assay with slight modifications [27]. Briefly, pure compounds were initially dissolved in DMSO (Sigma, St. Louis, MO, USA) at 400 times the desired final maximum test concentration. Cells were inoculated onto 96-well plates in a volume of 100 μL per well at densities of 2500 (A549, HBL-100, HeLa and SW1573) or 5 000 (T-47D and WiDr) cells per well, based on their doubling times. Control cells were exposed to an equivalent concentration of DMSO (0.25% v/v, negative control). Each agent was tested in triplicate at different dilutions in the range of 0.5–50 µM. The drug treatment was started on day 1 after plating. Drug incubation times comprised 48 h, after which time cells were precipitated with 25 µL ice-cold 50% (w/v) trichloroacetic acid (Merck, Darmstadt, Germany) and fixed for 60 min at 4 °C. Then the SRB assay was performed. The optical density (OD) of each well was measured at 530 nm, using BioTek’s PowerWave XS microplate reader. Values were corrected for background OD from wells containing only medium. The percentage growth (PG) was calculated in relation to untreated control cells (C) at each of the drug concentration levels based on the difference in OD at the start (T0) and end of drug exposure (T), according to NCI formulas Therefore, if T is greater than or equal to T0, the calculation is 100 × [(T − T0)/(C − T0)]. If T is less than T0, denoting cell killing, the calculation is 100 × [(T − T0)/(T0)]. The effect is defined as percentage of growth, where 50% growth inhibition (GI50) represents the concentration at which PG is +50. With these calculations, a PG value of 0 represents no difference from the start of drug exposure, while negative PG values denote net cell death.
Supplementary Materials
The following are available online at www.mdpi.com/1660-3397/16/1/5/s1. Scheme S1: Isolation procedure for new acetogenins; Figures S1–S31: NMR and HRESIMS spectra of isolated compounds; Figures S32 and S33: Correlations plots for 13C and 1H calculated vs. 13C and 1H experimental data for compounds 3 and 4; Figure S34: DP4 app for (3R,4S)-epoxy-pinnatifidenyne 3; Table S1: Calculated vs. experimental RMDS for compounds 3 and 4; Table S2: Scaled 13C and 1H chemical shifts for the two studied diasteroisomers on B3LYP-D3 in gas phase; Tables S3–S14: Computational calculation results; Table S15: Antiproliferative activity against human cancer cell lines.
Acknowledgments
This work was supported by grant CTQ2014-55888-C03-01-R (Ministry of Economy and Competitiveness of Spain, MINECO). A.M.-A. and C.R.d.V. acknowledge MINECO for a Ph.D. scholarship and O.M.-F. thanks ULL for a grant. Authors are grateful to M. C. Gil-Rodríguez (Departamento de Biología Vegetal, ULL) for the taxonomic classification of the alga.
Author Contributions
J.J.F., M.L.S. and M.N. conceived the study. A.M.-A., C.R.d.V. and O.M.-F. conducted the isolation experiments and the initial structure determinations. A.H.D., A.M.-A., J.J.F. and M.L.S. performed J-based configuration analysis. A.H.D. and A.M.-A carried out the NMR chemical shifts study by DFT calculations. J.M.P. conducted the bioassays. J.J.F. and M.L.S. wrote the paper. All authors contributed to the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinset, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294, (and earlier reviews in this series). [Google Scholar] [CrossRef] [PubMed]
- Harizani, M.; Ioannou, E.; Roussis, V. The Laurencia paradox: An endless source of chemodiversity. In Progress in the Chemistry of Organic Natural Products; Kinghorn, A.D., Galk, H., Gibbons, S., Kobayashi, J., Eds.; Springer: Berlin, Germany, 2016; Volume 102, pp. 91–252. [Google Scholar]
- Lin, R.; Cao, L.; West, F.G. Medium-sized cyclic ethers via Stevens [1,2]-shift of mixed monothioacetal-derived sulfonium ylides: Application to formal synthesis of (+)-laurencin. Org. Lett. 2017, 19, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Zhang, W.; Schienebeck, C.M.; Bennett, S.R.; Tang, W. Intermolecular bromoesterification of conjugated enynes: An efficient synthesis of bromoallenes. Org. Chem. Front. 2014, 1, 386–390. [Google Scholar] [CrossRef]
- Bonney, K.J.; Braddock, D.C. A unifying stereochemical analysis for the formation of halogenated C15-acetogenin medium-ring ethers from Laurencia species via intramolecular bromonium ion assisted epoxide ring-opening and experimental corroboration with a model epoxide. J. Org. Chem. 2012, 77, 9574–9584, (and references cited therein). [Google Scholar] [CrossRef] [PubMed]
- Braddock, D.C.; Sbircea, D.-T. Proof-of-principle direct double cyclisation of a linear C15-precursor to a brominated bicyclic medium-ring ether relevant to Laurencia species. Chem. Commun. 2014, 507, 12691–12693. [Google Scholar] [CrossRef] [PubMed]
- González, A.G.; Martín, J.D.; Martín, V.S.; Norte, M.; Pérez, R.; Ruano, J.Z.; Drexler, S.A.; Clardy, J. Non-terpenoid C-15 metabolites from the red seaweed Laurencia pinnatifida. Tetrahedron 1982, 38, 1009–1014. [Google Scholar] [CrossRef]
- Norte, M.; González, A.G.; Cataldo, F.; Rodríguez, M.L.; Brito, I. New examples of acyclic and cyclic C-15 acetogenins from Laurencia pinnatifida. Reassignment of the absolute configuration for E and Z pinnatifidienyne. Tetrahedron 1991, 47, 9411–9418. [Google Scholar] [CrossRef]
- Kim, D. Intramolecular enolate alkylation from steroids through cladiellins to isolaurallene. Synlett 2014, 25, 33–57. [Google Scholar] [CrossRef]
- Snyder, S.A.; Brucks, A.P.; Treitler, D.S.; Moga, I. Concise synthetic approaches for the Laurencia family: Formal total syntheses of (±)-laurefucin and (±)-E- and (±)-Z-pinnatifidenyne. J. Am. Chem. Soc. 2012, 134, 17714–17721. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cepeda, A.; Fernández, J.J.; Norte, M.; López-Rodríguez, M.; Brito, I.; Souto, M.L. Additional insights into the obtusallene family: Components of Laurencia marilzae. J. Nat. Prod. 2016, 79, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Cen-Pacheco, F.; Santiago-Benítez, A.J.; García, C.; Álvarez-Méndez, S.J.; Martín-Rodríguez, A.J.; Norte, M.; Martín, V.S.; Gavín, J.A.; Fernández, J.J.; Daranas, A.H. Oxasqualenoids from Laurencia viridis: Combined spectroscopic–computational analysis and antifouling potential. J. Nat. Prod. 2015, 78, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cepeda, A.; Hernández Daranas, A.; Fernández, J.J.; Norte, M.; Souto, M.L. Stereochemical determination of five-membered cyclic ether acetogenins using a spin-spin coupling constant approach and DFT calculations. Mar. Drugs 2014, 12, 4031–4044. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cepeda, A.; Fernández, J.J.; Norte, M.; Souto, M.L. New bicyclotridecane C15 nonterpenoid bromoallenes from Laurencia marilzae. Org. Lett. 2011, 13, 2690–2693. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, A.G.; Reddy, D.S. High-throughput in silico structure validation and revision of halogenated natural products is enabled by parametric corrections to DFT-computed 13C NMR chemical shifts and spin-spin coupling constants. J. Org. Chem. 2017, 82, 3368–3381. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Cen-Pacheco, F.; Rodríguez, J.; Norte, M.; Fernández, J.J.; Hernández Daranas, A. Connecting discrete stereoclusters by using DFT and NMR spectroscopy: The case of nivariol. Chem. Eur. J. 2013, 19, 8525–8532. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, H.J.; Crespín, G.D.; Santiago Benítez, A.J.; Gavín, J.A.; Norte, M.; Fernández, J.J.; Hernández Daranas, A. Stereochemistry of complex marine natural products by quantum mechanical calculations of NMR chemical shifts: Solvent and conformational effects on okadaic acid. Mar. Drugs 2014, 12, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.G.; Channon, J.A.; Paterson, I.; Goodman, J.M. The stereochemical assignment of acyclic polyols: A computational study of the NMR data of a library of stereopentad sequences from polyketide natural products. Tetrahedron 2010, 66, 6437–6444. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6–31G* basis set for third-row atoms. J. Comp. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, W.J.; Jung, J.; Kim, S.; Kim, D. Construction of eight-membered ether rings by olefin geometry-dependent internal alkylation: First asymmetric total synthesis of (+)-3-(E)- and (+)-3-(Z)-pinnatifidenyne. J. Am. Chem. Soc. 2003, 125, 10238–10240. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Li, X.-M.; Cui, C.-M.; Li, C.-S.; Sun, H.; Wang, B.-G. Sesquiterpene and acetogenin derivatives from the marine red alga Laurencia okamurai. Mar. Drugs 2012, 10, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Miao, F.P.; Li, K.; Ji, N.Y. Sesquiterpenes and acetogenins from the marine red alga Laurencia okamurai. Fitoterapia 2012, 83, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Castillo, Q.A.; Triana, J.; Eiroa, J.L.; Calcul, L.; Rivera, E.; Wojtas, L.; Padrón, J.M.; Boberieth, L.; Keramane, M.; Abel-Santos, E.; et al. ent-Labdane diterpenoids from the aerial parts of Eupatorium obtusissmum. J. Nat. Prod. 2016, 79, 907–913. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).