New 1,4-Dienonesteroids from the Octocoral Dendronephthya sp.

 , , ,
, , , 
Abstract
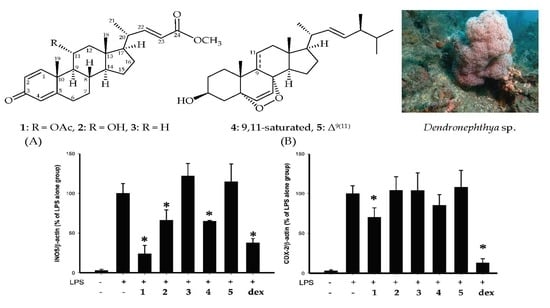
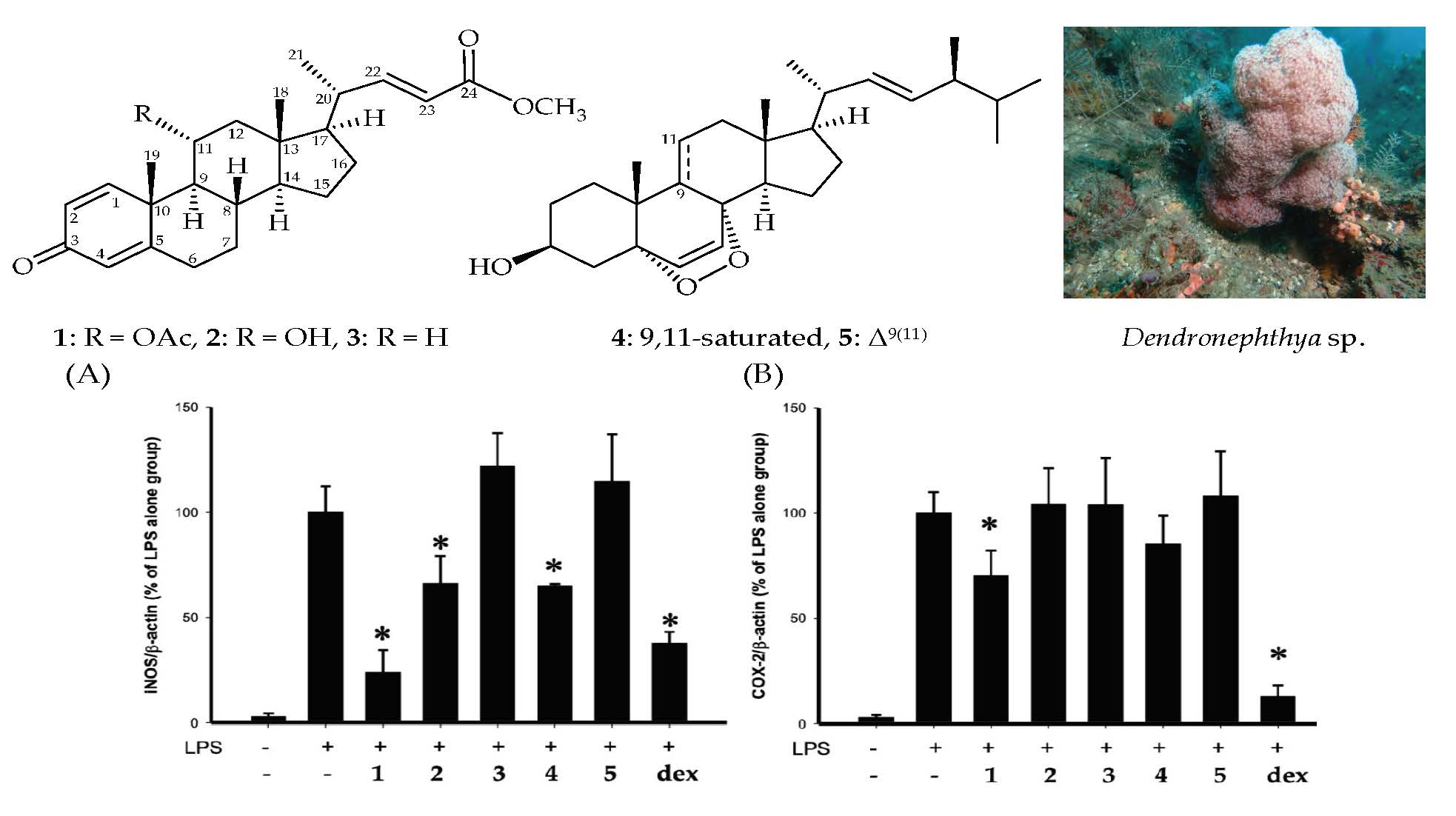
:
1. Introduction
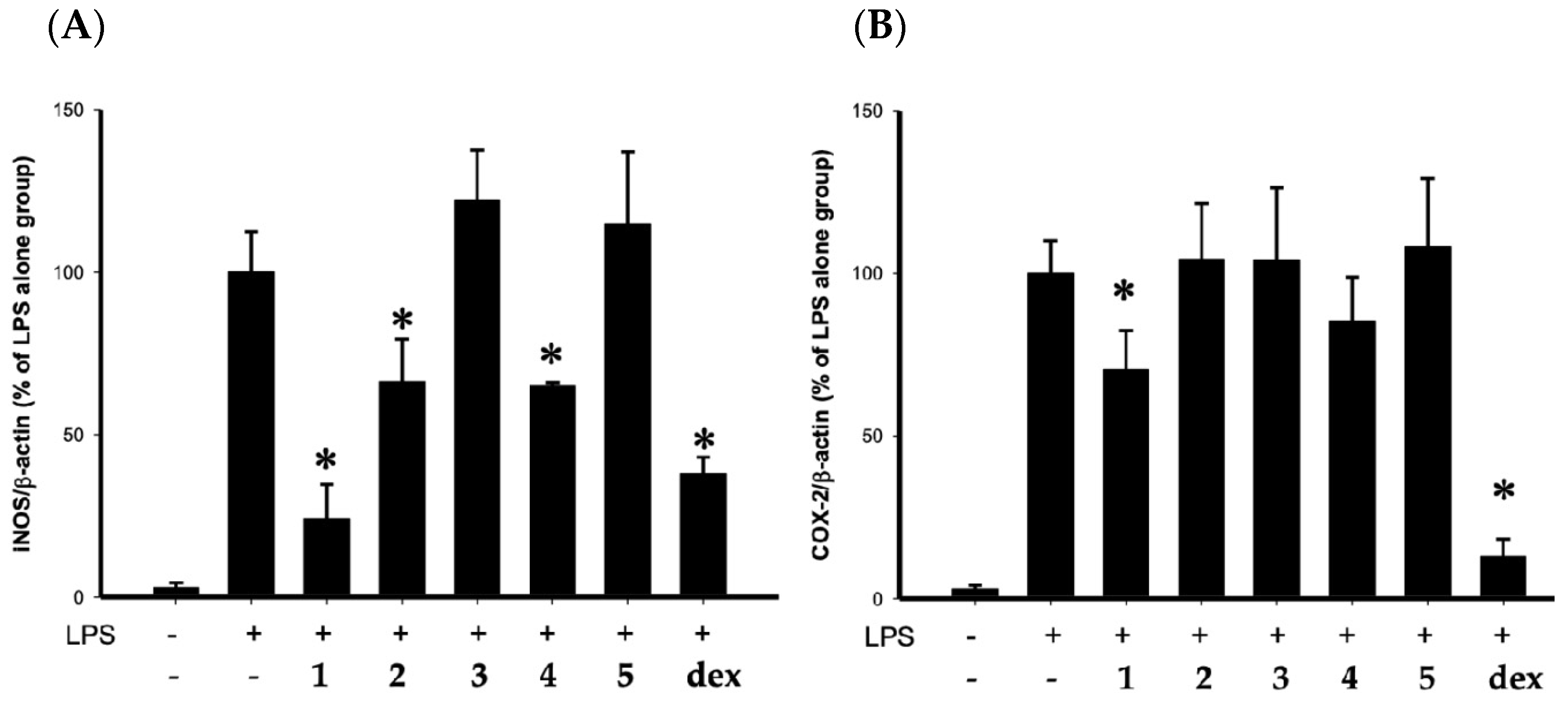
2. Results
3. Discussion
4. Experimental Section
4.1. General Experimental Procedures
4.2. Animal Material
4.3. Extraction and Separation
4.4. In Vitro Anti-Inflammatory Assay
4.5. Cell Viability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duh, C.Y.; El-Gamal, A.A.H.; Song, P.Y.; Wang, S.K.; Dai, C.F. Steroids and sesquiterpenoids from the soft corals Dendronephthya gigantea and Lemnalia cervicorni. J. Nat. Prod. 2004, 67, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.H.; Wen, Z.H.; Chen, I.M.; Su, J.H.; Huang, H.C.; Chiang, M.Y.; Sheu, J.H. Anti-inflammatory steroids from the octocoral Dendronephthya griffini. Tetrahedron 2008, 64, 3554–3560. [Google Scholar] [CrossRef]
- Chao, C.H.; Wen, Z.H.; Su, J.H.; Chen, I.M.; Huang, H.C.; Dai, C.F.; Sheu, J.H. Further study on anti- inflammatory oxygenated steroids from the octocoral Dendronephthya griffini. Steroids 2008, 73, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Tomono, Y.; Hirota, H.; Imahara, Y.; Fusetani, N. Four new steroids from two octocorals. J. Nat. Prod. 1999, 62, 1538–1541. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, E.; Abdel-Razik, A.F.; Zervou, M.; Christofidis, D.; Alexi, X.; Vagias, C.; Alexis, M.N.; Roussis, V. 5α,8α-Epidioxysterols from the gorgonian Eunicella cavolini and the ascidian Trididemnum inarmatum: Isolation and evaluation of their antiproliferative activity. Steroids 2009, 74, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Gunatilaka, A.A.L.; Gopichand, Y.; Schmitz, F.J.; Djerassi, C. Minor and trace sterols in marine invertebrates. 26. Isolation and strucutre elucidation of nine new 5α,8α-epidioxy sterols from four marine organisms. J. Org. Chem. 1981, 46, 3860–3866. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chang, C.W.; Tseng, Y.J.; Lee, J.; Sung, P.J.; Su, J.H.; Hwang, T.L.; Dai, C.F.; Wang, H.C.; Sheu, J.H. Bioactive steroids from the Formosan soft coral Umbellulifera petasites. Mar. Drugs 2016, 14, 180. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Liaw, C.C.; Chen, B.W.; Chen, P.C.; Su, J.H.; Sung, P.-J.; Dai, C.F.; Chiang, M.Y.; Sheu, J.H. Withanolide-based steroids from the cultured soft coral Sinularia brassica. J. Nat. Prod. 2013, 76, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.H.; Liu, H.L.; Huang, H.; Li, X.B.; Guo, Y.W. Steroids with aromatic A-rings from the Hainan soft coral Dendronephthya studeri Ridley. J. Nat. Prod. 2011, 74, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Tomono, Y.; Hirota, H.; Fusetani, N. Isogosterones A–D, antifouling 13,17-secosteroids from an octocoral Dendronephthya sp. J. Org. Chem. 1999, 64, 2272–2275. [Google Scholar] [CrossRef]
- Shin, K.; Chin, J.; Hahn, D.; Lee, J.; Hwang, H.; Won, D.H.; Ham, J.; Choi, H.; Kang, E.; Kim, H.; et al. Sterols from a soft coral, Dendronephthya gigantea as farnesoid X-activated receptor antagonists. Steroids 2012, 77, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Fernando, I.P.S.; Sanjeewa, K.K.S.; Kim, H.S.; Kim, S.Y.; Jeon, Y.J. Identification of sterols from the soft coral Dendronephthya gigantea and their anti-inflammatory potential. Environ. Toxicol. Pharm. 2017, 55, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.A.; Ding, Y.; Yang, H.W.; Heo, S.J.; Lee, S.H. Soft coral Dendronephthya puetteri extract ameliorates inflammations by suppressing inflammatory mediators and oxidative stress in LPS-stimulated zebrafish. Int. J. Mol. Sci. 2018, 19, 2695. [Google Scholar] [CrossRef] [PubMed]
- Fabricius, K.; Alderslade, P. Soft Corals and Sea Fans–A Comprehensive Guide to the Tropical Shallow-Water Genera of the Central-West Pacific, the Indian Ocean and the Red Sea, 1st ed.; Australian Institute of Marine Science: Townsville, Queensland, Australia, 2001; pp. 50–51; 112–115. [Google Scholar]
- Hung, H.C.; Feng, C.W.; Lin, Y.Y.; Chen, C.H.; Tsui, K.H.; Chen, W.F.; Pan, C.Y.; Sheu, J.H.; Sung, C.S.; Wen, Z.H. Nucleophosmin modulates the alleviation of atopic dermatitis caused by the marine-derived compound dihydroaustrasulfone alcohol. Exp. Mol. Med. 2018, 50, e446. [Google Scholar] [CrossRef] [PubMed]
- Jean, Y.H.; Chen, W.F.; Duh, C.Y.; Huang, S.Y.; Hsu, C.H.; Lin, C.S.; Sung, C.S.; Chen, I.M.; Wen, Z.H. Inducible nitric oxide synthase and cyclooxygenase-2 participate in anti-inflammatory and analgesic effects of the natural marine compound lemnalol from Formosan soft coral Lemnalia cervicorni. Eur. J. Pharmacol. 2008, 578, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.Y.; Lin, S.C.; Feng, C.W.; Chen, P.C.; Su, Y.D.; Li, C.M.; Yang, S.N.; Jean, Y.H.; Sung, P.J.; Duh, C.Y.; et al. Anti-inflammatory and analgesic effects of the marine-derived compound excavatolide B isolated from the culture-type Formosan gorgonian Briareum excavatum. Mar. Drugs 2015, 13, 2559–2579. [Google Scholar] [CrossRef] [PubMed]

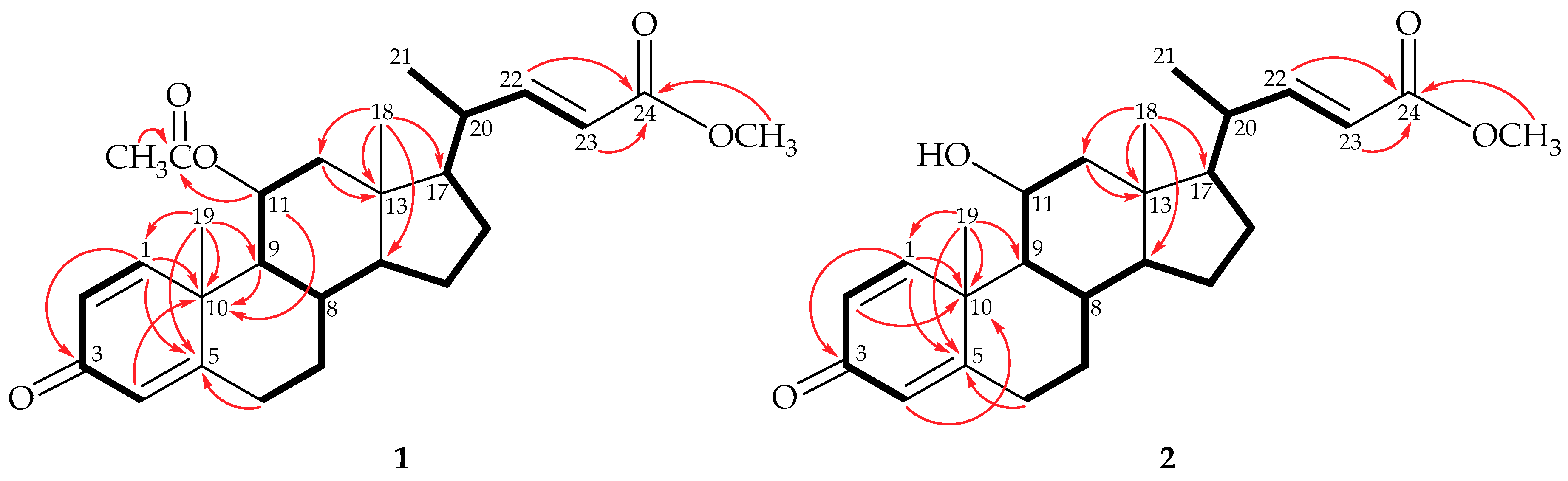
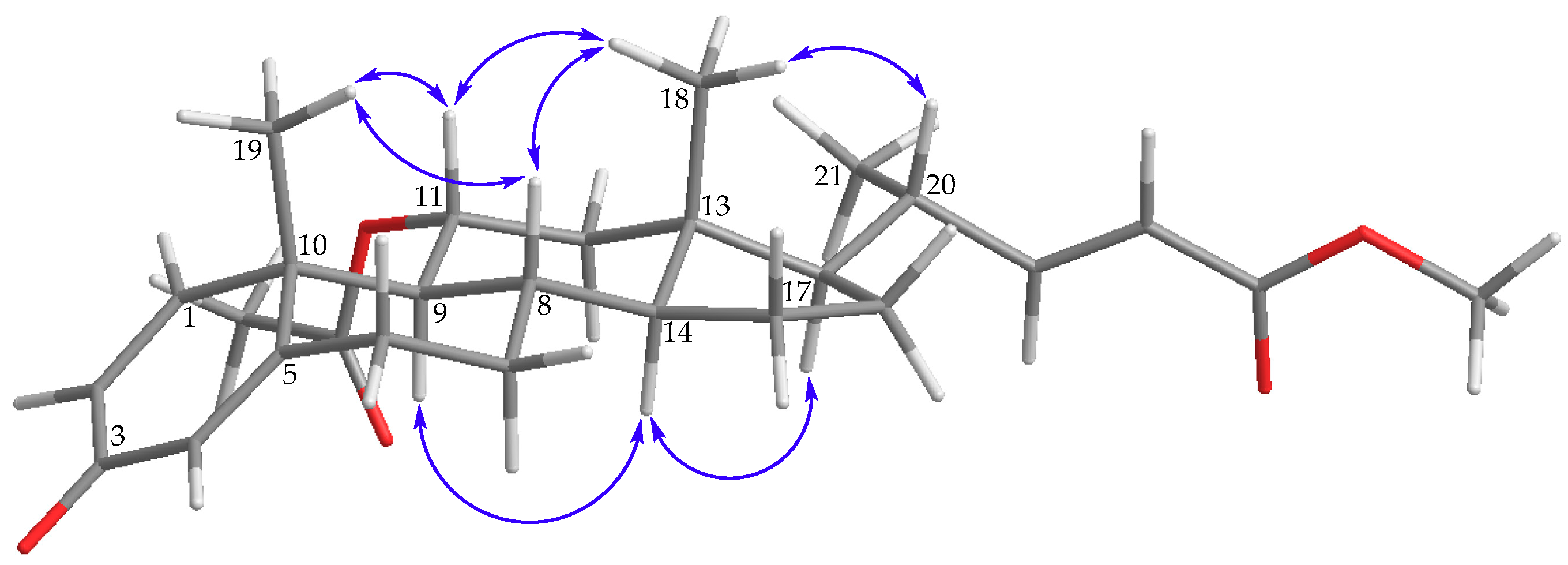
 ) correlations and selective HMBC (
) correlations and selective HMBC (  ) of steroids 1 and 2.
) of steroids 1 and 2.
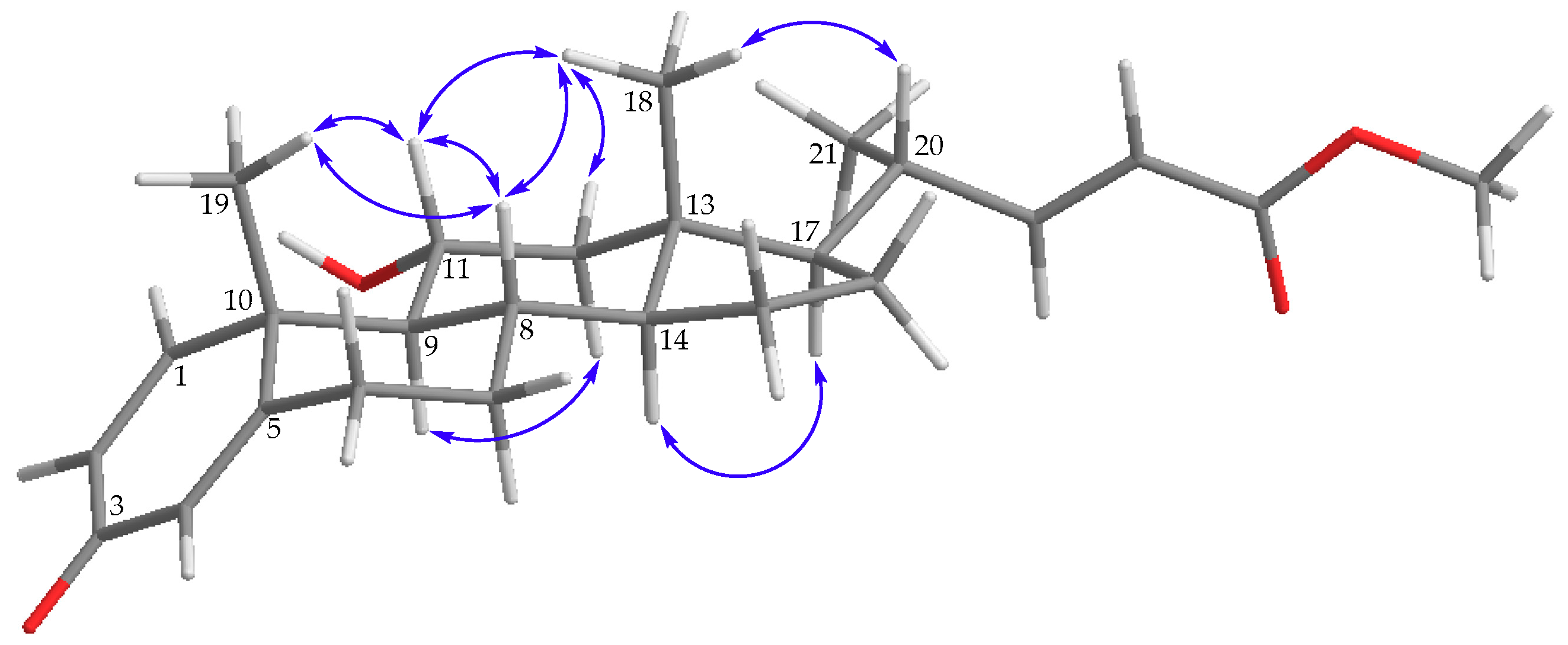
 ) of 1.
) of 1. ) of 2.
) of 2.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | ||||
|---|---|---|---|---|---|
| C/H | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | |
| 1 | 6.78 d (10.8) | 156.2 (CH) | 7.74 d (10.8) | 158.8 (CH) | |
| 2 | 6.13 dd (10.8, 2.0) | 125.7 (CH) | 6.15 dd (10.8, 2.0) | 125.1 (CH) | |
| 3 | 186.2 (C) | 183.8 (C) | |||
| 4 | 6.10 dd (2.0, 1.6) | 124.6 (CH) | 6.09 dd (2.0, 1.2) | 124.6 (CH) | |
| 5 | 167.1 (C) | 167.9 (C) | |||
| 6α β | 2.38 ddd (13.2, 4.4, 2.4) 2.48 ddd (13.2, 13.2, 4.8, 0.8) | 32.8 (CH2) | 2.36 ddd (13.2, 4.4, 2.8) 2.45 ddd (13.2, 13.2, 5.2, 1.6) | 33.2 (CH2) | |
| 7α/β | 1.14 m; 1.97 m | 33.3 (CH2) | 1.09 m; 1.96 m | 33.4 (CH2) | |
| 8 | 1.72 m | 34.4 (CH) | 1.61 m | 34.3 (CH) | |
| 9 | 1.39 dd (10.8, 10.8) | 56.3 (CH) | 1.09 dd (10.4, 10.4) | 60.2 (CH) | |
| 10 | 43.4 (C) | 44.0 (C) | |||
| 11 | 5.17 ddd (10.8, 10.8, 5.6) | 69.8 (CH) | 3.99 m | 67.9 (CH) | |
| 12α/β | 1.00 dd (12.4, 10.8); 2.13 dd (12.4, 5.6) | 44.7 (CH2) | 1.00 m; 2.10 dd (12.0, 4.8) | 50.0 (CH2) | |
| 13 | 42.5 (C) | 42.9 (C) | |||
| 14 | 1.14 m | 53.9 (CH) | 1.09 m | 54.5 (CH) | |
| 15α/β | 1.67 m; 1.16 m | 23.9 (CH2) | 1.63 m; 1.18 m | 24.0 (CH2) | |
| 16α/β | 1.92 m; 1.36 m | 27.4 (CH2) | 1.93 m; 1.38 m | 27.7 (CH2) | |
| 17 | 1.30 dd (9.2, 9.2) | 55.3 (CH) | 1.32 m | 55.3 (CH) | |
| 18 | 0.76 s | 12.9 (CH3) | 0.73 s | 13.3 (CH3) | |
| 19 | 1.26 s | 18.7 (CH3) | 1.25 s | 18.7 (CH3) | |
| 20 | 2.24 m | 39.5 (CH) | 2.25 m | 40.0 (CH) | |
| 21 | 0.97 d (6.4) | 19.8 (CH3) | 0.99 d (6.4) | 20.0 (CH3) | |
| 22 | 6.74 dd (15.6, 10.0) | 154.0 (CH) | 6.84 dd (15.6, 10.4) | 154.8 (CH) | |
| 23 | 5.79 d (15.6) | 119.3 (CH) | 5.81 d (15.6) | 119.2 (CH) | |
| 24 | 166.8 (C) | 167.2 (C) | |||
| OAc-11 | 2.01 s | 169.7 (C) 21.6 (CH3) | |||
| OMe-24 | 3.72 s | 51.3 (CH3) | 3.74 s | 51.5 (CH3) | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huynh, T.-H.; Chen, P.-C.; Yang, S.-N.; Lin, F.-Y.; Su, T.-P.; Chen, L.-Y.; Peng, B.-R.; Hu, C.-C.; Chen, Y.-Y.; Wen, Z.-H.; et al. New 1,4-Dienonesteroids from the Octocoral Dendronephthya sp. Mar. Drugs 2019, 17, 530. https://doi.org/10.3390/md17090530
Huynh T-H, Chen P-C, Yang S-N, Lin F-Y, Su T-P, Chen L-Y, Peng B-R, Hu C-C, Chen Y-Y, Wen Z-H, et al. New 1,4-Dienonesteroids from the Octocoral Dendronephthya sp. Marine Drugs. 2019; 17(9):530. https://doi.org/10.3390/md17090530
Chicago/Turabian StyleHuynh, Thanh-Hao, Pei-Chin Chen, San-Nan Yang, Feng-Yu Lin, Tung-Pin Su, Lo-Yun Chen, Bo-Rong Peng, Chiung-Chin Hu, You-Ying Chen, Zhi-Hong Wen, and et al. 2019. "New 1,4-Dienonesteroids from the Octocoral Dendronephthya sp." Marine Drugs 17, no. 9: 530. https://doi.org/10.3390/md17090530