Antitumor Potential of Marine and Freshwater Lectins




Abstract
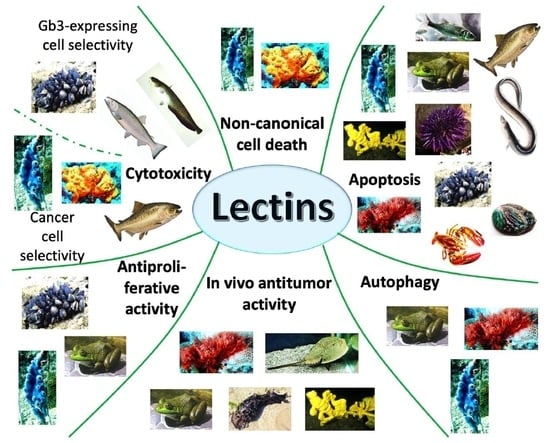
:
1. Introduction
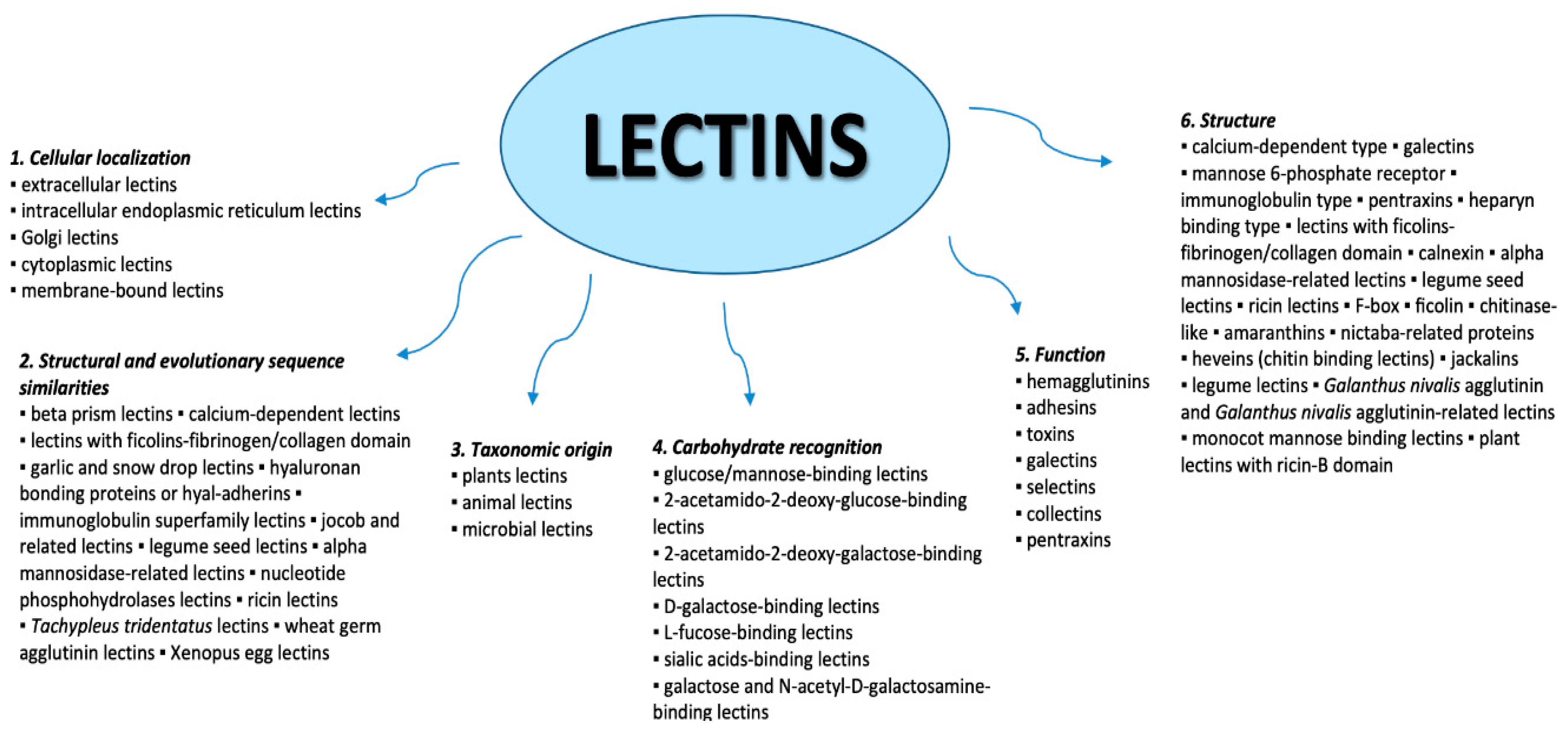
2. Lectins
3. Lectins from Marine and Freshwater Algae
4. Lectins from Marine and Freshwater Invertebrates
4.1. Phylum Mollusca and Arthropoda
4.2. Phylum Porifera
4.3. Phylum Chordata
5. Lectins from Marine and Freshwater Vertebrates
5.1. Amphibians
5.2. Fish
6. Conclusions
Funding
Conflicts of Interest
References
- Fondation Tara Océan. Available online: https://oceans.taraexpeditions.org/en/m/agenda/tara-museum-national-histoire-naturelle/ (accessed on 18 August 2019).
- World Register of Marine Species (WORMS). Available online: http://www.marinespecies.org (accessed on 18 August 2019).
- Vermeulen, N. From Darwin to the census of marine life: marine biology as big science. PLoS ONE 2013, 8, e54284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erwin, P.M.; López-Legentil, S.; Schuhmann, P.W. The pharmaceutical value of marine biodiversity for anti-cancer drug discovery. Ecol. Econ. 2010, 70, 445–451. [Google Scholar] [CrossRef]
- Calcabrini, C.; Catanzaro, E.; Bishayee, A.; Turrini, E.; Fimognari, C. Marine sponge natural products with anticancer potential: An updated review. Mar. Drugs 2017, 15, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyshlovoy, S.A.; Honecker, F. Marine Compounds and Cancer: 2017 Updates. Mar. Drugs 2018, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Postovit, L.; Widmann, C.; Huang, P.; Gibson, S.B. Harnessing oxidative stress as an innovative target for cancer therapy. Oxid. Med. Cell. Longev 2018, 2018, 6135739. [Google Scholar] [CrossRef] [Green Version]
- Molecular and Chemical Targets for Tumor-Selective Cancer Treatment. Available online: https://www.frontiersin.org/research-topics/2268/molecular-and-chemical-targets-for-tumor-selective-cancer-treatment (accessed on 20 August 2019).
- Akhtar, M.J.; Ahamed, M.; Alhadlaq, H.A. Therapeutic targets in the selective killing of cancer cells by nanomaterials. Clin. Chim. Acta 2017, 469, 53–62. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef]
- Widodo, N.; Priyandoko, D.; Shah, N.; Wadhwa, R.; Kaul, S.C. Selective killing of cancer cells by Ashwagandha leaf extract and its component Withanone involves ROS signaling. PLoS ONE 2010, 5, e13536. [Google Scholar] [CrossRef] [Green Version]
- Kaltner, H.; Toegel, S.; Caballero, G.G.; Manning, J.C.; Ledeen, R.W.; Gabius, H.-J. Galectins: Their network and roles in immunity/tumor growth control. Histochem. Cell Biol. 2017, 147, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Kizuka, Y. Glycans and cancer: role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv. Cancer Res. 2015, 126, 11–51. [Google Scholar] [PubMed]
- Vajaria, B.N.; Patel, P.S. Glycosylation: A hallmark of cancer? Glycoconj. J. 2017, 34, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, K.S.; Cunha, A.I.; Nascimento, K.S.; Cavada, B.S.; Azevedo, A.M.; Aires-Barros, M.R. An overview of lectins purification strategies. J. Mol. Recognit. 2012, 25, 527–541. [Google Scholar] [CrossRef] [PubMed]
- De Araújo, R.M.S.; da Ferreira, R.S.; Napoleão, T.H.; das Carneiro-da-Cunha, M.G.; Coelho, L.C.B.B.; dos Correia, M.T.S.; Oliva, M.L.V.; Paiva, P.M.G. Crataeva tapia bark lectin is an affinity adsorbent and insecticidal agent. Plant Sci. 2012, 183, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Francis, F.; Jaber, K.; Colinet, F.; Portetelle, D.; Haubruge, E. Purification of a new fungal mannose-specific lectin from Penicillium chrysogenum and its aphicidal properties. Fungal Biol. 2011, 115, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Bhutia, S.K.; Panda, P.K.; Sinha, N.; Praharaj, P.P.; Bhol, C.S.; Panigrahi, D.P.; Mahapatra, K.K.; Saha, S.; Patra, S.; Mishra, S.R.; et al. Plant lectins in cancer therapeutics: Targeting apoptosis and autophagy-dependent cell death. Pharmacol. Res. 2019, 144, 8–18. [Google Scholar] [CrossRef]
- Wang, H.; Ng, T.B. A lectin with some unique characteristics from the samta tomato. Plant Physiol. Biochem. 2006, 44, 181–185. [Google Scholar] [CrossRef]
- Santos, A.F.S.; da Silva, M.D.C.; Napoleão, T.H.; Paiva, P.M.G.; Correia, M.T.S.; Coelho, L.C.B.B. Lectins: Function, structure, biological properties and potential applications. Curr. Top. Pep. Protein Res. 2014, 15, 41–62. [Google Scholar]
- Višnjar, T.; Romih, R.; Zupančič, D. Lectins as possible tools for improved urinary bladder cancer management. Glycobiology 2019, 29, 355–365. [Google Scholar] [CrossRef]
- Shi, Z.; Li, W.; Tang, Y.; Cheng, L. A novel molecular model of plant lectin-induced programmed cell death in cancer. Biol. Pharm. Bull. 2017, 40, 1625–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, T.; Dan, X.; Ng, C.C.W.; Ng, T.B. Lectins with potential for anti-cancer therapy. Molecules 2015, 20, 3791–3810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Alarcón, D.; Blanco-Labra, A.; García-Gasca, T. Expression of lectins in heterologous systems. Int. J. Mol. Sci. 2018, 19, 616. [Google Scholar] [CrossRef] [Green Version]
- Drickamer, K. Two distinct classes of carbohydrate-recognition domains in animal lectins. J. Biol. Chem. 1988, 263, 9557–9560. [Google Scholar] [PubMed]
- De Oliveira Figueiroa, E.; Albuquerque da Cunha, C.R.; Albuquerque, P.B.S.; de Paula, R.A.; Aranda-Souza, M.A.; Alves, M.S.; Zagmignan, A.; Carneiro-da-Cunha, M.G.; Nascimento da Silva, L.C.; Dos Santos Correia, M.T. Lectin-carbohydrate interactions: implications for the development of new anticancer agents. Curr. Med. Chem. 2017, 24, 3667–3680. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Munkley, J.; Elliott, D.J. Hallmarks of glycosylation in cancer. Oncotarget 2016, 7, 35478–35489. [Google Scholar] [CrossRef] [Green Version]
- Häuselmann, I.; Borsig, L. Altered tumor-cell glycosylation promotes metastasis. Front. Oncol. 2014, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, F.; Aebi, M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Yen, H.-Y.; Chen, C.-Y.; Chen, C.-H.; Cheng, P.-F.; Juan, Y.-H.; Chen, C.-H.; Khoo, K.-H.; Yu, C.-J.; Yang, P.-C.; et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11332–11337. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Huang, B.; Deng, L.; Hu, Z. Progress in gene therapy using oncolytic vaccinia virus as vectors. J. Cancer Res. Clin. Oncol. 2018, 144, 2433–2440. [Google Scholar] [CrossRef]
- Anam, C.; Chasanah, E.; Perdhana, B.P.; Fajarningsih, N.D.; Yusro, N.F.; Sari, A.M.; Nursiwi, A.; Praseptiangga, D.; Yunus, A. Cytotoxicity of Crude Lectins from Red Macroalgae from the Southern Coast of Java Island, Gunung Kidul Regency, Yogyakarta, Indonesia. In Proceedings of the IOP Conference Series: Materials Science and Engineering, Jawa Tengah, Indonesia, 2017; Volume 193, p. 012017. [Google Scholar]
- Fukuda, Y.; Sugahara, T.; Ueno, M.; Fukuta, Y.; Ochi, Y.; Akiyama, K.; Miyazaki, T.; Masuda, S.; Kawakubo, A.; Kato, K. The anti-tumor effect of Euchema serra agglutinin on colon cancer cells in vitro and in vivo. Anticancer Drugs 2006, 17, 943–947. [Google Scholar] [CrossRef]
- Hayashi, K.; Walde, P.; Miyazaki, T.; Sakayama, K.; Nakamura, A.; Kameda, K.; Masuda, S.; Umakoshi, H.; Kato, K. Active targeting to osteosarcoma cells and apoptotic cell death induction by the novel lectin Eucheuma serra agglutinin isolated from a marine red alga. J. Drug. Deliv. 2012, 2012, 842785. [Google Scholar] [CrossRef] [Green Version]
- Sugahara, T.; Ohama, Y.; Fukuda, A.; Hayashi, M.; Kawakubo, A.; Kato, K. The cytotoxic effect of Eucheuma serra agglutinin (ESA) on cancer cells and its application to molecular probe for drug delivery system using lipid vesicles. Cytotechnology 2001, 36, 93–99. [Google Scholar] [CrossRef]
- Omokawa, Y.; Miyazaki, T.; Walde, P.; Akiyama, K.; Sugahara, T.; Masuda, S.; Inada, A.; Ohnishi, Y.; Saeki, T.; Kato, K. In vitro and in vivo anti-tumor effects of novel Span 80 vesicles containing immobilized Eucheuma serra agglutinin. Int. J. Pharm. 2010, 389, 157–167. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X. Recombinant Microcystis viridis lectin as a potential anticancer agent. Pharmazie 2010, 65, 922–923. [Google Scholar]
- Chaves, R.P.; da Silva, S.R.; Nascimento Neto, L.G.; Carneiro, R.F.; da Silva, A.L.C.; Sampaio, A.H.; de Sousa, B.L.; Cabral, M.G.; Videira, P.A.; Teixeira, E.H.; et al. Structural characterization of two isolectins from the marine red alga Solieria filiformis (Kützing) P.W. Gabrielson and their anticancer effect on MCF-7 breast cancer cells. Int. J. Biol. Macromol. 2018, 107, 1320–1329. [Google Scholar] [CrossRef]
- Li, G.; Zhao, Z.; Wu, B.; Su, Q.; Wu, L.; Yang, X.; Chen, J. Ulva pertusa lectin 1 delivery through adenovirus vector affects multiple signaling pathways in cancer cells. Glycoconj. J. 2017, 34, 489–498. [Google Scholar] [CrossRef]
- Hori, K.; Sato, Y.; Ito, K.; Fujiwara, Y.; Iwamoto, Y.; Makino, H.; Kawakubo, A. Strict specificity for high-mannose type N-glycans and primary structure of a red alga Eucheuma serra lectin. Glycobiology 2007, 17, 479–491. [Google Scholar] [CrossRef]
- Hori, K.; Miyazawa, K.; Ito, K. Hemagglutinins in Marine algae. Nippon Suisan Gakkaishi 1981, 47, 793–798. [Google Scholar] [CrossRef] [Green Version]
- UniProt. Available online: https://www.uniprot.org (accessed on 15 August 2019).
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed. Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Zhong, F.-D.; Zhang, Y.-J.; Wu, Z.-J.; Lin, Q.-Y.; Xie, L.-H. Molecular characterization of a new lectin from the marine alga Ulva pertusa. Acta Biochim. Biophys. Sin. (Shanghai) 2004, 36, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atta-ur-Rahman, M.; Choudhary Iqbal, M.; Khan, K. Frontiers in Natural Product Chemistry; Bentham Science Publishers: Sharjah, UAE, 2005. [Google Scholar]
- Turrini, E.; Calcabrini, C.; Tacchini, M.; Efferth, T.; Sacchetti, G.; Guerrini, A.; Paganetto, G.; Catanzaro, E.; Greco, G.; Fimognari, C. In Vitro study of the cytotoxic, cytostatic, and antigenotoxic profile of Hemidesmus indicus (L.) R.Br. (Apocynaceae) crude drug extract on T lymphoblastic cells. Toxins 2018, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Voultsiadou, E. Therapeutic properties and uses of marine invertebrates in the ancient Greek world and early Byzantium. J. Ethnopharmacol. 2010, 130, 237–247. [Google Scholar] [CrossRef]
- De Zoysa, M. Medicinal benefits of marine invertebrates: sources for discovering natural drug candidates. Adv. Food Nutr. Res. 2012, 65, 153–169. [Google Scholar]
- Kawsar, S.; Aftabuddin, S.; Yasumitsu, H.; Ozeki, Y. The cytotoxic activity of two d-galactose-binding lectins purified from marine invertebrates. Arch. biol. sci. (Beogr.) 2010, 62, 1027–1034. [Google Scholar] [CrossRef]
- Liao, J.-H.; Chien, C.-T.H.; Wu, H.-Y.; Huang, K.-F.; Wang, I.; Ho, M.-R.; Tu, I.-F.; Lee, I.-M.; Li, W.; Shih, Y.-L.; et al. A multivalent marine lectin from Crenomytilus grayanus possesses anti-cancer activity through recognizing globotriose Gb3. J. Am. Chem. Soc. 2016, 138, 4787–4795. [Google Scholar] [CrossRef]
- Chernikov, O.; Kuzmich, A.; Chikalovets, I.; Molchanova, V.; Hua, K.-F. Lectin CGL from the sea mussel Crenomytilus grayanus induces Burkitt’s lymphoma cells death via interaction with surface glycan. Int. J. Biol. Macromol. 2017, 104, 508–514. [Google Scholar] [CrossRef]
- Li, G.; Cheng, J.; Mei, S.; Wu, T.; Ye, T. Tachypleus tridentatus lectin enhances oncolytic vaccinia virus replication to suppress in vivo hepatocellular carcinoma growth. Mar. Drugs 2018, 16, 200. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wu, L.; Duan, X.; Cui, L.; Luo, J.; Li, G. Adenovirus carrying gene encoding Haliotis discus discus sialic acid binding lectin induces cancer cell apoptosis. Mar. Drugs 2014, 12, 3994–4004. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Mei, S.; Cui, L.; Zhao, Z.; Chen, J.; Wu, T.; Li, G. Marine lectins DlFBL and HddSBL fused with soluble coxsackie-adenovirus receptor facilitate adenovirus infection in cancer cells but have different effects on cell survival. Mar. Drugs 2017, 15, 73. [Google Scholar] [CrossRef] [Green Version]
- Fujii, Y.; Fujiwara, T.; Koide, Y.; Hasan, I.; Sugawara, S.; Rajia, S.; Kawsar, S.M.A.; Yamamoto, D.; Araki, D.; Kanaly, R.A.; et al. Internalization of a novel, huge lectin from Ibacus novemdentatus (slipper lobster) induces apoptosis of mammalian cancer cells. Glycoconj. J. 2017, 34, 85–94. [Google Scholar] [CrossRef]
- Fujii, Y.; Dohmae, N.; Takio, K.; Kawsar, S.M.A.; Matsumoto, R.; Hasan, I.; Koide, Y.; Kanaly, R.A.; Yasumitsu, H.; Ogawa, Y.; et al. A lectin from the mussel Mytilus galloprovincialis has a highly novel primary structure and induces glycan-mediated cytotoxicity of globotriaosylceramide-expressing lymphoma cells. J. Biol. Chem. 2012, 287, 44772–44783. [Google Scholar] [CrossRef] [Green Version]
- Hasan, I.; Sugawara, S.; Fujii, Y.; Koide, Y.; Terada, D.; Iimura, N.; Fujiwara, T.; Takahashi, K.G.; Kojima, N.; Rajia, S.; et al. MytiLec, a mussel R-type lectin, interacts with surface glycan Gb3 on Burkitt’s lymphoma cells to trigger apoptosis through multiple pathways. Mar. Drugs 2015, 13, 7377–7389. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Yang, X.; Duan, X.; Cui, L.; Li, G. Exogenous expression of marine lectins DlFBL and SpRBL induces cancer cell apoptosis possibly through PRMT5-E2F-1 pathway. Sci. Rep. 2014, 4, 4505. [Google Scholar] [CrossRef] [Green Version]
- Odintsova, N.A.; Belogortseva, N.I.; Khomenko, A.V.; Chikalovets, I.V.; Luk’yanov, P.A. Effect of lectin from the ascidian on the growth and the adhesion of HeLa cells. Mol. Cell. Biochem. 2001, 221, 133–138. [Google Scholar] [CrossRef]
- Wu, T.; Xiang, Y.; Liu, T.; Wang, X.; Ren, X.; Ye, T.; Li, G. Oncolytic Vaccinia Virus Expressing Aphrocallistes vastus Lectin as a Cancer Therapeutic Agent. Mar. Drugs 2019, 17, 363. [Google Scholar] [CrossRef] [Green Version]
- Rabelo, L.; Monteiro, N.; Serquiz, R.; Santos, P.; Oliveira, R.; Oliveira, A.; Rocha, H.; Morais, A.H.; Uchoa, A.; Santos, E. A lactose-binding lectin from the marine sponge Cinachyrella apion (Cal) induces cell death in human cervical adenocarcinoma cells. Mar. Drugs 2012, 10, 727–743. [Google Scholar] [CrossRef] [Green Version]
- Queiroz, A.F.S.; Silva, R.A.; Moura, R.M.; Dreyfuss, J.L.; Paredes-Gamero, E.J.; Souza, A.C.S.; Tersariol, I.L.S.; Santos, E.A.; Nader, H.B.; Justo, G.Z.; et al. Growth inhibitory activity of a novel lectin from Cliona varians against K562 human erythroleukemia cells. Cancer Chemother. Pharmacol. 2009, 63, 1023–1033. [Google Scholar] [CrossRef]
- Do Nascimento-Neto, L.G.; Cabral, M.G.; Carneiro, R.F.; Silva, Z.; Arruda, F.V.S.; Nagano, C.S.; Fernandes, A.R.; Sampaio, A.H.; Teixeira, E.H.; Videira, P.A. Halilectin-3, a lectin from the marine sponge Haliclona caerulea, induces apoptosis and autophagy in human breast cancer MCF7 cells through caspase-9 pathway and LC3-II protein expression. Anticancer Agents Med. Chem. 2018, 18, 521–528. [Google Scholar] [CrossRef]
- Pajic, I.; Kljajic, Z.; Dogovic, N.; Sladic, D.; Juranic, Z.; Gasic, M.J. A novel lectin from the sponge Haliclona cratera: Isolation, characterization and biological activity. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2002, 132, 213–221. [Google Scholar] [CrossRef]
- Matsumoto, R.; Fujii, Y.; Kawsar, S.M.A.; Kanaly, R.A.; Yasumitsu, H.; Koide, Y.; Hasan, I.; Iwahara, C.; Ogawa, Y.; Im, C.H.; et al. Cytotoxicity and glycan-binding properties of an 18 kDa lectin isolated from the marine sponge Halichondria okadai. Toxins 2012, 4, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, I.; Ozeki, Y. Histochemical localization of N-acetylhexosamine-binding lectin HOL-18 in Halichondria okadai (Japanese black sponge), and its antimicrobial and cytotoxic anticancer effects. Int. J. Biol. Macromol. 2019, 124, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Ponder, W.; Lindberg, D.R. Phylogeny and Evolution of the Mollusca; University of California Press: Berkeley, CA, USA, 2008. [Google Scholar]
- Bouchet, P.; Duarte, C.M. The exploration of marine biodiversity scientific and technological challenges. Fundatiòn BBVA 2006, 33, 1–34. [Google Scholar]
- Fredrick, W.S.; Ravichandran, S. Hemolymph proteins in marine crustaceans. Asian Pac. J. Trop. Biomed 2012, 2, 496–502. [Google Scholar] [CrossRef] [Green Version]
- Kovalchuk, S.N.; Chikalovets, I.V.; Chernikov, O.V.; Molchanova, V.I.; Li, W.; Rasskazov, V.A.; Lukyanov, P.A. cDNA cloning and structural characterization of a lectin from the mussel Crenomytilus grayanus with a unique amino acid sequence and antibacterial activity. Fish Shellfish Immunol. 2013, 35, 1320–1324. [Google Scholar] [CrossRef]
- Bekri, S.; Lidove, O.; Jaussaud, R.; Knebelmann, B.; Barbey, F. The role of ceramide trihexoside (globotriaosylceramide) in the diagnosis and follow-up of the efficacy of treatment of Fabry disease: A review of the literature. Cardiovasc. Hematol. Agents Med. Chem. 2006, 4, 289–297. [Google Scholar] [CrossRef]
- Behnam-Motlagh, P.; Tyler, A.; Grankvist, K.; Johansson, A. Verotoxin-1 treatment or manipulation of its receptor globotriaosylceramide (gb3) for reversal of multidrug resistance to cancer chemotherapy. Toxins 2010, 2, 2467–2477. [Google Scholar] [CrossRef] [Green Version]
- Qi, M.; Elion, E.A. MAP kinase pathways. J. Cell. Sci. 2005, 118, 3569–3572. [Google Scholar] [CrossRef] [Green Version]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Gilboa-Garber, N.; Wu, A.M. Binding properties and applications of Aplysia gonad lectin. In The Molecular Immunology of Complex Carbohydrates—2; Wu, A.M., Ed.; Springer US: Boston, MA, USA, 2001; pp. 109–126. ISBN 978-1-4615-1267-7. [Google Scholar]
- Avichezer, D.; Leibovici, J.; Gilboa-Garber, N.; Michowitz, M.; Lapis, K. New galactophilic lectins reduce tumorigenicity and preserve immunogenicity of Lewis lung carcinoma cells. In Proceedings of the Lectures and Symposia 14th Int. Cancer Cong., II, Budapest, Hungary, 21–27 August 1986; Karger: Basel, Switzerland, 1987; pp. 79–85. [Google Scholar]
- Gokudan, S.; Muta, T.; Tsuda, R.; Koori, K.; Kawahara, T.; Seki, N.; Mizunoe, Y.; Wai, S.N.; Iwanaga, S.; Kawabata, S. Horseshoe crab acetyl group-recognizing lectins involved in innate immunity are structurally related to fibrinogen. Proc. Natl. Acad. Sci. USA 1999, 96, 10086–10091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kairies, N.; Beisel, H.G.; Fuentes-Prior, P.; Tsuda, R.; Muta, T.; Iwanaga, S.; Bode, W.; Huber, R.; Kawabata, S. The 2.0-A crystal structure of tachylectin 5A provides evidence for the common origin of the innate immunity and the blood coagulation systems. Proc. Natl. Acad. Sci. USA 2001, 98, 13519–13524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Whang, I.; Lee, J. A novel C-type lectin from abalone, Haliotis discus discus, agglutinates Vibrio alginolyticus. Dev. Comp. Immunol. 2008, 32, 1034–1040. [Google Scholar] [CrossRef]
- Carneiro, R.F.; de Melo, A.A.; de Almeida, A.S.; da Moura, R.M.; Chaves, R.P.; de Sousa, B.L.; do Nascimento, K.S.; Sampaio, S.S.; Lima, J.P.M.S.; Cavada, B.S.; et al. H-3, a new lectin from the marine sponge Haliclona caerulea: Purification and mass spectrometric characterization. Int. J. Biochem. Cell Biol. 2013, 45, 2864–2873. [Google Scholar] [CrossRef]
- Xiong, C.; Li, W.; Liu, H.; Zhang, W.; Dou, J.; Bai, X.; Du, Y.; Ma, X. A normal mucin-binding lectin from the sponge Craniella australiensis. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2006, 143, 9–16. [Google Scholar] [CrossRef]
- Miarons, P.B.; Fresno, M. Lectins from tropical sponges. Purification and characterization of lectins from genus Aplysina. J. Biol. Chem. 2000, 275, 29283–29289. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, D.S.; Medeiros, T.L.; Ribeiro, J.K.C.; Monteiro, N.K.V.; Migliolo, L.; Uchoa, A.F.; Vasconcelos, I.M.; Oliveira, A.S.; de Sales, M.P.; Santos, E.A. A lactose specific lectin from the sponge Cinachyrella apion: purification, characterization, N-terminal sequences alignment and agglutinating activity on Leishmania promastigotes. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2010, 155, 211–216. [Google Scholar] [CrossRef]
- Frisch, S.M.; Screaton, R.A. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001, 13, 555–562. [Google Scholar] [CrossRef]
- Mihaly, S.R.; Sakamachi, Y.; Ninomiya-Tsuji, J.; Morioka, S. Noncanonical cell death program independent of caspase activation cascade and necroptotic modules is elicited by loss of TGFβ-activated kinase 1. Sci. Rep. 2017, 7, 2918. [Google Scholar] [CrossRef] [Green Version]
- Gundacker, D.; Leys, S.P.; Schröder, H.C.; Müller, I.M.; Müller, W.E. Isolation and cloning of a C-type lectin from the hexactinellid sponge Aphrocallistes vastus: A putative aggregation factor. Glycobiology 2001, 11, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moura, R.M.; Queiroz, A.F.S.; Fook, J.M.S.L.L.; Dias, A.S.F.; Monteiro, N.K.V.; Ribeiro, J.K.C.; Moura, G.E.D.D.; Macedo, L.L.P.; Santos, E.A.; Sales, M.P. CvL, a lectin from the marine sponge Cliona varians: Isolation, characterization and its effects on pathogenic bacteria and Leishmania promastigotes. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2006, 145, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Bröker, L.E.; Kruyt, F.A.E.; Giaccone, G. Cell death independent of caspases: A review. Clin. Cancer Res. 2005, 11, 3155–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Investig. 2000, 106, 1127–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foghsgaard, L.; Wissing, D.; Mauch, D.; Lademann, U.; Bastholm, L.; Boes, M.; Elling, F.; Leist, M.; Jäättelä, M. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J. Cell Biol. 2001, 153, 999–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; McDonald, D.G.; Dixon-Mah, Y.N.; Jacqmin, D.J.; Samant, V.N.; Vandergrift, W.A.; Lindhorst, S.M.; Cachia, D.; Varma, A.K.; Vanek, K.N.; et al. RIP1 and RIP3 complex regulates radiation-induced programmed necrosis in glioblastoma. Tumour Biol. 2016, 37, 7525–7534. [Google Scholar] [CrossRef]
- Horton, T.; Kroh, A.; Ahyong, B.; Bailly, N.; Brandão, S.N.; Costello, M.J.; Gofas, S.; Hernandez, F.; Holovachov, O.; Boyko, C.B.; et al. World Register of Marine Species; WoRMS Editorial Board: Ostend, Belgium, 2018. [Google Scholar]
- Negi, B.; Kumar, D.; Rawat, D.S. Marine Peptides as Anticancer Agents: A Remedy to Mankind by Nature. Curr. Protein Pept. Sci. 2017, 18, 885–904. [Google Scholar] [CrossRef]
- Ankisetty, S.; Khan, S.I.; Avula, B.; Gochfeld, D.; Khan, I.A.; Slattery, M. Chlorinated didemnins from the tunicate Trididemnum solidum. Mar. Drugs 2013, 11, 4478–4486. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Currano, J.N.; Carroll, P.J.; Joullié, M.M. Didemnins, tamandarins and related natural products. Nat. Prod. Rep. 2012, 29, 404–424. [Google Scholar] [CrossRef]
- Belogortseva, N.; Molchanova, V.; Glazunov, V.; Evtushenko, E.; Luk’yanov, P. N-Acetyl-d-glucosamine-specific lectin from the ascidian Didemnum ternatanum. Biochim. Biophys. Acta 1998, 1380, 249–256. [Google Scholar] [CrossRef]
- Fang, Y.; Eglen, R.M. Three-dimensional cell cultures in drug discovery and development. SLAS Discov. 2017, 22, 456–472. [Google Scholar]
- Bourgine, P.E.; Klein, T.; Paczulla, A.M.; Shimizu, T.; Kunz, L.; Kokkaliaris, K.D.; Coutu, D.L.; Lengerke, C.; Skoda, R.; Schroeder, T.; et al. In vitro biomimetic engineering of a human hematopoietic niche with functional properties. Proc. Natl. Acad. Sci. USA 2018, 115, E5688–E5695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B.T. The natural history of amphibian skin secretions, their normal functioning and potential medical applications. Biol. Rev. Camb. Philos. Soc. 1997, 72, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-X.; Nan, K.-J.; Lei, Y. Agents from amphibians with anticancer properties. Anticancer Drugs 2008, 19, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Iwama, M.; Ogawa, Y.; Sasaki, N.; Nitta, K.; Takayanagi, Y.; Ohgi, K.; Tsuji, T.; Irie, M. Effect of modification of the carboxyl groups of the sialic acid binding lectin from bullfrog (Rana catesbeiana) oocyte on anti-tumor activity. Biol. Pharm. Bull. 2001, 24, 978–981. [Google Scholar] [CrossRef] [Green Version]
- Nitta, K.; Ozaki, K.; Ishikawa, M.; Furusawa, S.; Hosono, M.; Kawauchi, H.; Sasaki, K.; Takayanagi, Y.; Tsuiki, S.; Hakomori, S. Inhibition of cell proliferation by Rana catesbeiana and Rana japonica lectins belonging to the ribonuclease superfamily. Cancer Res. 1994, 54, 920–927. [Google Scholar]
- Tatsuta, T.; Hosono, M.; Sugawara, S.; Kariya, Y.; Ogawa, Y.; Hakomori, S.; Nitta, K. Sialic acid-binding lectin (leczyme) induces caspase-dependent apoptosis-mediated mitochondrial perturbation in Jurkat cells. Int. J. Oncol. 2013, 43, 1402–1412. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Sugawara, S.; Tatsuta, T.; Hosono, M.; Nitta, K.; Fujii, Y.; Kobayashi, H.; Fujimura, T.; Taka, H.; Koide, Y.; et al. Sialyl-glycoconjugates in cholesterol-rich microdomains of P388 cells are the triggers for apoptosis induced by Rana catesbeiana oocyte ribonuclease. Glycoconj. J. 2014, 31, 171–184. [Google Scholar] [CrossRef]
- Tatsuta, T.; Hosono, M.; Takahashi, K.; Omoto, T.; Kariya, Y.; Sugawara, S.; Hakomori, S.; Nitta, K. Sialic acid-binding lectin (leczyme) induces apoptosis to malignant mesothelioma and exerts synergistic antitumor effects with TRAIL. Int. J. Oncol. 2014, 44, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Kariya, Y.; Tatsuta, T.; Sugawara, S.; Kariya, Y.; Nitta, K.; Hosono, M. RNase activity of sialic acid-binding lectin from bullfrog eggs drives antitumor effect via the activation of p38 MAPK to caspase-3/7 signaling pathway in human breast cancer cells. Int. J. Oncol. 2016, 49, 1334–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; Tatsuta, T.; Sugawara, S.; Hara, A.; Hosono, M. Synergistic anti-tumor effect of bullfrog sialic acid-binding lectin and pemetrexed in malignant mesothelioma. Oncotarget 2017, 8, 42466–42477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsuta, T.; Satoh, T.; Sugawara, S.; Hara, A.; Hosono, M. Sialic acid-binding lectin from bullfrog eggs inhibits human malignant mesothelioma cell growth in vitro and in vivo. PLoS ONE 2018, 13, e0190653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsuta, T.; Sato, S.; Sato, T.; Sugawara, S.; Suzuki, T.; Hara, A.; Hosono, M. Sialic acid-binding lectin from bullfrog eggs exhibits an anti-tumor effect against breast cancer cells including triple-negative phenotype cells. Molecules 2018, 23, 2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiang, G.-T.; Yu, Y.-L.; Chou, P.-L.; Tsai, H.-F.; Chen, L.-A.; Chen, Y.H.; Su, K.-J.; Wang, J.-J.; Bau, D.-T.; Wei, C.-W. The cytotoxic protein can induce autophagocytosis in addition to apoptosis in MCF-7 human breast cancer cells. In Vivo 2012, 26, 403–409. [Google Scholar]
- Tseng, H.-H.; Yu, Y.-L.; Chen, Y.-L.S.; Chen, J.-H.; Chou, C.-L.; Kuo, T.-Y.; Wang, J.-J.; Lee, M.-C.; Huang, T.-H.; Chen, M.H.-C.; et al. RC-RNase-induced cell death in estrogen receptor positive breast tumors through down-regulation of Bcl-2 and estrogen receptor. Oncol. Rep. 2011, 25, 849–853. [Google Scholar]
- Hu, C.C.; Tang, C.H.; Wang, J.J. Caspase activation in response to cytotoxic Rana catesbeiana ribonuclease in MCF-7 cells. FEBS Lett. 2001, 503, 65–68. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.W.; Hu, C.C.A.; Tang, C.H.A.; Lee, M.C.; Wang, J.J. Induction of differentiation rescues HL-60 cells from Rana catesbeiana ribonuclease-induced cell death. FEBS Lett. 2002, 531, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-N.; Yiang, G.-T.; Lin, Y.-F.; Chou, P.-L.; Wu, T.-K.; Chang, W.-J.; Chen, C.; Yu, Y.-L. Rana catesbeiana ribonuclease induces cell apoptosis via the caspase-9/-3 signaling pathway in human glioblastoma DBTRG, GBM8901 and GBM8401 cell lines. Oncol. Lett. 2015, 9, 2471–2476. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.-H.A.; Hu, C.-C.A.; Wei, C.-W.; Wang, J.-J. Synergism of Rana catesbeiana ribonuclease and IFN-gamma triggers distinct death machineries in different human cancer cells. FEBS Lett. 2005, 579, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.C.; Lee, Y.H.; Tang, C.H.; Cheng, J.T.; Wang, J.J. Synergistic cytotoxicity of Rana catesbeiana ribonuclease and IFN-gamma on hepatoma cells. Biochem. Biophys. Res. Commun. 2001, 280, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Peng, H.; Zhang, R.; Chen, Y.; Zhao, L.; Tang, K. Recombinant hHscFv-RC-RNase protein derived from transgenic tobacco acts as a bifunctional molecular complex against hepatocellular carcinoma. Biotechnol. Appl. Biochem. 2012, 59, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Gao, Y.; Cui, L.; Wu, L.; Yang, X.; Chen, J. Anguilla japonica lectin 1 delivery through adenovirus vector induces apoptotic cancer cell death through interaction with PRMT5. J. Gene Med 2016, 18, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Pan, S.; Zhou, M. Structural characterization and antitumor and mitogenic activity of a lectin from the gill of bighead carp (Aristichthys nobilis). Fish Physiol. Biochem. 2012, 38, 1815–1824. [Google Scholar] [CrossRef]
- Shirai, T.; Watanabe, Y.; Lee, M.; Ogawa, T.; Muramoto, K. Structure of rhamnose-binding lectin CSL3: unique pseudo-tetrameric architecture of a pattern recognition protein. J. Mol. Biol. 2009, 391, 390–403. [Google Scholar] [CrossRef]
- Bah, C.S.F.; Fang, E.F.; Ng, T.B.; Mros, S.; McConnell, M.; Bekhit, A.E.-D.A. Purification and characterization of a rhamnose-binding chinook salmon roe lectin with antiproliferative activity toward tumor cells and nitric oxide-inducing activity toward murine macrophages. J. Agric. Food Chem. 2011, 59, 5720–5728. [Google Scholar] [CrossRef]
- Sugawara, S.; Hosono, M.; Ogawa, Y.; Takayanagi, M.; Nitta, K. Catfish egg lectin causes rapid activation of multidrug resistance 1 P-glycoprotein as a lipid translocase. Biol. Pharm. Bull. 2005, 28, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, S.; Im, C.; Kawano, T.; Tatsuta, T.; Koide, Y.; Yamamoto, D.; Ozeki, Y.; Nitta, K.; Hosono, M. Catfish rhamnose-binding lectin induces G0/1 cell cycle arrest in Burkitt’s lymphoma cells via membrane surface Gb3. Glycoconj. J. 2017, 34, 127–138. [Google Scholar] [CrossRef]
- Titani, K.; Takio, K.; Kuwada, M.; Nitta, K.; Sakakibara, F.; Kawauchi, H.; Takayanagi, G.; Hakomori, S. Amino acid sequence of sialic acid binding lectin from frog (Rana catesbeiana) eggs. Biochemistry 1987, 26, 2189–2194. [Google Scholar] [CrossRef]
- Nitta, K.; Takayanagi, G.; Kawauchi, H.; Hakomori, S. Isolation and characterization of Rana catesbeiana lectin and demonstration of the lectin-binding glycoprotein of rodent and human tumor cell membranes. Cancer Res. 1987, 47, 4877–4883. [Google Scholar]
- Nitta, K.; Ozaki, K.; Tsukamoto, Y.; Furusawa, S.; Ohkubo, Y.; Takimoto, H.; Murata, R.; Hosono, M.; Hikichi, N.; Sasaki, K. Characterization of a Rana catesbeiana lectin-resistant mutant of leukemia P388 cells. Cancer Res. 1994, 54, 928–934. [Google Scholar] [PubMed]
- Irie, M.; Nitta, K.; Nonaka, T. Biochemistry of frog ribonucleases. Cell. Mol. Life Sci. 1998, 54, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.D.; Huang, H.C.; Chan, H.J.; Kuo, S.J. Large-scale preparation of a ribonuclease from Rana catesbeiana (bullfrog) oocytes and characterization of its specific cytotoxic activity against tumor cells. Protein Expr. Purif. 1996, 7, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Hosono, M.; Miura, Y.; Sugawara, S.; Kariya, Y.; Hakomori, S.; Nitta, K. Involvement of ER stress in apoptosis induced by sialic acid-binding lectin (leczyme) from bullfrog eggs. Int. J. Oncol. 2013, 43, 1799–1808. [Google Scholar] [CrossRef] [Green Version]
- Tatsuta, T.; Hosono, M.; Ogawa, Y.; Inage, K.; Sugawara, S.; Nitta, K. Downregulation of Hsp70 inhibits apoptosis induced by sialic acid-binding lectin (leczyme). Oncol. Rep. 2014, 31, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Osborne, C.K.; Wakeling, A.; Nicholson, R.I. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br. J. Cancer 2004, 90 (Suppl. 1), S2–S6. [Google Scholar] [CrossRef]
- Petrovski, G.; Zahuczky, G.; Katona, K.; Vereb, G.; Martinet, W.; Nemes, Z.; Bursch, W.; Fésüs, L. Clearance of dying autophagic cells of different origin by professional and non-professional phagocytes. Cell Death Differ. 2007, 14, 1117–1128. [Google Scholar] [CrossRef] [Green Version]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-H.; Wei, C.-W.; Wang, J.-J.; Chiou, C.-T. Rana catesbeiana ribonuclease inhibits Japanese encephalitis virus (JEV) replication and enhances apoptosis of JEV-infected BHK-21 cells. Antiviral Res. 2011, 89, 193–198. [Google Scholar] [CrossRef]
- McLaughlin, M.B.; Jialal, I. Calcitonin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Jensen, L.E.; Thiel, S.; Petersen, T.E.; Jensenius, J.C. A rainbow trout lectin with multimeric structure. Comp. Biochem. Physiol. B, Biochem. Mol. Biol. 1997, 116, 385–390. [Google Scholar] [CrossRef]
- Ottinger, C.A.; Johnson, S.C.; Ewart, K.V.; Brown, L.L.; Ross, N.W. Enhancement of anti-Aeromonas salmonicida activity in Atlantic salmon (Salmo salar) macrophages by a mannose-binding lectin. Comp. Biochem. Physiol. C, Pharmacol. Toxicol. Endocrinol. 1999, 123, 53–59. [Google Scholar] [CrossRef]
- Dong, C.-H.; Yang, S.-T.; Yang, Z.-A.; Zhang, L.; Gui, J.-F. A C-type lectin associated and translocated with cortical granules during oocyte maturation and egg fertilization in fish. Dev. Biol. 2004, 265, 341–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasumi, S.; Yang, W.-J.; Usami, T.; Tsutsui, S.; Ohira, T.; Kawazoe, I.; Wilder, M.N.; Aida, K.; Suzuki, Y. Characteristics and primary structure of a galectin in the skin mucus of the Japanese eel, Anguilla japonica. Dev. Comp. Immunol. 2004, 28, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Sinha, B.; Bhattacharya, B.; Chatterjee, B.; Mazumder, S. Characterization of a galactose binding serum lectin from the Indian catfish, Clarias batrachus: possible involvement of fish lectins in differential recognition of pathogens. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2005, 141, 76–84. [Google Scholar] [CrossRef]
- Pan, S.; Tang, J.; Gu, X. Isolation and characterization of a novel fucose-binding lectin from the gill of bighead carp (Aristichthys nobilis). Vet. Immunol. Immunopathol. 2010, 133, 154–164. [Google Scholar] [CrossRef]
- Hosono, M.; Kawauchi, H.; Nitta, K.; Takayanagi, Y.; Shiokawa, H.; Mineki, R.; Murayama, K. Purification and characterization of Silurus asotus (catfish) roe lectin. Biol. Pharm. Bull. 1993, 16, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Suryadinata, R.; Sadowski, M.; Sarcevic, B. Control of cell cycle progression by phosphorylation of cyclin-dependent kinase (CDK) substrates. Biosci. Rep. 2010, 30, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Gartel, A.L.; Radhakrishnan, S.K. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005, 65, 3980–3985. [Google Scholar] [CrossRef] [Green Version]
- Shailesh, H.; Zakaria, Z.Z.; Baiocchi, R.; Sif, S. Protein arginine methyltransferase 5 (PRMT5) dysregulation in cancer. Oncotarget 2018, 9, 36705–36718. [Google Scholar] [CrossRef] [Green Version]
- Irwin, M.; Marin, M.C.; Phillips, A.C.; Seelan, R.S.; Smith, D.I.; Liu, W.; Flores, E.R.; Tsai, K.Y.; Jacks, T.; Vousden, K.H.; et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 2000, 407, 645–648. [Google Scholar] [CrossRef]
- Wu, X.; Levine, A.J. p53 and E2F-1 cooperate to mediate apoptosis. Proc. Natl. Acad. Sci. USA 1994, 91, 3602–3606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cammarata, M.; Vazzana, M.; Chinnici, C.; Parrinello, N. A serum fucolectin isolated and characterized from sea bass Dicentrarchus labrax. Biochim. Biophys. Acta 2001, 1528, 196–202. [Google Scholar] [CrossRef]
- Vasconcelos, I.M.; Oliveira, J.T.A. Antinutritional properties of plant lectins. Toxicon 2004, 44, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.K.; Ng, T.B. Lectins: production and practical applications. Appl. Microbiol. Biotechnol. 2011, 89, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, C.; Hsu, C.-H.; Wen, Z.-H.; Lin, C.-S. Anticancer drugs discovery and development from marine organism. Curr. Top. Med. Chem. 2009, 9, 1536–1545. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=Breast+Cancer&term=sunitinib&cntry=&state=&city=&dist= (accessed on 16 August 2019).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
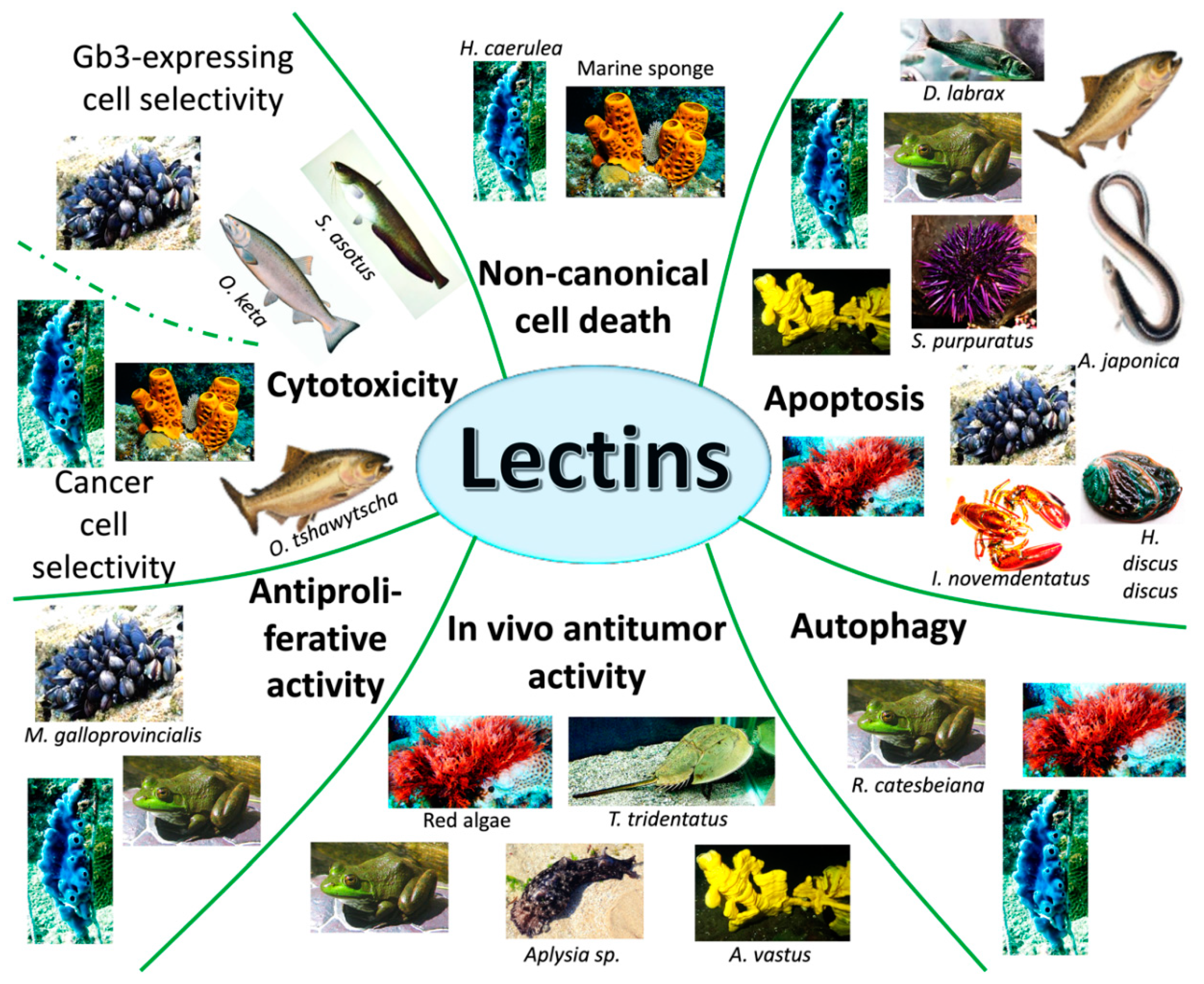{kind=link}
| Origin | Lectin | Recognition Glycans | Cell Lines | In Vivo Model | Treatment Times and Doses | Cellular and Molecular Target | Ref. |
|---|---|---|---|---|---|---|---|
| Acanthophora spicifera | Crude lectins fraction | MCF-7 | 24 h (100 µg/mL) | % of cell growth inhibition in A. spicifera: 1.78 µg/mL (MCF-7); 4.27 µg/mL (HeLa) | [35] | ||
| Acrocystis nana | HeLa | % of cell growth inhibition in A. nana: 9.10 µg/mL (MCF-7); 47.68 (HeLa) | |||||
| Eucheuma serra | Eucheuma serra agglutinin (ESA) | Mannose | Colon26 | Colon-26 cells injected in BALB/c mice | 48 h (0–1000 µg/mL) | ↓viability at concentrations > 8 µg/mL | [36] |
| 48 h (50 µg/mL) | % of AnnexinV+/propidium iodide−: 31.4% | ||||||
| ↑caspase-3 activity | |||||||
| 400 mg /200 mL PBS every 3 days up to 15 days (intravenously injection) | ↓tumor volume | ||||||
| TUNEL-positive cells in tumor | |||||||
| Eucheuma serra | Eucheuma serra agglutinin (ESA) | Mannose | OST | 24 h (10–50 µg/mL) | Cell viability (50 µg/mL): 41.7 ± 12.3% (LM8); 54.7 ± 11.4% (OST) | [37] | |
| LM8 | 3–4 h (50 µg/mL) | AnnexinV+/propidium iodide− (3 h): 68.2% (LM8); 74.8% (OST) AnnexinV+/propidium iodide− (24 h): 24.1% (OST); 68.8% (LM8) | |||||
| 16 h (50 µg/mL) | OST: ↑caspase-3 activity (2.3-fold increase) | ||||||
| PEGylated vesicles with immobilized ESA (EPV) | 24 h (1–5 µg/mL of ESA delivered by EPV) | Cell viability (1 µg/mL): ∼50% (OST) | |||||
| Eucheuma serra | Eucheuma serra agglutinin (ESA) | Mannose | Colo201 | 72 h (0.05–150 µg/mL) | ↓Viability at concentrations > 1.2 µg/mL (cancer cells) | [38] | |
| HeLa | No cytotoxicity at 10 µg/mL (MCF10-2A) | ||||||
| MCF-7 | |||||||
| 24 h (64 µg/mL) | DNA degradation (Colo201) | ||||||
| MCF10-2A | 16 h (10.8 µg/mL) | ↑caspase-3 activity (Colo201) | |||||
| Eucheuma serra | Span 80 vesicles containing immobilized ESA (EV) Span 80 vesicles containing DSPE-PEG2000 and immobilized ESA (EPV) Span 80 vesicles containing DSPE-PEG2000, immobilized ESA and entrapped ESA (EEPV) | Mannose | Colo201 | 0–24 h (54 µg/mL of ESA delivered by EV) | Cell viability (24 h): 17.2% (Colo201); no effect (MCF10-2A) | [39] | |
| MCF-7 | |||||||
| Colon26 | 8 h (54 µg/mL of ESA delivered by EV) | DNA fragmentation in Colo201 and MCF-7 | |||||
| MCF10-2A | |||||||
| Colo201 cells transplanted in Balb/c-nu/nu mice | EPVs (containing 2.0 µg/mL of ESA) or EEPVs (containing 2.5 µg/mL of ESA) (0.01 mL/g b.w.) injected every 3 days up to 15 days | ↓tumor volume (9th day): 51.1% (EEPV); 58.0% (EPV) | |||||
| 3 days after EPVs (0.01 mL/g b. w.) injection | TUNEL-positive cells around the blood vessels | ||||||
| Gloiocladia repens | Crude lectins fraction | MCF-7 | 24 h (100 µg/mL) | % of cell growth inhibition in G. repens: 14.19 µg/mL (HeLa); 28.54 µg/mL (MCF-7) | [35] | ||
| Helminthora divaricata | HeLa | % of cell growth inhibition in H. divaricata: HeLa cells: 3.63 µg/mL (HeLa); 12.25 µg/mL (MCF-7) | |||||
| Microcystis viridis | Recombinant Microcystis viridis lectin (R-MVL) | Mannose | HT-29 | 72 h (2–64 µg/mL) | IC50 1: 40.20 µg/mL (SCG-7904); 42.67 µg/mL (HepG2); 49.87 µg/mL (HT-29); 53.40 µg/mL (SKOV3) | [40] | |
| HepG2 | |||||||
| SKOV3 | |||||||
| SCG-7904 | |||||||
| Nitophylium punctatum | Crude lectins fraction | MCF-7 | 24 h (100 µg/mL) | % of cell growth inhibition: 2.97 (HeLa); 15.53 (MCF-7) | [35] | ||
| HeLa | |||||||
| Solieria filiformis | Solieria filiformis lectin (SfL) (mixture of isoforms 1 and 2) | Mannopentose | MCF-7 | 24 h (0–500 µg/mL) | [41] | ||
| HDA | |||||||
| 24 h (125 µg/mL) | AnnexinV+/propidium iodide−: 25.07%; AnnexinV+/propidium iodide+: 35.16% | ||||||
| ↓Bcl-2; ↑Bax, ↑caspase-3, ↑caspase-8, ↑caspase-9 | |||||||
| Ulva pertusa | Adenovirus-Ulva pertusa lectin 1 (Ad-UPL1) | N-acetyl d-glucosamine | Huh7 | 48 h (100 MOI 2 Ad-UPL1 ± 10 µM U0126 3) | Cell viability: ∼50% (Ad-UPL1 + U0126, Huh7); ∼90% (Ad-UPL1, BEL-7404 or Huh7) | [42] | |
| BEL-7404 | 48 h (50–100 MOI) | Huh-7: ↑pERK1/2, p-p38; ↓Akt | |||||
| BEL-7404: ↑pERK1/2; ↓Akt | |||||||
| Huh-7: ↓Beclin-1; ↑LC3-II | |||||||
| BEL-7404: ↑Beclin-1; ↓LC3-II |
| Origin | Lectin | Recognition Glycans | Cell Lines | In vivo Model | Treatment Times and Doses | Cellular and Molecular Target | Ref. |
|---|---|---|---|---|---|---|---|
| Arthropoda and Mollusca | |||||||
| Aplysia kurodai eggs | Aplysia kurodai egg lectin (AKL) | Galactose | Brine shrimp nauplii | 24 h (2–32 µg/mL) | Mortality (32 µg/mL): 33.33% (PnL); 63.33% (AKL) | [52] | |
| Crenomytilus grayanus | Crenomytilus grayanus lectin (CGL) | Galactose-lactosylceramide | MCF-7 | 24 h (50–200 µg/mL) | Cell viability: 33% at 200 µg/mL | [53] | |
| Crenomytilus grayanus | Crenomytilus grayanus lectin (CGL) | Galactose-lactosylceramide | Raji | 48 h (0–100 µg/mL) | IC50: 6.81 ± 0.83 µg/mL (Raji); unaffected (K562) | [54] | |
| K562 | 24 h (10 µg/mL CGL + 100 mM glucose, lactose, melibiose, raffinose or galactose) | Cell viability (Raji): ∼55% (CGL + Lactose); ∼70% (CGL + Glucose); ∼100% (CGL + Melbiose, raffinose or galactose) | |||||
| 24 h (2.5–20 µg/mL) | ↑% of cells in sub-G1 and G2/M phases and ↓G1 and S phases | ||||||
| 24 h (2.5–20 µg/mL) | AnnexinV+/propidium iodide (5 µg/mL): ∼70% | ||||||
| 24 h (2.5–10 µg/mL) | ↑ caspase-9, caspase-3 and PARP cleavage | ||||||
| Haliotis discus discus | Oncolytic vaccinia virus (oncoVV)-Haliotis discus discus sialic acid-binding lectin (HddSBL) | Sialic acid | C6 | C6 tumor-bearing athymic BALB/c nude mice | 60 days (107 pfu 1/twice) | Mice survival: oncoVV-HddSBL > onco-VV | [55] |
| 15 days (107 pfu) | IL-2 secretion: oncoVV-HddSBL < onco-VV | ||||||
| 24 h (5 MOI) | mRNA IL-2: oncoVV-HddSBL < onco-VV | ||||||
| NF-kB and AP-1 activity: oncoVV-HddSBL > onco-VV | |||||||
| IFIT2, IFIT3; DDX58: oncoVV-HddSBL < onco-VV | |||||||
| 2–36 h (2 MOI) | OncoVV-HddSBL replication > onco-VV | ||||||
| Haliotis discus discus | Adenovirus (Ad.FLAG)-Haliotis discus discus sialic acid binding lectin (Ad.FLAG-HddSBL) | Sialic acid | Hep3B | 96 h (1–20 MOI) | Cell viability (20 MOI): ∼40% (Hep3B); ∼50% (A549); ∼60% (H1299); ∼80% (SW400) | [56] | |
| A549 | 48 h (20 MOI) | AnnexinV+/propidium iodide (Hep3B): 19.8% (Ad.FLAG-HddSBL) vs 4.79% (Ad.FLAG) | |||||
| H1299 | ↓Bcl-2; | ||||||
| SW480 | |||||||
| Haliotis discus discus | Haliotis discus discus sialic acid binding lectin (HddSBL) + coxsackie-adenovirus receptor (sCAR-HddSBL) | Sialic acid | K562/adr | 48 h (5–30 MOI Ad-EGFP + 10 µg/mL sCAR-HddSBL) | Viral infection and replication: 13% sCAR-HddSBL vs 3.19% Ad-EGFP (K562/ADR); 48.6% sCAR-HddSBL vs 23.1% Ad-EGFP (U87MG) | [57] | |
| U87MG | 96 h (8.2 MOI Ad-DlFBL + 10.6–31.8 µg/mL sCAR-HddSBL) | Cell viability (U87MG): ∼40% (Ad-DlFBL); ∼90% (31.8 µg/mL Ad-DlFBL-sCAR-HddSBL) | |||||
| 48 h (8.2 MOI Ad-DlFBL + 31.8 µg/mL sCAR-HddSBL) | U87MG: AnnexinV+/propidium iodide- 10.2% (Ad-DlFBL-sCAR-DlFBL) vs 7.91% (Ad-DlFBL) | ||||||
| 48 h (8.2 MOI Ad-DlFBL + 31.8 µg/mL sCAR-HddSBL) | U87MG: ↑pERK (Ad-DlFBL-sCAR-HddSBL); ↑E2F1 (sCAR-HddSBL; Ad-DlFBL-sCAR-HddSBL) | ||||||
| Ibacus novemdentatus | N-acetyl sugar-binding lectin (iNol) | N-acetylated glycan | MCF-7 | 48 h (0–100 µg/mL) | IC50: 12.5 µg/mL (Caco2); 25 µg/mL (HeLa); 50 µg/mL (MCF-7); 100 µg/mL (TD47D) | [58] | |
| T47D | 24 h (0–100 µg/mL) | HeLa: ↑caspase-9 | |||||
| HeLa | 12–48h (100 µg/mL) | HeLa: ↑caspase-3 activity | |||||
| Caco2 | 24 h (100 µg/mL) | HeLa: ↑DNA degradation; chromatin condensation | |||||
| Mytilus galloprovincialis | 𝜶-d-galactose-binding lectin (MytiLec) | Galactose-lactosylceramide | Raji | 24 h (0-50 µg/mL) | Cell viability (50 µg/mL): ∼40% (Raji); ∼100% (K562) | [59] | |
| K562 | 24 h (20 µg/mL MytiLec + 100 mM Sucrose, Lactose or Melbiose) | Cell viability (Raji): ∼40% (MytiLec + Sucrose or Lactose); ∼100% (MytiLec + Melbiose) | |||||
| Mytilus galloprovincialis | 𝜶-d-galactose-binding lectin (MytiLec) | Galactose-lactosylceramide | Ramos | 24 h (0.5–50 µg/mL) | Cell viability (50 µg/mL): ∼45% (Raji); ∼100% (K562) | [60] | |
| K562 | 12–24 h (0.5–50 µg/mL) | ↑pMEK, pERK and p21; ↓CDK6, ↓cyclinD3 | |||||
| 12–24 h (20 µg/mL) | ↑pJNK, ↑pp38, ↑pERK | ||||||
| 12–24 h (20 µg/mL) | ↑caspase-9, ↑caspase-3, ↑TNF𝜶 | ||||||
| 12–24 h (20 µg/mL + 10 µM U0126- pMEK inhibitor) | ↑caspase-9, ↑caspase-3 | ||||||
| Perinereis nuntia | Perinereis nuntia lectin (PnL) | Galactose | Brine shrimp nauplii | 24 h (2–32 µg/mL) | Mortality (32 µg/mL): 33.33% | [52] | |
| Strongylocentrotus purpuratus | Adenovirus FLAG (Ad.FLAG)-Strongylocentrotus purpuratus rhamnose-binding lectin (SpRBL) | Rhamnose | Hep3B | 48–96 h (1–20 MOI) | Cell viability (20MOI; 96 h; Ad.FLAG-SpRBL): ∼20% (Hep3B); ∼30% (BEL-7404, A549); ∼40% (SW480) | [61] | |
| BEL-7404 | 48 h (20 MOI) | Hep3B: Annexin V+/propidium iodide-: 25.4% (Ad.FLAG-SpRBL) vs 1.35% (Ad.FLAG) | |||||
| A549 | Hep3B: = cleaved PARP; ↓Bcl-2, ↓XIAP | ||||||
| SW480 | 48 h (20 MOI) | Hep3B: ↓E2F-1 | |||||
| Tachypleus tridentatus | Oncolytic vaccinia virus (oncoVV)-Tachypleus tridentatus Lectin (TTL) | Rhamnose | MHCC97-H | MHCC97-H tumor-bearing athymic BALB/c nude mice | 44 days (107 pfu/twice) | ↓tumor volume | [55] |
| 36 h (5 MOI) | OncoVV-TTL replication > onco VV | ||||||
| BEL-7404 | 24 h (5 MOI) | ↑pERK (onco VV = oncoVV-TTL) | |||||
| MAVS, IFI16, IFNβ: ↑ (oncoVV); = (onco VV-TTL) | |||||||
| (5 MOI ± U0126) | U0126 ↓ oncoVV-TTL replication | ||||||
| Chordata | |||||||
| Didemnum ternatanum | Didemnum ternatanum lectin (DTL) | N-acetyl-D-glucosamine | HeLa in adhesion plates | 72 h (2.5 µg/mL) | Cell proliferation: ∼50% | [62] | |
| HeLa in soft agar | 2 weeks (2.5 µg/mL) | Colony formation: 7 ± 1 (control); 25 ± 2 (DTL in agar); 60 ± 4 (DTL in plates and in agar) | |||||
| Porifera | |||||||
| Aphrocallistes vastus | Oncolytic vaccinia virus (oncoVV)-Aphrocallistes vastus lectin (AVL) Adenovirus (Ad)-Aphrocallistes vastus lectin (AVL) | Galactose | HCT116 | 48–72 h (10–20 MOI Ad-AVL) | Cell Viability (20 MOI Ad-AVL; 72h): ∼40% (HCT116, U251); ∼50% (HT-29, MHCC97-H, BEL-7404) | [63] | |
| U251 | BEL-7404 or HCT116 tumor-bearing athymic BALB/c nude mice | 48-72 h (1-10 MOI Ad-AVL) | Cell Viability (5 MOI oncoVV-AVL; 72h): ∼20% (HCT116); ∼40% (U87, 4T1-LUC, BEL-7404) | ||||
| BEL-7404 | 2–36 h (5 MOI) | OncoVV-AVL replication > onco VV | |||||
| MHCC97-H | 24 h (2 MOI) | AnnexinV+/propidium iodide (HCT116): 6.49% (oncoVV-AVL) vs 1.26% (oncoVV) | |||||
| HT-29 | 24 h (2 MOI) on HCT116 | MDA5: no effect | |||||
| 4T1-LUC | ↓caspase-3 (oncoVV-AVL); ↓caspase-8, ↓Bax (oncoVV, oncoVV-AVL) | ||||||
| U87 | ↓NIK, pNF-𝜅B2 (oncoVV-AVL); ↑NIK (oncoVV) | ||||||
| ↑pERK (onco VV = oncoVV-TTL) | |||||||
| 24 h (5 MOI ± 10 µM U0126) | U0126 ↓ oncoVV-AVL replication | ||||||
| 25 (BEL-7404) or 35 (HCT116) days (107 pfu) | ↓tumor volume | ||||||
| Cinachyrella apion | Lactose-Binding Lectin (CaL) | Lactose | HeLa | 24-48 h (0.5 - 10 µg/mL) | Cell Viability (10 µg/mL; 24h): ∼50% (HeLa); ∼60% (PC3); ∼75% (3T3) | [64] | |
| PC3 | (10 - 20 µg/mL) | No cytotoxicity in peripheral blood cells | |||||
| 3T3 | 1 h (10 µg/mL) | No hemolysis in erythrocytes | |||||
| Erythrocytes and peripheral blood cells | 24 h (10 µg/mL) | Membrane blebbing and nuclear condensation | |||||
| 24 h (10 µg/mL ± 0.02 mM Z-VAD-FMK 2) | % of cells in S phase (HeLa): ∼40% (control); ∼ 50% (CaL + Z-VAD-FMK); 57.6% (CaL) | ||||||
| AnnexinV+/propidium iodide (HeLa): 3.84% (control); 15.5% (CaL + Z-VAD-FMK); 23.2% (CaL) | |||||||
| 6–24 h (10 µg/mL) | HeLa: ↑Bax, ↑pNF-κB (105 kDa), ↑JNK; =Bcl2, =pAKT; ↓pNFkB (50 kDa) | ||||||
| Cliona varians | Cliona varians lectin (CvL) | Galactose | Jurkat | 72 h (1–150 µg/mL) | IC50: 70 µg/mL (K562); 100 µg/mL (Jurkat); no effect on lymphocytes | [65] | |
| K562 | 24 h (1–150 µg/mL) | No effect (B16, 786-O, PC3) | |||||
| blood lymphocytes | 72 h (70 µg/mL) | K562: ↑subG1 (28% CvL vs 14.1% control) | |||||
| B16 | 72 h (50–70 µg/mL) | Apoptotic cells (K562; 70 µg/mL): 43% CvL vs 10% control | |||||
| 786-O | 72 h (70 µg/mL) | K562: 25.3% (Annexin V-/propidium iodide+); 60.4% (Annexin V+/propidium iodide+); | |||||
| PC3 | 72 h (50–70 µg/mL) | No increase in caspase-8, -9, and -3 activity | |||||
| 72 h (70 µg/mL) | Cathepsin B founded in cytoplasm and nucleus | ||||||
| 2 h (5 µM E-64) + 72 h (50-80 µg/mL CvL) | Cell viability (80 µg/mL, K562): ∼30% (CvL); ∼100% (CvL + E-64) | ||||||
| 72 h (50–70 µg/mL) | K562: ↑TNFR1, ↓NF-κB (p65 sub) | ||||||
| K562: ↑Bax, ↑Bcl-2 | |||||||
| K562: ↑p21, ↓pRb | |||||||
| Haliclona caerulea | Halilectin-3 (H3) | Mucin | MCF7 | 6-48 h (7.81–500 µg/mL) | Cell viability (250 µg/mL): 42% (MCF7); 75% (HDF); IC50: 100 μg/ml (MCF7) | [66] | |
| HDF | 24 h (100 µg/mL) | ↑% of cells in the G1 phase | |||||
| 24-48 h (100 µg/mL) | ↑early apoptosis cells: 46% (24h); 55.4% (48h) | ||||||
| 6-24 h (100 µg/mL) | 24h: ↑caspase-3, ↑caspase-8, ↑caspase-9, ↑Bax, ↑TP53; ↓Bcl-2 | ||||||
| 8-24 h (100 µg/mL) | ↑agglutination of MCF-7 cells; ↓cell adhesion | ||||||
| 6 h (100 µg/mL) | ↑LC3; ↓BECLIN-1 | ||||||
| ↑LC3II/LC3I | |||||||
| Autophagosoma vescicles | |||||||
| Haliclona cratera | Haliclona cratera Lectin (HCL) | Galactose, N-Acetyl-d-galactosamine, Lactose | HeLa | 48 h (0–40 µg/mL) | IC50: 9 µg/mL (HeLa); 11 µg/mL (FemX) | [67] | |
| FemX | 72 h (0–15 µg/mL) | Lymphocytes: no toxicity | |||||
| human T-lymphocytes | 2 h (5 µg/mL PHA) + 72 h (0–15 µg/mL HCL) | Lymphocytes: 23% (PHA + HCL 15 µg/mL) | |||||
| Halichondria okadai | 18 kDa Lectin (HOL-18) | N-acetylhexosamine | Jurkat | 24 h (1–25 µg/mL) | Cell Viability (25 µg/mL): ∼30% (Jurkat); ∼60% (K562) | [68] | |
| K562 | 24 h (25 µg/mL HOL-18 ± 50 mM d-GlcNAc 3, d-GalNAc 4 or Mannose) | Cell Viability: ∼30% (Jurkat) - 50% (K562) (HOL-18 + Mannose); ∼80% (HOL-18 +d-GalNAc); ∼90% (HOL-18 + d-GlcNAc) | |||||
| Halichondria okadai | 18 kDa Lectin (HOL-18) | N-acetylhexosamine | HeLa | 48 h (6.25–100 µg/mL) | IC50: 40 µg/mL (HeLa); 52 µg/mL (MCF7); 63 µg/mL (T47D); no effect (Caco-2) | [69] | |
| MCF7 | 48 h (50 µg/mL HOL-18 + 20 mM Glucose, GlcNAc, Mannose, ManNAc 5) | Cell Viability: ∼45% (HOL-18 ± Glucose or Mannose); ∼75% (HOL-18 ± GlcNAC); ∼90% (HOL-18 ± ManNAC) | |||||
| T47D | 48 h (6.25–100 µg/mL) | HeLa: ↑pERK, ↑caspase-3 | |||||
| Caco2 | |||||||
| Origin | Lectin | Recognition Glycans | Cell Lines | In Vivo Model | Treatment Times and Doses | Cellular and Molecular Target | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Amphibians | ||||||||
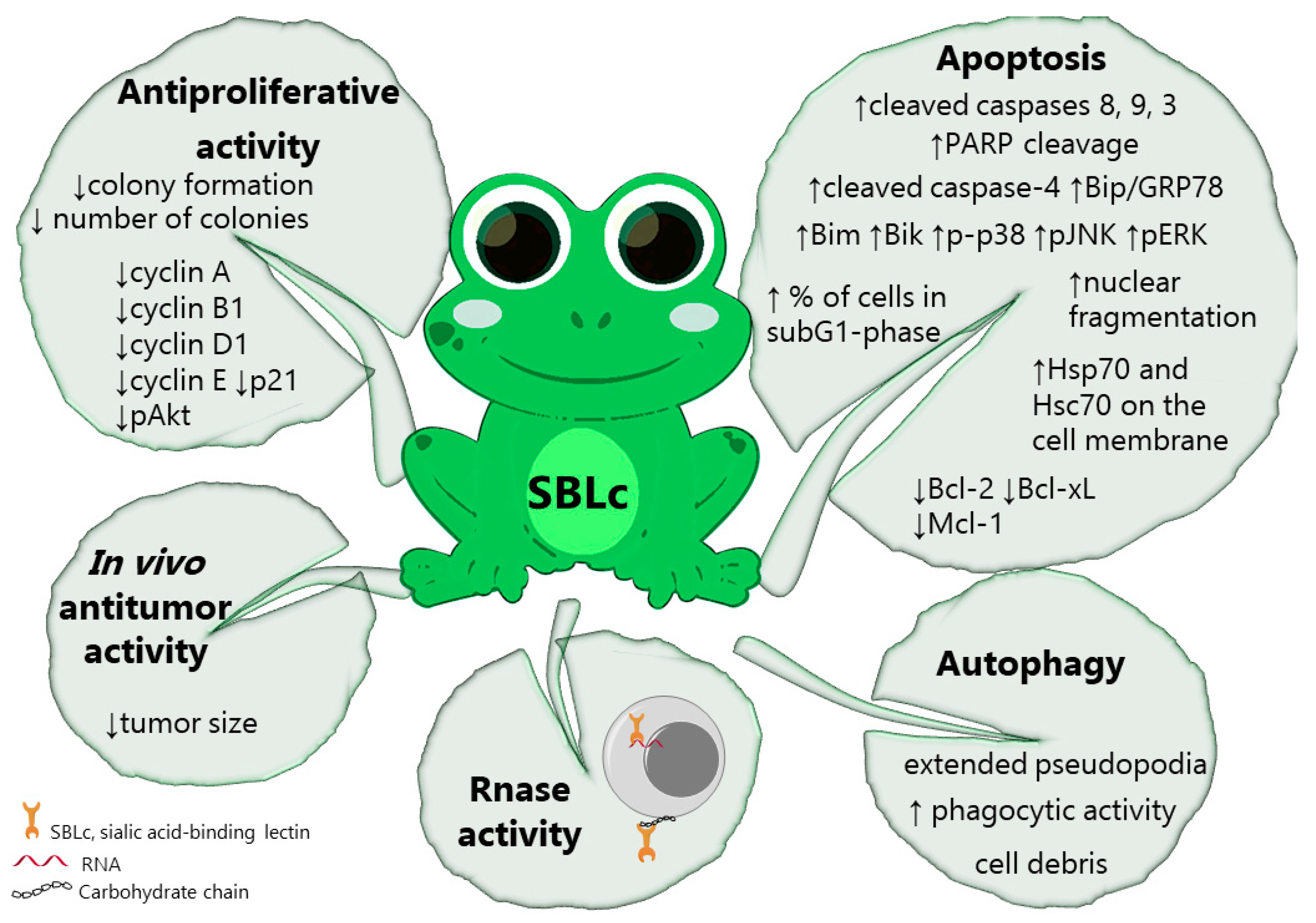
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | P388 | 48 h (0.1–5 µM) | IC50: 0.3 µM (EDC-ED SBLc); 1.0 µM (EDC-GM SBLc); 1.5 µM (EDC-TA SBLc; SBLc) | [106] | ||
| EDC-TA SBLc | ||||||||
| EDC-GM SBLc | ||||||||
| EDC-ED SBLc | ||||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | P388 | 48 h (0.1–5 µM) | GI50 1 (P388): 1.56 (SBLj); 6.25 µM (SBLc) | [107] | ||
| Rana japonica | Sialic acid-binding lectin (SBLj) | L1210 | GI1002 (L1210): 1.56 µM (SBLc and -j) | |||||
| Sarcoma 180-bearing ddY mice | A single SBLc injection (2.5–10 mg/kg) | IC50: 5 mg/kg (Sarcoma 180-bearing mice) after 45 days | ||||||
| MepII-bearing ddI mice | IC50: 10 mg/kg (MepII-bearing mice) after 45 days | |||||||
| Sarcoma 180-bearing ddY mice | Continuous SBLc injection (0.5–2 mg/kg) for 10 days | IC50: <0.5 mg/kg (Sarcoma 180-bearing mice) after 45 days | ||||||
| MepII-bearing ddI mice | IC50: 0.5 mg/kg (MepII-bearing mice) after 45 days | |||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | Jurkat | 48 h (2 µM) | 44% of cells in sub-G1 phase | [108] | ||
| 1–48 h (2 µM) | ↑cleaved caspase-8, -9, -3 | |||||||
| 3–48 h (2 µM) | ↑cleaved caspase-4, ↑Bip/GRP78 | |||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | P388 | 24 h (3 µM) | Cell viability: ∼20% (P388); ∼30% (K562); ∼40% (HL60); ∼80% (MCF-7); ∼100% (Daudi; Raji; NHDF; NHEM; NHEK) | [109] | ||
| K562 | ||||||||
| HL60 | 24 h (3, 20 µM) | No DNA fragmentation in Raji and NHDF cells | ||||||
| MCF-7 | DNA fragmentation in P388 and K562 cells | |||||||
| Daudi | 24 h (3 µM) | ↑caspase-8, ↑caspase-3 | ||||||
| Raji | 1h (1 µM) | ↑Hsp70 and Hsc70 on the cell membrane | ||||||
| NHDF | ||||||||
| NHEM | ||||||||
| NHEK | ||||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | H28 | 48 h (0.2–20 µM) of treatment and 12 days of posttreatment | Colony formation (5 µM): <5% (H28); 20% (MESO-4); <70% (MESO-1) | [110] | ||
| MESO-1 | Colony formation (5 µM): >90% (Met-5A) | |||||||
| MESO-4 | 24–72 h (5 µM) | Annexin V+ (72h): ∼5% (Met-5A); ∼15% (MESO-1 and -4); ∼50% (H28) | ||||||
| Met-5A | 6–48 h (5 µM) | H28: ↑caspase-8, ↑caspase-9, ↑caspase-3 | ||||||
| H28: ↑Bim, ↑Bik, ↑p-p38, ↑pJNK, ↑pERK | ||||||||
| 24 h (SBLc 5 µM ± TRAIL 2 ng/mL) on H28 | ↑cytotoxicity (∼30%) vs single treatment (∼70%) | |||||||
| ↑ Annexin V+ (∼50%) cells vs single treatment (∼50%) | ||||||||
| ↑mitochondrial membrane depolarization vs single treatments | ||||||||
| ↑caspase-8, -9, -3 protein expression vs single treatment | ||||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | MCF-7 | 72 h (2 µM SBLc) | Cell viability: 25.5% (MDA-MB-231); 30.4% (MCF-7); 65.3% (SK-BR-3) | [111] | ||
| SK-BR-3 | ↑p-p38 | |||||||
| MDA-MB-231 | ↑ caspase-3/7 activity | |||||||
| SBLc mutant lacking RNase activity (H103A) | 72 h (10 µM H103A) | Cell viability: 100% (MDA-MB-231) | ||||||
| 72 h (2 µM H103A) | No effect on pp38, PARP expression | |||||||
| No effect on caspase-3/7 | ||||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | H28 | 72 h (1–30 µM) | IC50: 0.46 µM (H28); 0.52 µM (H2452); 1.54 µM (MESO-4); 5.05 µM (MSTO); 5.51 µM (MESO-1); 52.22 (Met-5A) | [112] | ||
| MESO-1 | 72 h (1 µM) | ↑ % of cells in subG1-phase | ||||||
| MESO-4 | 72 h (1 µM) | ↓cyclin A, ↓cyclin B1, ↓cyclin D1, ↓cyclin E, ↓p21, ↓pAkt | ||||||
| H2452 | 72 h (1 µM SBLc + 20 µM pemetrexed or 40 µM cisplatin) | CI3 (H28): 0.05 (SBLc + pemetrexed); 0.47 (SBLc + cisplatin) | ||||||
| Met-5A | Annexin V+/propidium iodide- (H28): no difference vs SBLc treatment | |||||||
| Caspase-3/7 activity (H28): no difference vs SBLc treatment | ||||||||
| H28: ↑ % of cells in S- and subG1-phases (SBLc + pemetrexed); ↑ % of cells in S-, G2- and subG1-phases (SBLc + cisplatin) | ||||||||
| H28: ↓cyclin B1, ↓p21, ↓pAkt (SBLc + pemetrexed or cisplatin) | ||||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | NCI-H2452 | H2452 or MSTO injected in BALB/C nu/nu Slc | 24–72 h (H2452: 1 µM; MSTO: 0.4 µM) | Annexin V+ (72 h): 16.13% (H2452); 40.05% (MSTO) | [113] | |
| MSTO-211H | 6–72 h (H2452: 5 µM; MSTO 2 µM) | ↑nuclear fragmentation (72h): ∼2.5-fold (H2452); ∼4-fold (MSTO) | ||||||
| 1–72 h (H2452: 5 µM; MSTO 2 µM) | ↑activity and expression of caspase-9, -8, -3 | |||||||
| 72 h [(1 pM–1 µM SBLc) + (0.8 nM–800 µM pemetrexed)] | CI (H2452) < 1 at all combinations (SBLc + pemetrexed or SBLc + cisplatin) | |||||||
| 72 h [(1 pM–1 µM SBLc) + (0.1 nM–100 µM cisplatin)] | CI (MSTO) < 1 up to 1 µM SBLc + 1.5 µM pemetrexed or 10 µM cisplatin | |||||||
| Pemetrexed (100 ng/kg) on days 1–5 and 15–19 | ↓tumor size after 47 (H2452) days of treatment | |||||||
| SBLc (2.5 mg/kg) 2/week for 4 weeks | ↓tumor size after 36 (H2452) or 29 (MSTO) days of treatment | |||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | ZR-75-1 | 72 h (1–20 µM) | Cell viability (20 µM): 40% (MDA-MB-468); 45% (MCF-7); 46% (SK-BR-3); 51% (BT-474); 52% (MDA-MB-231); 69% (ZR-75-1); 85% (MCF10A) | [114] | ||
| BT474 | 72 h (1–10 µM) + 7–28 days in drug-free medium | ↓cell number (except for MCF10A) | ||||||
| MCF-7 | 72 h (10 µM) | ↓ number of colonies (except for MCF10A) | ||||||
| SK-BR-3 | 72–96 h (10 µM) | chromatin condensation and nuclear collapse (except in MCF10A) | ||||||
| MDA-MB-231 | ↑cleaved caspase-9 and PARP cleavage (except in MCF10A) | |||||||
| MDA-MB-468 | 72 h (10 µM) | ↑pp38, ↑pJNK (ZR-75-1); ↑pp38, ↓JNK (MCF-7) | ||||||
| MCF10A | ↓Bcl-2, ↓Bcl-xL, ↓Mcl-1 (MCF-7); ↓Bcl-2, ↑Bcl-xL; Mcl-1 (ZR-75-1) | |||||||
| ↓ER𝛂, ↓PgR, ↓HER2 (MCF-7); ↓ER𝛂, ↓HER2 (ZR-75-1) | ||||||||
| ↓ErbB family in each cancer cells | ||||||||
| 3–24 h (10 µM) | Triple negative cells: ↑pp38 (MDA-MB-231 and -468); ↓EGFR/HER1, ↓pAKT (only in MDA-MB-231 cells) | |||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | ZR-75-1 | 120 h (20 µg/mL) | Cell survival (72h): ∼10% (MCF-7); ∼30% (ZR-75-1) | [115] | ||
| MCF-7 | 72 h (20 µg/mL) | MCF-7, ZR-75-1: ↑caspase-3 activity | ||||||
| MCF-7: extended pseudopodia, increase in phagocytic activity, cell debris | ||||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | ZR-75-1 | 96 h (20 µg/mL) | Cell survival (120 h): <50% (MCF-7; ZR-75-1); >80% (MDA-MB-231; ZR-75-30) | [116] | ||
| MCF-7 | 72 h (20 µg/mL) | ↑ caspase-3 activity (MCF-7; ZR-75-1) | ||||||
| MDA-MB-231 | 96 h (0–40 µg/mL) | ↓ER, ↓Bcl-2 (MCF-7) | ||||||
| ZR-75-30 | ||||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | MCF-7 | 0–120 h (20 µg/mL) | Cell survival (120 h; SBLc): 13% (MCF-7); 31.3% (MCF-7/Bcl-xL) | [117] | ||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | Undifferentiated HL-60 | 120 h (2, 20 µg/mL) | Cell survival (120 h; 20 µM): 5.5% (undifferentiated) | [118] | ||
| retinoic acid-differentiated HL-60 | 5–7–9 days of differentiation + 120 h (2, 20 µg/mL) | Cell viability (120 h; 20 µM; differentiated cells): 65% (5 days); 82.5% (7 and 9 days) | ||||||
| 7 days of differentiation + 48, 96 h (20 µg/mL); 48, 96 h (20 µg/mL) | ↑caspase-9, -3 and cleaved PARP (undifferentiated cells) | |||||||
| ↑caspase-9 and -3 activity (undifferentiated cells) | ||||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | DBTRG | 0–96 h (20 µg/mL) | Cell inhibition rate (96 h): ∼10% (RG2); ∼25% (GBM8401); ∼45% (DBTRG; GBM8901) | [119] | ||
| GBM8901 | 0–96 h (2–50 µg/mL) | Cell inhibition rate (50 µg/mL; 96 h): ∼15% (RG2); ∼40% (DBTRG); ∼65% (GBM8901) | ||||||
| GBM8401 | 24, 72 h (50 µg/mL) | ↑% of cells in sub-G1-phase (~30%; DBTRG) | ||||||
| RG2 | 72 h (50 µg/mL) | ↑caspase-9 and -3 activity, not caspase-8 (DBTRG) | ||||||
| DBTRG cells subcutaneously injected in nude mice | a single injection (5 µg) | ↓tumor size after 18 days of treatment | ||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | HL60 | 120 h (20 µg/mL SBLc; 10 ng/mL IFN-𝛾) | Cell viability: SBLc + IFN-𝛾 < SBLc (MCF-7; SK-Hep-1); SBLc + IFN-𝛾 = SBLc (HL60) | [120] | ||
| MCF-7 | 48, 96 h (20 µg/mL SBLc; 10 ng/mL IFN-𝛾) | HL60: ↑ caspase-3, -8, and -9 activity (SBLc + IFN-𝛾 = SBLc) | ||||||
| SK-Hep-1 | MCF-7: ↑caspase-7 activity (SBLc + IFN-𝛾 > SBLc); | |||||||
| SK-Hep-1: caspase-3, -8, and 9 activity = control | ||||||||
| 48, 96 h (20 µg/mL SBLc + 10 ng/mL IFN-𝛾) | ↑cleaved caspase-3 and PARP (HL60); ↑ cleaved caspase-7 and PARP (MCF-7) | |||||||
| Rana catesbeiana | Sialic acid-binding lectin (SBLc) | Sialic acid | SK-Hep-1 | 0–96 h (20 µM SBLc + 10 ng/mL TNF-𝛼 or -𝛽) | Cell survival (96 h): ∼40% (J5); ∼50% (SK-Hep-1); ∼90% (HepG2) | [121] | ||
| J5 | 0–120 h (20 µM SBLc + 10 ng/mL IFN-𝛾) | Cell survival (120 h, SK-Hep-1): 13.3% (SBLc+IFN-𝛾) vs 64.7% (SBLc) | ||||||
| HepG2 | Cell survival (120 h, J5): 27.8% (SBLc+IFN-𝛾) vs 76.8% (SBLc) | |||||||
| BHK21 | Cell survival (120 h, HepG2): 64.2% (SBLc+IFN-𝛾) vs 93.9% (SBLc) | |||||||
| Cell survival (120 h, BHK21): 91.52% (SBLc+IFN-𝛾) vs 96.67% (SBLc) | ||||||||
| Rana catesbeiana | Tobacco-derived his-HR Recombinant hHscFv–RC-RNase protein | Sialic acid | SMMC7721 | 24 h (0.7–3.5 nM) | IC50: 2 nM (SMMC7721); 2.4 nM (HepG2); 4.8 nM (DV145) | [122] | ||
| HepG2 | Cell inhibition rate (3.5 nM): ∼15% (HL-7702) | |||||||
| DV145 | ||||||||
| HL-7702 | ||||||||
| Rana japonica | Sialic acid-binding lectin (SBLj) | Sialic acid | P388 | 48 h (0.1–5 µM) | GI501 (P388): 1.56 (SBLj) | [107] | ||
| L1210 | GI1001 (L1210): 1.56 (SBLj) | |||||||
| Fish | ||||||||
| Anguilla japonica | Adenovirus FLAG (Ad.FLAG) Anguilla japonica lectin 1 (AJL1) | β-galactoside | Hep3B | 48–120 h (50–100 MOI) | Cell viability (100MOI; 96h): ∼10% (SMMC-7721); ∼20% (Hep3B, BEL-7404, QSG-7701); ∼40% (Huh7); ∼60% (A549) | [123] | ||
| BEL-7404 | 48 h (50 MOI) | Hep3B: AnnexinV+/propidium iodide- 19.5% (Ad.FLAG-AJL1) vs 5.04% (Ad.FLAG) | ||||||
| Huh7 | Hep3B: ↑cleaved PARP, Bcl-XL; ↓ procaspase-9, Bcl-2, XIAP | |||||||
| SMMC7721 | 48 h (50–100 MOI) | Hep3B: ↑PMRT5; ↓E2F-1 | ||||||
| A549 | 48 h (50–100 MOI) | Hep3B: ↓ERK, ↓pERK, ↓p38 | ||||||
| QSG-7701 | ||||||||
| Aristichthys nobilis | Bighead carp gill lectin (GANL) | SMMC7721 | 24 h (0.5–64 µg/mL) | Cell inhibition rate (64 µg/mL): ∼0% (SMMC7721, BGC803); ∼20% (SKOV3, HepG2); ∼-30% (LoVo); ∼80% (HeLa) | [124] | |||
| HepG2 | 72 h (0.5–64 µg/mL) | Cell survival (16 µg/mL): ∼115% (splenocytes) | ||||||
| SKOV3 | ||||||||
| HeLa | ||||||||
| BGC803 | ||||||||
| LoVo | ||||||||
| BALB/c mice splenocytes | ||||||||
| Dicentrarchus labrax | Adenovirus FLAG (Ad.FLAG)-Dicentrarchus labrax fucose-binding lectin (DlFBL) | Fucose | Hep3B | 48–96 h (1–20 MOI) | Cell viability (20MOI; 96h; Ad.FLAG-DlFBL): ∼30% (Hep3B); ∼40% (BEL-7404, A549, SW480) | [61] | ||
| BEL-7404 | 48 h (20 MOI) | Hep3B: Annexin V+/propidium iodide- 21.5% (Ad.FLAG-DlFBL) vs 1.35% (Ad.FLAG) | ||||||
| A549 | Hep3B: = cleaved PARP; ↓Bcl-2, ↓XIAP | |||||||
| SW480 | Hep3B: ↓E2F-1 | |||||||
| Dicentrarchus labrax | Dicentrarchus labrax fucose-binding lectin (Ad-DlFBL) + coxsackie-adenovirus receptor (sCAR)-DlFBL | Fucose | K562/adr | 48 h (5–30 MOI Ad-EGFP + 10 µg/mL sCAR-DlFBL) | Viral infection and replication: K562/ADR: 20% sCAR-DlFBL vs 3.19% Ad-EGFP; U87MG: 40.6% sCAR-DlFBL vs 23.1% Ad-EGFP | [57] | ||
| U87MG | 96 h (8.2 MOI Ad-DlFBL + 14-42 µg/mL sCAR-DlFBL) | Cell viability (U87MG): ∼20% (42 µg/mL Ad-DlFBL-sCAR-DlFBL); ∼50% (Ad-DlFBL) | ||||||
| 48 h (8.2 MOI Ad-DlFBL + 42 µg/mL sCAR-DlFBL) | U87MG: AnnexinV+/propidium iodide- 4.87% (Ad-DlFBL-sCAR-DlFBL) vs 7.91% (Ad-DlFBL) | |||||||
| 48 h (8.2 MOI Ad-DlFBL + 42 µg/mL sCAR-DlFBL) | U87MG: ↑pERK (Ad-DlFBL-sCAR-DlFBL) | |||||||
| Oncorhynchus keta | l-rhamnose-binding lectin (CSEL) | Rhamnose | Caco-2 | 24 h (1–100 µg/mL) | Cell viability: ~35% (Caco-2); no effects on DLD-1 or HCT-15 | [125] | ||
| DLD-1 | 24 h (1–100 µg/mL CSEL ± 0.1 M l-rhamnose or 2 µM PPMP) | Cell viability (Caco-2): ~35% (CSEL); ~90% (CSEL ± L-rhamnose or PPMP) | ||||||
| HCT-15 | 24 h (50–100 µg/mL) | DNA fragmentation in Caco-2 | ||||||
| Annexin V+/propidium iodide− (100 µg/mL; Caco-2): 32.8% (CSEL) vs 3.1% (control) | ||||||||
| Oncorhynchus tshawytscha | Rhamnose-binding roe chinook salmon lectin (CSRL) | Rhamnose | MCF-7 | 24–48 h (3.9–250 μM) | IC50 (24–48 h): 93–45 µM (HepG2); 220–68 µM (MCF-7); | [126] | ||
| Hep G2 | 48 h (68 µM) | WRL68: no effect | ||||||
| WRL68 | 24 h (0.625–20 µM) | NO production at 0.62 µM (at 20 µM CSRL) | ||||||
| Mouse peritoneal macrophages | ||||||||
| Silurus asotus | Rhamnose-binding lectin (SAL) | Rhamnose | Raji | 5–30 min (2.5–10 μg/mL) | Raji (30 min; 10 µg/mL): ↑ AnnexinV+/propidium iodide- (16.7% SAL vs 4.31% control) and AnnexinV+/ propidium iodide+ (77.25% SAL vs 4.79% control) | [127] | ||
| K562 | Raji (30 min; 10 µg/mL): ↑ 30-fold shrunken cell population | |||||||
| K562/DXR | K562; K562/DXR: no effects | |||||||
| 30 min (10 μg/mL SAL + 4 µM CsA) | Raji: ↓ necrotic cells (58.03% SAL + CsA vs 77.92% SAL) | |||||||
| Silurus asotus | Rhamnose-binding lectin (SAL) | Rhamnose | Raji | 24–120 h (0–100 μg/mL) | Cell viability: no effects | [128] | ||
| 24–48 h (100 µg/mL) | Cell proliferation: block at 50 µg/mL | |||||||
| 72 h (100 µg/mL) + 48 h SAL-free medium | Cell proliferation: restored | |||||||
| 24 h (100 µg/mL) | ↑% of cells in G0/G1-phase (20%); ↓% of cells in S-phase (20%) | |||||||
| 12–24 h (100 µg/mL SAL ± 20 mM Saccharide) | ↓ CDK4, C-MYC (40%), CCND3 (30%); = CDK2; ↑ p21 (130%), p27 (70%); + saccharide reverts effects (except for p27) | |||||||
| 12–24 h (100 µg/mL) | ↓CDK4, ↓c-Myc, ↓cyclin D3, ↑ p21, ↑p27 | |||||||
| 0.5–24 h (100 µg/mL) | ↑ GTP-Ras, pMEK, pERK; = pp38 and cJNK | |||||||
| (100 µg/mL) in A4GALT 4siRNA Raji cells | ↓pMEK, ↓pERK induced by SAL | |||||||
| 2 h (10 µM U0126) ± 12 h (100 µg/mL SAL) | ↓p21, ↓pERK induced by SAL (cell-cycle arrest depending on Ras-MEK-ERK pathway) | |||||||
| ↑cell proliferation rate | ||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catanzaro, E.; Calcabrini, C.; Bishayee, A.; Fimognari, C. Antitumor Potential of Marine and Freshwater Lectins. Mar. Drugs 2020, 18, 11. https://doi.org/10.3390/md18010011
Catanzaro E, Calcabrini C, Bishayee A, Fimognari C. Antitumor Potential of Marine and Freshwater Lectins. Marine Drugs. 2020; 18(1):11. https://doi.org/10.3390/md18010011
Chicago/Turabian StyleCatanzaro, Elena, Cinzia Calcabrini, Anupam Bishayee, and Carmela Fimognari. 2020. "Antitumor Potential of Marine and Freshwater Lectins" Marine Drugs 18, no. 1: 11. https://doi.org/10.3390/md18010011
APA StyleCatanzaro, E., Calcabrini, C., Bishayee, A., & Fimognari, C. (2020). Antitumor Potential of Marine and Freshwater Lectins. Marine Drugs, 18(1), 11. https://doi.org/10.3390/md18010011